

Физикохимия поверхности и защита материалов, 2019, T. 55, № 1, стр. 48-58
Фотоэлектрохимическая активность в видимой области спектра наноразмерных пленок диоксида титана, легированного висмутом и свинцом
В. А. Гринберг 1, *, В. В. Емец 1, Н. А. Майорова 1, Д. А. Маслов 2, А. А. Аверин 1, С. Н. Поляков 2, 3, 4, С. П. Молчанов 2, И. С. Левин 2, М. В. Цодиков 2
1 Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский просп., 31, Россия
2 Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Ленинский просп., 29, Россия
3 Технологический институт сверхтвердых и новых углеродных материалов
142190 Москваг., Троицк, ул. Центральная, 7а, Россия
4 Московский физико-технический институт (государственный университет),
141701 Московская обл., г. Долгопрудный, Институтский пер., 9, Россия
* E-mail: vitgreen@mail.ru
Поступила в редакцию 08.02.2018
После доработки 19.04.2018
Принята к публикации 24.05.2018
Аннотация
На основе золь-гель синтеза разработан метод получения пленочных покрытий из диоксида титана, легированного ионами висмута (Bi3+) и свинца (Pb2+), введенных как по отдельности, так и совместно. По данным рентгенофазового анализа синтезированные пленки представляют собой однофазную систему диоксида титана в модификации анатаза. Показано, что легирование диоксида титана ионами висмута и свинца приводит к сдвигу максимума поглощения в видимую область спектра электромагнитного излучения, при этом наибольший сдвиг наблюдается для образца, содержащего по 2.5 мас. % висмута и свинца. Полученные покрытия исследованы в качестве катализаторов фотоэлектроокисления метанола, муравьиной кислоты и фенола. Показано, что наибольший каталитический эффект наблюдается на образцах, содержащих одновременно и висмут, и свинец, однако наибольшее влияние на скорость окисления субстратов оказывает легирование диоксида титана висмутом. Высказано предположение, что возможность осуществления фотоэлектрохимического окисления указанных модельных систем видимым светом связана со снижением ширины запрещенной зоны легированного диоксида титана до 2.7 эВ.
ВВЕДЕНИЕ
Диоксид титана – твердое белое вещество, широко применявшееся в производстве лакокрасочных материалов с начала 20-го века [1]. Фудзишима и др. первыми использовали n-TiO2 для фотоэлектрохимического разложения воды при освещении ультрафиолетовым светом [2]. В настоящее время наноматериалы на основе диоксида титана интенсивно применяются в фотовольтаических устройствах (цветосенсибилизированные солнечные ячейки) [3–8], сенсорах [9], а также в таких областях как фотоэлектрокаталитическое получение водорода из воды [10–17], деградация загрязнений окружающей среды [18–25], селективное превращение органических соединений [26, 27], восстановление диоксида углерода с целью получения топлив [11, 28–34] и др. Реакции фотоэлектроокисления органических соединений на электродах из n-TiO2 (в модификации анатаза) исследуются уже более 30 лет [35–49]. Однако ширина запрещенной зоны анатаза (~3.2 эВ) обеспечивает его активность только в УФ области спектра, составляющей всего 3–5% от всего спектра солнечного света [46–49], что оставляет неиспользованной почти половину всей солнечной энергии. Повышение эффективности реакций фотокаталитического и фотоэлектрокаталитического окисления на n-TiO2 связывают, в первую очередь, с увеличением поглощения фотокатализатором видимого света (λ = 400 – 780 нм) для более полного использования солнечной радиации, а также с уменьшением скорости рекомбинации фотогенерируемых пар электрон−дырка. Для решения этих задач в течение последних десятилетий разработаны второе (легированные металлами) и третье (легированные неметаллами) поколения наноматериалов на основе диоксида титана для применения в различных областях, связанных с фотокатализом и фотоэлектрохимией [50–60]. Так, в работе [51] была предпринята попытка провести фотоэлектрохимическое окисление метиленового синего на фотоаноде из легированного железом диоксида титана, поглощающего свет в области частот видимого спектра [52]. Эффективность преобразования энергии фотона в ток (IPCE) на таком электроде составила 0.5% (λ = 450 нм). При этом авторы другого исследования [53] не наблюдали фотоэлектрохимического окисления некоторых модельных систем (метанол и др.) на легированных железом фотоанодах, несмотря на заметный сдвиг поглощения в видимую область спектра. По их мнению, отсутствие фотоэлектрокаталитической активности в этом случае могло быть связано с усилением рекомбинационных процессов.
В последнее время было показано, что легирование n-TiO2 такими металлами как висмут, кадмий, хром, свинец также сдвигает область поглощения света полупроводником в видимую область спектра и увеличивает его фотокаталитическую активность в реакциях окисления органических соединений [54–57]. Предметом изучения в настоящей работе был выбран легированный ионами висмута и свинца диоксид титана, синтезированный с использованием модифицированного золь-гель метода как в виде высокодисперсных порошков, так и пленочных покрытий. Исследовано поглощение света фотоанодами с синтезированными пленками и их фотоэлектрокаталитическая активность в реакции окисления модельных соединений (метанола, муравьиной кислоты и фенола) в видимой области спектра.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для изготовления фотоанодов использовали стекла марки ТСО 22–15 с электропроводящим покрытием из фтор-стабилизированного диоксида олова (SnO2 : F; Solaronix; удельное поверхностное сопротивление ~15 Ом). Стекла предварительно были очищены выдержкой в растворе бихромата калия и серной кислоты, промыты дистиллированной водой и ацетоном и высушены при 100°С.
Металлоксидные системы получали с использованием алкоксо-метода. Для этого в 2.1 см3н‑бутилата титана Ti(OС4Н9)4 добавляли эквимольное количество ацетилацетона (acacH) в качестве стабилизирующей добавки и перемешивали полученный раствор в течение 30 минут (Раствор 1). Для введения легирующих добавок пентагидрат нитрата висмута Bi(NO3)3 ⋅ 5H2O (0.031 г) и нитрат свинца Pb(NO3)3 (0.021 г), как по отдельности, так и совместно, растворяли в смеси ледяной уксусной кислоты, воды и этанола, взятых в мольном соотношении 1 : 0.25 : 0.075, до достижения рН 3.5 (Растворы 2, 3 и 4 соответственно). Затем к Раствору 1 по каплям и при интенсивном перемешивании добавляли Раствор 2, 3 или 4 в количестве, соответствующем мольному соотношению Bi(Pb) : Ti = 0.01 моль/моль, а также стехиометрическому количеству воды (H2O : Ti = 4 моль/моль), необходимому для гидролиза н-тетрабутоксититана. После недлительного интенсивного перемешивания полученных растворов наблюдалось их помутнение без выделения осадка. Для интенсификации процесса формирования золя растворы нагревали в колбе с обратным холодильником до кипения и кипятили (1 ч) до образования прозрачного коллоидного раствора – золя (Растворы 5, 6 и 7 соответственно).
На следующем этапе соответствующий золь, с добавленными нитратами либо висмута, либо свинца, либо обоих металлов, наносили на поверхность стекла ТСО 22-15 методом spin-coating при скорости вращения ротора 2000 об мин–1. Скорость нанесения коллоидных растворов на вращающуюся пластину подбирали таким образом, чтобы раствор равномерно заполнял всю ее поверхность. После формирования на поверхности слоя геля проводили термическую обработку полученного образца в электрической печи (условия термообработки аналогичны описанным в [53]). После медленного остывания образца процедуры нанесения и термообработки проводили еще несколько раз до образования равномерного плотного покрытия. Число циклов описанных процедур (до 10) подбиралось таким образом, чтобы массовая доля нанесенного поликристаллического слоя во всех полученных образцах составляла 0.1% от массы пластины анода. С целью удаления остаточных углеродсодержащих веществ образцы перед дальнейшими исследованиями подвергали дополнительному прокаливанию при 500°С в течение 5 ч. Таким способом были изготовлены образцы фотоанодов со сформированными на их поверхности пленочными покрытиями из диоксида титана, легированного либо висмутом, либо свинцом, либо обоими металлами.
Для оценки влияния разработанной методики синтеза на структурные свойства получаемых металлоксидов были также приготовлены образцы в виде дисперсных порошков из тех же коллоидных Растворов 5, 6 и 7. Для этого из каждого из указанных растворов отгоняли органическую фракцию, а полученные металлсодержащие гели подвергали сушке под вакуумом (3 Т) при 120°С (1 ч) и последующему прокаливанию в печи при 500°С (5 ч) до образования порошков TiO2, легированных ионами Bi3+, Pb2+ или Bi3+ и Pb2+ соответственно.
Содержание введенных ионов каждого из металлов составляло 2.5 мас. % как в пленочных покрытиях фотоанодов, так и в порошковых образцах.
Общая схема методики приготовления образцов представлена на рис. 1.
Рис. 1.
Схема синтеза образцов состава Bi3+(Pb2+)-TiO2 в виде порошков и пленочных покрытий, нанесенных на FTO-стекла.

Рентгеновская дифрактометрия
Съемку дифрактограмм проводили на дифрактометре Empyrean (Panalytical, Голландия), оснащенном высокочувствительным полупроводниковым детектором PIXel-3D. Для реализации геометрии Брэгга-Брентано использовали рентгенооптический модуль Bragg-BrentanoHD (многослойное параболоидное зеркало), осуществляющий монохроматизацию первичного пучка и обеспечивающий высокую интенсивность падающего на образец рентгеновского пучка. Кроме того, данный оптический модуль улучшает соотношение сигнал/шум на экспериментальных дифрактограммах и обеспечивает более низкое значение интенсивности возбуждаемого в образцах флуоресцентного излучения. Режим работы рентгеновского источника (отпаянная рентгеновская трубка, CuKα-излучение λ = 1.542 Å) при съемке образцов: U = 40 кВ, I = 40 мА, шаг сканирования 0.013 угл. град. Детектор работал в линейном (1D) режиме с угловым диапазоном 3.3 угл. град., определяемым входной апертурой детектора (14 × 14 мм2). Для обработки дифрактограмм использовали программу Jade9++ (Material Data Incorporation, США). Съемку дифрактограмм в асимметричной геометрии при скользящем падении рентгеновского пучка (grazing incidence asymmetric Bragg diffraction) проводили с использованием гибридного монохроматора, представляющего из себя оптический модуль, в состав которого входит параболоидное зеркало в комбинации с германиевым щелевым монохроматором (channel-cut crystal monochromator) 4х Ge(220). Такой модуль выполняет две функции: формирует коллимированный пучок с малой расходимостью и осуществляет монохроматизацию первичного пучка с сохранением его высокой интенсивности. Спектральная ширина рентгеновского пучка, падающего на образец, составляет величину Δλ/λ ~ 10–4, что, в итоге, позволяет проводить фазовый анализ покрытий по глубине с высоким разрешением и высокой чувствительностью
Атомно-силовая микроскопия (АСМ)
Для исследования морфологии рельефа поверхности применялся метод атомно-силовой микроскопии. Измерения проводили на сканирующем зондовом микроскопе SolverBio (NT-MDT, Россия) кремниевым зондом с радиусом острия <10 nm в полуконтактном режиме.
Спектральные характеристики
Спектральные характеристики полученных пленок изучали в диапазоне 300–900 нм при комнатной температуре с использованием спектрометра Lambda35 “PerkinElmer”.
Фотоэлектроокисление
При проведении фотоэлектрохимических исследований использовали компьютеризированную установку, которая включала в себя фотоэлектрохимическую трехэлектродную ячейку PECC-2 (Zahner Elektrik, Германия), симулятор солнечного спектра 96 000 (Newport, США) мощностью 150 Вт и потенциостат IPC-Pro MF (ИФХЭ РАН, Россия). Рабочий электрод в ячейке представлял собой фотоанод из n-TiO2 или из n-TiO2, легированного висмутом и/или свинцом, в виде пленочного покрытия с площадью поверхности 1 см2. Вспомогательным электродом служила платиновая проволока с площадью поверхности ≈3 см2. Серебряную проволоку использовали в качестве электрода сравнения, относительно которого приведены все потенциалы в данной работе. Освещение осуществляли с обратной стороны фотоанода, а мощность освещения на различных расстояниях от источника света определяли с помощью прибора Nova (OPHIR-SPIRICONInc., Израиль). Фотоэлектрохимическое окисление модельных соединений на приготовленных фотоанодах проводили либо монохроматическим светом (длина волны 464 нм) при мощности освещения 10 мВт см–2, либо светом видимой области спектра.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
На рис. 2 представлен общий вид фотоанода с пленочным покрытием. Нанесенные пленки всех синтезированных образцов характеризовались высокой адгезией к поверхности стекла, покрытого слоем токопроводящего материала.

На рис. 3 представлены экспериментальные дифрактограммы для трех образцов диоксида титана, легированного либо висмутом, либо свинцом, либо обоими металлами одновременно, полученных в виде порошков (а) и пленочных покрытий (б). Как видно из рис. 3а, дифрактограммы порошкообразных образцов практически не отличаются друг от друга, что свидетельствует об одинаковом фазовом составе и близких параметрах их субструктуры.
Рис. 3.
Экспериментальные дифрактограммы Bi, Pb и Bi-Pb-содержащих образцов n-TiO2 в виде порошков (а) и пленочных покрытий фотоанода (б).

С использованием базы данных порошковых дифракционных стандартов ICDD PDF-2 было установлено, что все три образца представляют собой фазу диоксида титана в модификации анатаза (номер карточки в базе PDF-2 01-073-1764; тетрагональная кристаллическая решетка с параметрами a = 3.75 Å и с = 9.51 Å) (рис. 4а). Других фаз, содержащих атомы висмута или свинца, ни в одном из образцов обнаружено не было.
Рис. 4.
Результаты рентгенофазового анализа образцов в виде порошка (а) и пленочного покрытия фотоанода (б).

На рис. 3б видно, что помимо пиков, соответствующих фазе анатаза (наиболее интенсивные из них, характеризующиеся угловыми положениями 2θ, град. = 25.3, 48.0, 53.7 и 63.0, отмечены пунктирными линиями), на экспериментальных дифрактограммах образцов пленочных покрытий присутствуют более узкие, острые дифракционные максимумы. Рентгенофазовый анализ, проведенный с использованием базы данных PDF-2, подтвердил, что они соответствуют фазе диоксида олова(IV), составляющего материал электропроводящего покрытия (номер карточки в базе PDF-2 00-046-1088; тетрагональная кристаллическая решетка с параметрами a = 4.75 Å и с = 3.198 Å) (рис. 4б).
На основе данных анализа уширения дифракционных линий по методу Вильямсона–Холла был оценен размер кристаллитов в исследованных образцах, который составил: 〈D〉 ~ 8 нм для фазы анатаза как в порошкообразных образцах, так и в пленочных покрытиях; 〈D〉 ~ 25 нм для фазы диоксида олова(IV).
Для пленочного образца, легированного висмутом и свинцом, с целью отстройки от дифракционных рефлексов материала фотоанода были сняты дифрактограммы в асимметричной геометрии брэгговской дифракции при скользящем падении пучка. Сравнительные дифрактограммы при углах падения рентгеновского пучка от 0.5 до 10 градусов представлены на рис. 5.
Рис. 5.
Сравнительные дифрактограммы, снятые в геометрии скользящего падения пучка, при различных значениях угла падения рентгеновского излучения на образец с пленочным покрытием из диоксида титана, легированного висмутом и свинцом.
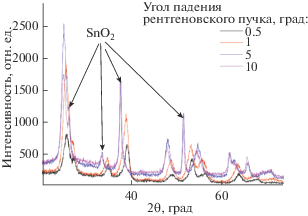
Как видно из рис. 5, по мере уменьшения угла падения, помимо значительного снижения интенсивности пиков, соответствующих оксиду олова, наблюдается сдвиг углового положения максимумов, соответствующих анатазу, в сторону больших углов. Согласно формуле Вульфа–Брэгга, сдвигу дифракционных линий в сторону больших углов соответствует уменьшение межплоскостных расстояний за счет деформации сжатия решетки анатаза в приповерхностных слоях. Движущей силой возникновения остаточных напряжений, приводящих к деформации сжатия кристаллической решетки анатаза, по всей вероятности, могло быть испарение растворителя из объема образца в процессе измерений.
Данные, полученные методом рентгеновской дифракции, хорошо коррелируют с результатами исследования пленочных образцов методом атомно-силовой микроскопии, иллюстрируемыми рис. 6. Данные АСМ показали, что морфология пленочного покрытия идентична для всех образцов, вне зависимости от легирующего компонента. Поверхность покрытия представляет собой систему, состоящую из раздельных блоков размером 2–3 мкм (рис. 6а). При более высоком масштабировании четко видна достаточно однородная структура, состоящая из сферических зерен размером 50–60 нм (рис. 6б)

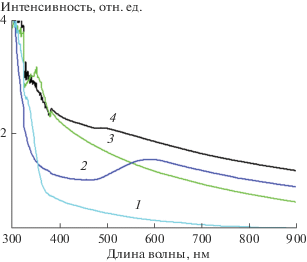
Результаты исследования характеристик поглощения нанесенных пленочных покрытий иллюстрирует рис. 7, из которого видно, что образцы диоксида титана, легированного свинцом и висмутом, характеризуются поглощением в области длин волн более 400 нм, в отличие от пленки диоксида титана, не содержащего легирующих добавок (кривая 1 на рисунке). Наибольший сдвиг в видимую область спектра (450–550 нм) наблюдается для образца диоксида титана, содержащего 2.5% Вi и 2.5% Pb (кривая 4), а также для образца, легированного 2.5% висмута (кривая 3). Сдвиг поглощения в видимую область может быть связан со снижением ширины запрещенной зоны диоксида титана (3.2 эВ) до ~2.7 эВ для TiO2, легированного висмутом или одновременно висмутом и свинцом. Аналогичный сдвиг поглощения в видимую область спектра для диоксида титана, содержащего добавки висмута, наблюдали в работе [55].
Рис. 7.
Спектры поглощения пленок: (1) TiO2; (2) ТiО2 + 2.5% Pb; (3) ТiО2 + 2.5% Bi; (4) ТiО2 + 2.5% Pb + + 2.5% Bi.
Фотоэлектрохимические процессы окисления различных субстратов при освещении пленок нелегированного диоксида титана видимым светом были исследованы нами ранее [53]. При освещении фотоанода на поверхности пленки инициируется фотохимический процесс. TiO2 абсорбирует свет и генерирует пары электрон/дырка в соответствии с уравнением
(1)
${\text{Ti}}{{{\text{O}}}_{{\text{2}}}}{\text{ + }}h\nu {\text{ }} \to {\text{ Ti}}{{{\text{O}}}_{{\text{2}}}}{\text{(e}}_{{{\text{cb}}}}^{ - } + h_{{{\text{vb}}}}^{ + }{\text{),}}$а фотогенерируемые дырки способны окислять широкий спектр органических соединений:
(2)
${\text{Ti}}{{{\text{O}}}_{{\text{2}}}}{\text{(}}{{h}^{{\text{ + }}}}{\text{)}} + {\text{S}} \to {\text{Ti}}{{{\text{O}}}_{{\text{2}}}} + {{{\text{S}}}^{{\text{ + }}}}.$Поглощаемое диоксидом титана излучение вызывает увеличение концентрации свободных носителей заряда в приповерхностном слое электрода, а также снижение поверхностного потенциального барьера (изгиб зон). Когда потенциал полупроводника положительнее потенциала плоских зон, то зоны загнуты вверх, и фотогенерированные дырки (h+) мигрируют к поверхности электрода, а электроны движутся к коллектору и при замкнутой цепи далее к катоду. Это предотвращает рекомбинацию фотогенерированных носителей. Низко расположенные поверхностные состояния являются сильными окислителями и способны окислять различные органические соединения.
В настоящей работе мы исследовали фотоэлектрокаталитическую активность пористых пленок диоксида титана, легированного свинцом и/или висмутом, в реакции окисления метанола, муравьиной кислоты и фенола при освещении как монохроматическим светом с длиной волны 464 нм, так и видимым светом (400–780 нм).
На рис. 8 представлены вольтамперограммы, снятые на фотоанодах с пленками n-TiO2, содержащего 2.5% свинца (кривые 1, 1'), 2.5% висмута (кривые 2, 2 ') и совместно свинца и висмута в концентрации 2.5% каждый (кривые 3, 3'), в диапазоне потенциалов – 0.9–0.5 В при освещении монохроматическим светом с длиной волны 464 нм в водных растворах 0.1 М КOH в отсутствие и в присутствии метанола. Как видно из рисунка, для всех исследованных образцов введение в раствор метанола приводит к смещению бестокового потенциала фотоанода в область более отрицательных значений. При этом во всей области потенциалов регистрируемые в присутствии метанола плотности тока превышают таковые в фоновом растворе, что является свидетельством фотоокисления метанола. Фототоки, регистрируемые в отсутствие метанола, по-видимому, обусловлены реакцией разложения растворителя (воды) при освещении. “Темновые” токи, т.е. токи на неосвещенном фотоаноде, во всей изученной области потенциалов отсутствуют.
Рис. 8.
Вольтамперные характеристики пленочных фотоанодов n-TiO2 + 2.5% Pb (1, 1') n-TiO2 + 2.5% Bi (2, 2') и n-TiO2 + 2.5% Bi + 2.5% Pb (3, 3') в водном растворе: (1) 0.1 М KOH; (2) 0.1 М KOH + 200 мкл (4.9 ммоль) CН3OH. Скорость развертки потенциала 0.01 В с–1. Условия освещения: ксеноновая лампа (150 Вт) с фильтром 464 нм; мощность освещения на фотоаноде – 10 мВт см–2.

На основе данных вольтамперометрических измерений были построены зависимости парциальных токов фотоэлектроокисления метанола от потенциала фотоанода, которые приведены на рис. 9. Из рисунка видно, что в ряду фотоанодов парциальный фототок окисления растет в последовательности: (n-TiO2 + 2.5% Pb) < (n-TiO2 + + 2.5% Bi) < (n-TiO2 + 2.5% Bi + 2.5% Pb). При этом фототок, регистрируемый на пленке (n-TiO2 + + 2.5% Bi + 2.5% Pb), в первом приближении равен сумме фототоков на пленках (n-TiO2 + 2.5% Pb) и (n-TiO2 + 2.5% Bi). Таким образом, легирование n-TiO2 свинцом и висмутом позволяет окислять метанол светом видимой части спектра. Величина парциального фототока окисления метанола на пленке (n-TiO2 + 2.5% Bi + 2.5% Pb) при мощности освещения 10 мВт см–2 составила 43 мкА см–2. что соответствует степени использования фотонов, т.е. фотоэлектрокаталитической активности (IPCE), равной 1.15%. IPCE вычисляли по формуле:
(3)
$\begin{gathered} {IPCE(\% ) = } \\ {\text{ = }}\,\,{{{\text{1240}}{{J}_{{{\text{sc}}}}}({\text{mA c}}{{{\text{m}}}^{{{\text{--2}}}}})} \mathord{\left/ {\vphantom {{{\text{1240}}{{J}_{{{\text{sc}}}}}({\text{mA c}}{{{\text{m}}}^{{{\text{--2}}}}})} {\lambda ({\text{nm}})}}} \right. \kern-0em} {\lambda ({\text{nm}})}}P({\text{mW c}}{{{\text{m}}}^{{ - {\text{2}}}}}) \times {\text{100,}} \\ \end{gathered} $Рис. 9.
Парциальные вольтамперные характеристики фотоэлектроокисления метанола на пленках n-TiO2, легированного свинцом и/или висмутом, в водном растворе 0.1 М KOH + 200 мкл (4.9 ммоль) CН3OH при освещении ксеноновой лампой (150 Вт) с фильтром 464 нм. Мощность освещения на фотоаноде – 10 мВт см–2.

Следует отметить, что и в предыдущей работе [53] мы не наблюдали фотоактивности в видимой области спектра пленок нелегированного диоксида титана, нанесенного из водного раствора. Это согласуется с отсутствием поглощения в видимой области спектра для подобных пленок, исследованиых в данной работе (рис. 7, кривая 1).
На рис. 10а и 10б показаны транзиенты фотопотенциала (а) и фототока (б) при включении и выключении освещения для образца n-TiO2 + + 2.5% Bi + 2.5% Pb.
Рис. 10
(а). Транзиенты фотопотенциала анода с пленкой n-TiO2 + 2.5% Bi + 2.5% Pb в растворе 0.1 М KOH + 200 мкл (4.9 ммоль) CН3OH в темновых условиях и при включении освещения. Условия освещения: ксеноновая лампа (150 Вт) с фильтром 464 нм; мощность освещения на фотоаноде – 10 мВт см–2. (б) Транзиенты фототока на аноде с пленкой n-TiO2 + + 2.5% Bi + 2.5% Pb при Е = –0.68 В в растворе 0.1 М KOH + 200 мкл (4.9 ммоль) CН3OH в темновых условиях и при включении освещения. Условия освещения: ксеноновая лампа (150 Вт) с фильтром 464 нм; мощность освещения на фотоаноде – 10 мВт см–2.
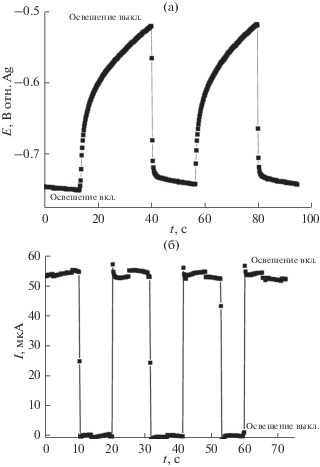
Как видно из рис. 10а, при освещении пленки диоксида титана, легированного висмутом и свинцом, монохроматическим светом с длиной волны 464 нм потенциал фотоанода смещается в отрицательную сторону на ~230 мВ, что связано с генерацией пары e−/h+ и обогащением зоны проводимости электронами, а валентной зоны – дырками. Прямоугольная форма транзиента тока (рис. 10б) указывает на быстрый отклик фотоанода на монохроматическое освещение.
Что касается механизма реакции окисления метанола на исследованных пленках, то, ввиду близости потенциалов фотоэлектроокисления растворителя и самого метанола, вполне вероятен механизм, предложенный в [61] для фотоэлектроокисления метанола на диоксиде титана при освещении ультрафиолетовым светом, согласно которому генерируемые дырки сначала окисляют анионы ОН− (активные акцепторы дырок), а затем уже образующиеся ОН• – радикалы окисляют метанол до диоксида углерода (уравнения (4) и (5) соответственно). Комбинация реакций (4) и (5) дает суммарное уравнение процесса (6). Приходящие на катод электроны в присутствии кислорода воздуха восстанавливают его до исходных ОН− – анионов согласно уравнению (7):
(4)
${\text{6O}}{{{\text{H}}}^{ - }} + {\text{6}}{{h}^{{\text{ + }}}} \to {\text{6O}}{{{\text{H}}}^{{ \bullet }}},$(5)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH + 6O}}{{{\text{H}}}^{{ \bullet }}} \to {\text{C}}{{{\text{O}}}_{{\text{2}}}} + {\text{5}}{{{\text{H}}}_{{\text{2}}}}{\text{O,}}$(6)
${\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH + 6O}}{{{\text{H}}}^{ - }}{\text{ + 6}}{{h}^{{\text{ + }}}} \to {\text{C}}{{{\text{O}}}_{{\text{2}}}} + {\text{5}}{{{\text{H}}}_{{\text{2}}}}{\text{O,}}$(7)
${{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{1/2}}{{{\text{O}}}_{{\text{2}}}} + {\text{2}}{{{\text{e}}}^{ - }} \to {\text{2O}}{{{\text{H}}}^{ - }}.$Вольтамперные зависимости фотоэлектрокаталитического окисления муравьиной кислоты на пленке (n-TiO2 + 2.5% Bi + 2.5% Pb) приведены на рис. 11а. Из рисунка видно, что при освещении фотоанода с пленкой n-TiO2 + 2.5% Bi + 2.5% Pb монохроматическим светом с длиной волны 464 нм добавка HCOOH к фоновому электролиту приводит к смещению бестокового потенциала анода в положительную сторону и к увеличению значений плотности тока во всей области потенциалов, что указывает на фотоэлектроокисление формиат-ионов. Парциальный ток окисления НCOO– на пленке n-TiO2 + 2.5% Bi + 2.5% Pb составил 56 мкА см–2 при мощности освещения 10 мВт см–2, а рассчитанное по уравнению (3) значение IPCE – 1.5%.
Рис. 11.
(а). Вольтамперные характеристики фотоанода с пленкой n-TiO2 + 2.5% Bi + 2.5% Pb в водном растворе: (1) 0.1 М KOH; (2) 0.1 М KOH + 9.2 ммоль НCOOH. Скорость развертки потенциала 0.01 В с–1. Условия освещения: ксеноновая лампа (150 Вт) с фильтром 464 нм; мощность освещения на фотоаноде – 10 мВт см–2. (б) Транзиенты фотопотенциала анода с пленкой n-TiO2 + 2.5% Bi + 2.5% Pb в растворе 0.1 М KOH + 9.2 ммоль НCOOH в темновых условиях и при включении освещения. Условия освещения: ксеноновая лампа (150 Вт) с фильтром 464 нм; мощность освещения на фотоаноде – 10 мВт см–2. (в) Транзиенты фототока на аноде с пленкой n-TiO2 + 2.5% Bi + 2.5% Pb при Е = 0 В в растворе 0.1 М KOH + 9.2 ммоль НCOOH в темновых условиях и при включении освещения. Условия освещения: ксеноновая лампа (150 Вт) с фильтром 464 нм; мощность освещения на фотоаноде – 10 мВт см–2.
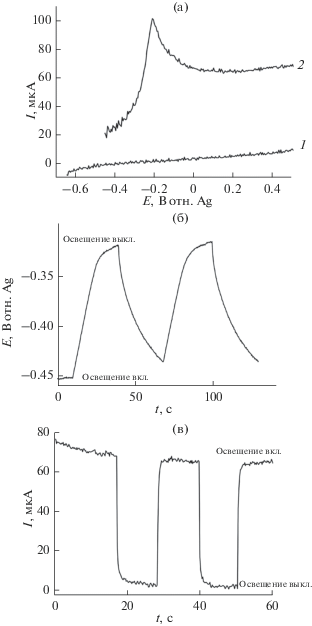
Появление пика тока на рис. 11а (кривая 2) может быть связано с тем, что формиат-ион HCOO– окисляется не только непосредственно из раствора, но и из адсорбированного на поверхности фотоанода состояния. В целом, механизм фотоэлектроокисления как муравьиной кислоты, так и метанола требует отдельного изучения.
Характер изменения транзиентов фотопотенциала (рис. 11б) и фототока (рис. 11в) в присутствии муравьиной кислоты указывает на то, что реакция фотоэлектроокисления анионов муравьиной кислоты протекает более медленно, чем фотоэлектроокисление анионов алкоголята. Это также согласуется с более низким значением фотопотенциала анода и замедленной релаксацией фототока в присутствии муравьиной кислоты. Следует уточнить, что более высокие значения парциальных токов фотоэлектроокисления формиата по сравнению с алкоголятом связаны с более высокой концентрацией первого в растворе.
На рис. 12а приведены вольтамперные зависимости фотоэлектрокаталитического окисления фенола (PhOH) на пленке n-TiO2 + 2.5% Bi + 2.5% Pb при освещении имитатором видимого света мощностью 450 мВт см–2. Из рисунка видно, что добавка PhOH к фоновому электролиту смещает бестоковый потенциал анода в положительную сторону и приводит к быстрому росту плотности тока уже при небольшом смещении потенциала, что указывает на фотоокисление PhOH. Пик тока на рис. 12а (кривая 2) и в данном случае, по-видимому, связан с адсорбцией аниона (PhO–) на поверхности фотоанода. Парциальный ток окисления PhO– на пленке n-TiO2 + 2.5% Bi + 2.5% Pb составил 250 мкА см–2, а значение фотопотенциала при освещении – 210 мВ. Если сопоставить результаты вольтамперометрических измерений для трех исследованных модельных систем, то можно сделать вывод, что фотоэлектроокисление субстратов затрудняется в ряду: метанол < муравьиная кислота < фенол.
Рис.12.
(а). Вольтамперные характеристики фотоанода с пленкой n-TiO2 + 2.5% Bi + 2.5% Pb в водном растворе: (1) 0.1 М KOH; (2) 0.1 М KOH + 2 ммоль PhOH. Скорость развертки потенциала 0.01 В с–1. Условия освещения: имитатор видимого света (ксеноновая лампа с УФ фильтром); мощность освещения на фотоаноде – 450 мВт см–2. (б) Транзиенты фотопотенциала анода с пленкой n-TiO2 + 2.5% Bi + 2.5% Pb в растворе 0.1 М KOH + 2 ммоль PhOH в темновых условиях и при включении освещения. Условия освещения: имитатор видимого света (ксеноновая лампа с УФ фильтром); мощность освещения на фотоаноде – 450 мВт см–2. (в) Транзиенты фототока на аноде с пленкой n-TiO2 + + 2.5% Bi + 2.5% Pb при Е = –0.2 В в растворе 0.1 М KOH + + 2 ммоль PhOH в темновых условиях и при включении освещения. Условия освещения: имитатор видимого света (ксеноновая лампа с УФ фильтром); мощность освещения на фотоаноде – 450 мВт см–2.

Полученные данные по фотоэлектрокаталитическому окислению модельных систем на пленке n-TiO2 + 2.5% Bi + 2.5% Pb согласуются с результатами оптических измерений по поглощению пленкой света видимой области спектра (рис. 7, кривая 4). Проведенные исследования показали, что использование солнечной энергии и легирование n-TiO2 висмутом и свинцом позволяет осуществлять на нем реакции фотокаталитического электроокисления ряда органических соединений светом видимой области солнечного спектра, что несомненно представляет интерес с точки зрения очистки воды от органических загрязнителей. Из вольтамперных характеристик видно, что наибольший каталитический эффект наблюдается при фотоэлектроокислении на образце n-TiO2, содержащем одновременно висмут и свинец, однако весомый вклад в скорость фотоэлектроокисления субстратов оказывает введение в диоксид титана висмута.
Таким образом, синтез сложных оксидов на основе TiO2 с использованием алкоксо-метода позволяет получать наноразмерные однофазные оксиды со структурой анатаза как в виде дисперсных порошков, так и пленочного покрытия. Включение ионов висмута и свинца в структуру диоксида титана приводит к сдвигу поглощения в видимую область (по сравнению с нелегированным диоксидом титана) и проявлению фотоэлектрокаталитической активности в окислении модельных органических соединений при облучении светом видимой области спектра. Наиболее весомый вклад в изменение оптических и фотоэлектрокаталитических свойств вносит легирование диоксида титана ионами висмута, однако легирование одновременно двумя металлами (висмутом и свинцом) приводит к аддитивному возрастанию электрокаталитической активности, что, вероятно, связано со снижением ширины запрещенной зоны диоксида титана.
Спектры поглощения наноразмерных пленок диоксида титана, легированного висмутом и свинцом выполнены на оборудовании ЦКП ФМИ ИФХЭ РАН.
Работа выполнена при финансовой поддержке Программы фундаментальных исследований Президиума РАН 1.8 П “Фундаментальные аспекты химии углеродной энергетики” и в рамках государственного задания ИФХЭ РАН (Тема 47.23).
Список литературы
Chen X.B., Mao S.S. // Chem. Rev. 2007. V. 107. P. 2891.
Fujishima A., Honda K. // Nature. 1972. V. 238. P. 37.
Yu J.G., Fan J.J., Lü K.L. // Nanoscale. 2010. V. 2. P. 2144.
Yu J.G., Fan J.J., Zhao L. // Electrochim. Acta. 2010. V. 55. P. 597.
Yu J.G., Li Q.L., Shu Z. // Electrochim. Acta. 2011. V. 56. P. 6293.
Xu J., Wang G.X., Fan J.J., Liu B.S., Cao S.W., Yu J.G. // J. Power Sources. 2015. V. 274. P. 77.
O'Regan B., Gratzel M. // Nature. 1991. V. 353. P. 737.
Bai Y., Mora-Seró I., De Angelis F., Bisquert J., Wang P. // Chem. Rev. 2014. V. 114. P. 10095.
Bai J., Zhou B.X. // Chem. Rev. 2014. V. 114. P. 10131.
Ni M., Leung M.K.H., Leung D.Y.C., Sumathy K. // Renew. Sust. Energy Rev. 2007. V. 11. P. 401.
Ma Y., Wang X.L., Jia Y.Y., Chen X.B., Han H.X., Li C. // Chem. Rev. 2014. V. 114. P. 9987.
Li X., Yu J.G., Low J.X., Fang Y.P., Xiao J., Chen X.B. // J. Mater. Chem. A. 2015. V. 3. P. 2485.
Chen X.B., Shen S.H., Guo L.J., Mao S.S. // Chem. Rev. 2010. V. 110. P. 6503.
Walter M.G., Warren E.L., McKone J.R., Boettcher S.W., Mi Q.X., Santori E.A., Lewis N.S. // Chem. Rev. 2010. V. 110. P. 6446.
Fujishima A., Zhang X.T., Tryk D.A. // Int. J. Hydrogen Energy. 2007. V. 32. P. 2664.
Kudo A., Miseki Y. // Chem. Soc. Rev. 2009. V. 38. P. 253.
Maeda K. // J. Photochem. Photobiol. C. 2011. V. 12. P. 237.
Han F., Kambala V.S.R, Srinivasan M., Rajarathnam D., Naidu R. // Appl. Catal. A. 2009. V. 359. P. 25.
Konstantinou I.K., Albanis T.A. // Appl. Catal. B. 2004.V. 49. P. 1.
Pelaez M., Nolan N.T., Pillai S.C., Seery M.K., Falaras P., Kontos A.G., Dunlop P.S.M., Hamilton J.W.J., Byrne J.A., O’Shea K., Entezari M.H., Dionysiou D.D. // Appl. Catal. B. 2012. V. 125. P. 331.
Hoffmann M.R., Martin S.T., Choi W., Bahnemann D.W. // Chem. Rev. 1995. V. 95. P. 69.
Akpan U.G., Hameed B.H. // J. Hazard. Mater. 2009. V. 170. P. 520.
Fujishima A., Rao T.W., Tryk D.A. // J. Photochem. Photobiol. C. 2000. V. 1. P. 1.
Chen C.C., Ma W.H., Zhao J.C. // Chem. Soc. Rev. 2010. V. 39. P. 4206.
Park H., Park Y., Kim W., Choi W. // J. Photochem. Photobiol. C. 2013. V. 15. P. 1.
Shiraishi Y., Hirai T. // J. Photochem. Photobiol. C. 2008. V. 9. P. 157.
Linsebigler A.L., Lu G.Q., Yates Jr J.T. // Chem. Rev. 1995. V. 95. P. 735.
Xu Q.L., Yu J.G., Zhang J., Zhang J.F., Liu G. // Chem. Commun. 2015. V. 51. P. 7950.
Yu J.G., Low J.X., Xiao W., Zhou P., Jaroniec M. // J. Am. Chem. Soc. 2014. V. 136. P. 8839.
Li X., Wen J.Q., Low J.X., Fang Y.P., Yu J.G. // Sci. China Mater. 2014. V. 57. P. 70.
Fu J.W., Cao S.W., Yu J.G., Low J.X., Lei Y.P. // Dalton Trans. 2014. V. 43. P. 9158.
Li X., Liu H.L., Luo D.L., Li J.T., Huang Y., Li H.L., Fang Y.P., Xu Y.H., Zhu L. // Chem. Eng. J. 2012. V. 180. P. 151.
Dhakshinamoorthy A., Navalon S., Corma A., Garcia H. // Energy Environ. Sci. 2012. V. 5. P. 9217.
Zhang Q.H., Han W.D., Hong Y.J., Yu J.G. // Catal. Today. 2009. V. 148. P. 335.
Hemminger J.C., Carr R., Somorjai G.A. // Chem. Phys. Lett. 1978. V. 57. P. 100.
Inoue T., Fujishima A., Konish S., Honda K. // Nature. 1979. V. 227. P. 637.
Hirano K., Bard A.J.Z. // J. Electrochem. Soc. 1980. V. 127. P. 1056.
Reiche H., Bard A.J. // J. Amer. Chem. Soc. 1979. V. 101. P. 3127.
Kanno T., Oguchi T., Sakuragi H., Tokumaru K. // Tetrahedron Lett. 1980. V. 21. P. 467.
Taniguchi I., Nakashima K., Yamaguchi H., Yasukouchi K. // J. Electroanal. Chem. 1982. V. 134. P. 191.
Плесков Ю.В. // Электрохимия. 1981. Т. 17. С. 3.
Гринберг В.А., Джавришвили Т.В., Васильев Ю.Б., Ротенберг З.А., Казаринов В.Е., Майорова Н.А. // Электрохимия. 1984. Т. 20. Вып. 1. С. 121.
Гуревич Ю.Я., Плесков Ю.В. // Итоги науки и техники. Электрохимия. Т. 18. М.: ВИНИТИ, 1982. С. 3–189.
Mills A., Le Hunte S. // J. Photochem. Photobiol. A: Chemistry. 1997. V. 108. P. 1.
Mills A., Lee S.K. // J. Photochem. Photobiol. A: Chemistry. 2002. V. 152. P. 233.
Anpo M., Takeuchi M. // J. Catalysis. 2003. V. 216. P. 505.
Antoniadou M., Lianos P. // J. Nanosci. Nanotechnol. 2010. V. 10. P. 6240.
Li L., Zhang S., Li G., Zhao H. // Anal. Chim. Acta. 2012. V. 754. P. 47.
Гринберг В.А., Емец В.В., Модестов А.Д., Майорова Н.А., Овсянникова Е.В., Бухтенко О.В., Маслов Д.А. // Электрохимия. 2017. Т. 53. № 2. С. 241.
Wen J., Li X., Liu W., Fang Y., Xie J., Xu Y. // Chinese J. Catal. 2015. V. 36. P. 2049.
Su R., Bechstein R., Kibsgaard J., Vang R.T., Besenbacher F. // J. Mater. Chem., 2012. V. 22. P. 23755.
Tu Y.F., Huang S.Y., Sang J.P., Zou X.W. // Mater. Res. Bull. 2010. V. 45. P. 224.
Гринберг В.А., Емец В.В, Майорова Н.А., Аверин А.А., Бухтенко О.В., Маслов Д.А. // Физикохимия поверхности и защита материалов. 2018. Т. 54. С. 55.
Baifu Xin, Liqiang Jing, Zhiyu Ren, Baiqi Wang, Honggang Fu // J. Phys. Chem. 2005. V. 109. P. 2805.
Sajjad S., Leghari S.A.K., Chen F., Zhang J. // Chem. Eur. J. 2010. V. 16. P. 13795.
Andronic L., Enesca A., Vladuta C., Duta A. // Chem. Eng. J. 2009. V. 152. P. 64.
Hajjaji A., Atyaoui A., Trabelsi K. et al. // Am. J. Anal. Chem. 2014. V. 5. P. 473.
Hoang S., Berglund S.P., Hahn N.T., Bard A.J., Mullins C.B. // J. Am. Chem. Soc. 2012. V. 134. P. 3659.
Hoang S., Guo S.W., Hahn N.T., Bard A.J., Mullins C.B. // Nano Lett. 2011. V. 12. P. 26.
Wang G.M., Wang H.Y., Ling Y.C., Tang Y.C., Yang X.Y., Fitzmorris R.C., Wang C.C., Zhang J.Z., Li Y. // Nano Lett. 2011. V. 11. P. 3026.
Lianos P. // J. Hazard. Mater. 2011. V. 185. P. 575.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов