

Физикохимия поверхности и защита материалов, 2019, T. 55, № 2, стр. 195-203
Электроактивные полимерные покрытия на модифицированной графитовой фольге в качестве электродов для суперконденсаторов
В. В. Абаляева 1, *, Г. В. Николаева 1, Н. Н. Дремова 1, Е. И. Кнерельман 1, Г. И. Давыдова 1, О. Н. Ефимов 1, С. Г. Ионов 2
1 Учреждение Российской академии наук Институт проблем химической физики Российской академии наук
142432 Московская обл, Черноголовка, просп. Академика Семенова, 1, Россия
2 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: avva@icp.ac.ru
Поступила в редакцию 22.03.2018
После доработки 23.05.2018
Принята к публикации 06.06.2018
Аннотация
В настоящей работе разработан способ нанесения электроактивных покрытий на основе проводящих полимеров на графитовую фольгу и изучено электрохимическое поведение таких электродов. Показано, что предварительная анодная обработка графитовой фольги значительно улучшает адгезию покрытий на ее поверхности и позволяет создавать электроактивные полимерные покрытия путем электрохимического окисления мономера или поливом из раствора химически полимеризованного мономера. Использование модифицированной графитовой фольги приводит к повышению электрохимической емкости электродов и стабильности покрытия.
1. ВВЕДЕНИЕ
Проводящие полимеры применяются при создании химически модифицированных электродов в электрохимических системах для запасания и преобразования энергии (аккумуляторы, суперконденсаторы) и электрокаталитических системах (сенсоры) [1]. Обычно такие покрытия наносят на инертную токопроводящую подложку – металлы, проводящее стекло (оксиды олова и индия), а также углеродные материалы, например стеклоуглерод и высокоориентированный пиролитический графит. Достоинством последних является коррозионная стойкость. Однако эти материалы не в полной мере удовлетворяют требованиям, предъявляемым к электродам в таких источниках тока как сернокислотные суперконденсаторы. С нашей точки зрения особый интерес представляет терморасширенный графит (ТРГ), получаемый, как правило, термической деструкцией гидролизованных интеркалированных соединений графита с рядом сильных кислот Бренстеда. Такое внимание к ТРГ обусловлено его интересными физико-химическими свойствами (большая удельная поверхность, достаточно высокая термическая и химическая стойкости, низкая теплопроводность, высокая пористость, большое аспектное отношение частиц и др.) [2, 3]. Одним из важных свойств ТРГ является способность при холодной прокатке без связующего образовывать графитовую фольгу (ГФ), которая находит широкое применение в качестве уплотнительного материала; экранов от электромагнитного излучения; резистивных элементов в гибких электрических нагревателях; газодиффузионных слоях и биполярных пластинах в водородно-воздушных топливных элементах и др. [4, 5]. С точки зрения возможности использования в электрохимии обращают на себя внимание хорошая электропроводность, низкая плотность 1 г/см3, развитая поверхность и однородная пористая структура, образованная щелевидными мезопорами с характеристическим радиусом около 20 Å [6, 7]. В литературе мы не нашли сведений о химически модифицированных электродах на основе гибкой графитовой фольги.
Можно было предположить, что нанесение проводящего полимера на графитовую фольгу (ГФ) приведет к повышению электрохимической емкости покрытия и большей стабильности при циклировании.
В данной работе, мы использовали ГФ в качестве подложки (токоотвода) для приготовления электродов, покрытых двумя типами электроактивных полимеров – гибкоцепным полианилином (ПАни), который наносили электросинтезом, и разветвленным полимером производного трифениламина с высокой плотностью свободных радикалов – 4,4',4-трис (N,N-дифениламино) трифениламин (TDATA) – ПТДАТА [8]. Этот полимер наносили поливом из раствора.
В таком электроде благодаря хорошей смачиваемости активированной ГФ при пропитке полимер не только образует прочное покрытие, но и проникает внутрь углеродного материала, что обеспечивает эффективный транспорт электронов при сохранении хорошей ионной проводимости.
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
2.1. Получение графитовой фольги
Гибкая ГФ получалась холодной прокаткой без связующего терморасширенного графита на лабораторной установке, в которой соблюдены все технологические особенности обычной промышленной линии по выпуску графитовой фольги-Графлекс. Установка подробно описана в работе [9]. ТРГ получали термической деструкцией гидролизованных интеркалированных соединений графита с азотной кислотой второй ступени при температуре 1200 K.
2.2. Анодирование графитовой фольги
Анодирование пластины из ГФ толщиной 0.6 мм проводили в 0.1% (NH4)2SO4 при 3 В в течение 1–5 мин. Рабочим анодом является пластина из ГФ, катодом служит пластина из нержавеющей стали. После анодирования ГФ промывали дистиллированной водой, высушивали при 60°С до постоянного веса.
2.3. Электросинтез (ЭС) ПАни
Проводили в потенциодинамическом режиме в интервале потенциалов –0.2 …+0.7 В (относительно Ag/AgCl) в трехкамерной стеклянной электрохимической ячейке (используемый объем электролита 15 мл) при комнатной температуре (21–22°С) из 0.2 М раствора анилин сульфата (АС) в 1 М H2SO4. Все потенциалы в тексте статьи приведены относительно Ag/AgCl. Пространства рабочего и вспомогательного электродов были разделены пористой стеклянной перегородкой. Рабочим электродом служила (исходная или анодированная) пластина ГФ размером 1 × 5 см2, вспомогательным электродом служила стеклоуглеродная пластина той же площади.
2.4. Синтез 4,4',4''-триc(N,N-дифенил-амино)трифениламин (ПТДАТА)
В отличие от работы [8] полимеризацию мономера проводили перекисью бензоила в муравьиной кислоте (МК).

1.2 × 10–4 моля ТДАТА были введены при интенсивном перемешивании в 20 мл 99.8% МК при комнатной температуре, при этом растворение мономера не наблюдалось. После добавления раствора 2.4 × 10–4 моля перекиси бензоила в 1 мл ацетона к суспензии мономера в МК (мольное соотношение мономера к окислителю 1 : 2) происходило растворение мономера, в ходе реакции цвет реакционной смеси изменялся от бледно-желтого до коричневого. Содержимое реактора перемешивали магнитной мешалкой в течение 2-х ч и оставляли на ночь. Образовавшийся вязкий раствор использовали для приготовления покрытий на графитовой фольге.
2.5. Приготовление рабочего электрода путем нанесения полимерного покрытия на графитовую фольгу из растворов полученных композитных материалов в МК с дальнейшим высушиванием и взвешиванием
Раствор полимера (ПТДАТА) в МК выдерживали в течение суток при комнатной температуре, затем фильтровали и наносили на электродный материал – исходную графитовую фольгу (ИГФ) или анодированную графитовую фольгу-(АГФ) в количестве одного или нескольких слоев. Покрытые полимером электроды (после ЭС ПАни и после синтеза in situ ПТДАТА) высушивали в течение суток при 40°С и взвешивали. Электрохимические исследования электродов проводили на потенциостате PS-7 (фирма Элинс, Россия) с компьютерным программированием.
Электронномикроскопические исследования проводили на растровом электронном автоэмиссионном микроскопе Supra 25 производства Zeiss с рентгеноспектральной энергодисперсионной приставкой INCA Energy производства Oxford Instruments для определения элементного состава образцов. Разрешение составляло 1–2 nm. ИК-спектры снимали на инфракрасном Фурье-спектрометре Perkin Elmer Spectrum 100, оснащенном приставкой многократного нарушенного полного внутреннего отражения с германиевой призмой в диапазоне 4000–675 см–1. Сухие образцы помещали на призму приставки и прижимали с помощью механического прижимного устройства. Обзорные рентгеновские фотоэлектронные спектры (РФС) низкого разрешения в диапазоне 0–1000 эВ с рассчитанным поверхностным содержанием элементов и спектры высокого разрешения С 1s, O 1s сняты на электронном спектрометре для химического анализа Specs PHOIBOS 150 MCD, с рентгеновской трубкой с магниевым анодом (MgKα – излучение 1253, 6 эВ).
Обзорный спектр записывался с шагом 1 эВ, спектры отдельных линий – с шагом 0.05 эВ.
Удельную площадь поверхности Sуд определяли по низкотемпературной адсорбции азота на приборе Autosorb-1 (Quantachrome, США). Значение Sуд рассчитывали по методу БЭТ [10].
3. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
3.1. Подготовка поверхности графитовой фольги
ИГФ представляет собой гладкий материал черного цвета, структуру которого можно рассмотреть только при очень сильном увеличении. На рис. 1а–г приведены микрофотографии такой поверхности при разном увеличении. Съемка электрода с торца (рис. 1б–г) позволяет рассмотреть слоистую структуру, состоящую из множества очень тонких пластин.

Целью анодирования была модификация лиофобной поверхности ГФ. Без такой обработки нанести покрытие из раствора не удавалось, капли не впитывались и скатывались с поверхности. Необходимо отметить, что воспроизводимость операции анодирования требовала точного соблюдения условий электролиза. На рис. 2а–г можно видеть изменение вида поверхности на начальных стадиях электролиза, снятые последовательно при выдержке образцов через интервалы времени 1, 2, 3, 4 мин. Сначала на поверхности появляются пузырьки (рис. 2а, б), которые затем схлопываются (рис. 2в), что приводит к разрушению сплошной поверхности ГФ. Разорванные фрагменты поверхности скручиваются (рис. 2г). При длительном электролизе происходит постепенное разрушение образца с отслоением темных частиц, которые осаждаются на дно. Этот процесс проходит исключительно с верхними слоями материала, вследствие чего поверхность исследуемого образца меняется. Попытку продолжить “крошить” поверхность далее можно продолжить, при этом внутренние слои действительно продолжают разламываться, но зато начинается активное разрушение верхних слоев с отрывом их от основного материала и выпадения в осадок. Поэтому главной целью установки режима анодирования было образование пористой поверхности и недопущение ее разрушения и отделения фрагментов от основного материала. Для оценки изменения удельной площади поверхности образцы были исследованы методом низкотемпературной адсорбции азота.

На рис.3а представлены изотермы адсорбции-десорбции исходной ГФ (кривая 1), АГФ после 5 мин анодирования (кривая 2), а также соскоба – слоя, снятого с поверхности (кривая 3). Все изотермы относятся к 4-му типу, характерному для мезопористых тел, и имеют гистерезис между адсорбционной и десорбционной ветвями, который можно отнести к типу H3 по классификации ИЮПАК [10]. Такая форма гистерезиса характерна для щелевидных мезопор или плоскопараллельных пластин, что соответствует слоистой чешуйчатой структуре ИГФ и АГФ а также структуре снятого с поверхности слоя. Под порами, по-видимому, следует подразумевать пространство между слоями материала или расстояние между частицами соскоба. Значения удельной площади поверхности Sуд ИГФ и АГФ, рассчитанные по методу BET, оказались близки и составили, соответственно, 22 и 16 м2/г. Некоторое снижение величины Sуд для АГФ, по-видимому, связано с уменьшением числа слоев материала при анодировании, вследствие чего уменьшается поверхность, доступная для адсорбции молекул азота. Для снятого с поверхности слоя значение Sуд составило 13 м2/г.
Рис. 3.
Изотермы адсорбции-десорбции (а) и кривые распределения пор по размерам (б) для ИГФ (1), АГФ, анодированной в течение 5 мин (2), соскоба с поверхности АГФ (3).

На рис. 3б представлено распределение удельного объема пор в зависимости от размера для исследуемых образцов (соответственно, кривые 1, 2 и 3). Видно, что для образцов ИГФ и АГФ кривые практически совпадают, подтверждая, что в материале, в основном, присутствуют мезопоры (пространство между слоями) толщиной 1–10 нм. Кривые имеют четко выраженный максимум в области 2 нм. Максимум на кривой распределения для снятого с поверхности слоя более широкий и немного смещен в область пор большего размера.
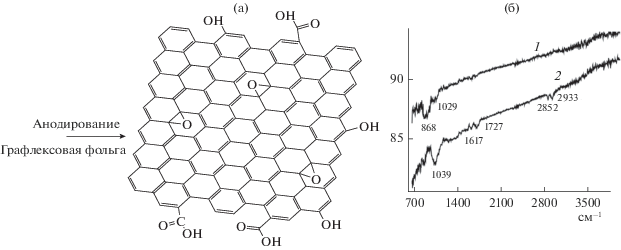
На рис. 4 приведены ИК спектры двух поверхностей ИГФ (спектр 1) и АГФ (спектр 2). После анодирования поверхности ГФ (рис. 4) в ИК-спектре появляются полосы соответствующие находящимся на поверхности кислородным С–О–, С=О и –ОН группам соответственно в области 1600–1700 см–1 [11] и также полосы, связанные с образованием на поверхности ГФ водородных связей между группами О–Н и С–Н [12], содержащими водородные и кислородные фрагменты в области 2800–2900 см–1. Также в этой области (2850–2920 см–1) достаточно активно проявляются полосы O–H и С–Н колебаний ароматического кольца [13, 14]. Полоса поглощения при 1039 см–1, скорей всего, относится к аниону ${\text{SO}}_{4}^{{ - 2}}$ [11], в среде которого проходило анодирование.
Также возрастает общее количество кислорода от 1.6 ат. % в ИГФ (рис. 5а, вкладка) до 6.12 ат. % в АГФ (рис. 5а), как это можно видеть из РФС-спектров. В РФС-спектре АГФ (рис. 5а) также после окисления появляются пики N и S, как остатки электролита, в котором проводится окисление ИГФ. В спектрах С 1s (рис. 5г, д) до и после анодирования никаких изменений не происходит: в обоих образцах С остается в равных долях гибридизации sp3 и sp2. В спектре О 1s (рис. 5б, в) доминируют пики с энергиями связи 532–533 eV, но на рис. 5б также можно видеть значительное количество продуктов с энергиями связи 535–536 eV, которые полностью исчезают после анодирования (рис. 5в). Можно сказать, что процесс анодирования стабилизирует количество и качество кислород-содержащих групп до С–ОН (531.9 eV), C=O и O=C–OH (530 eV). Также исчезают пики, которые характеризуют наличие в образце (535 eV) связанной воды [15].
Рис. 5.
(а) Обзорный RFS спектр АГФ и (на вкладке) ИГФ. (б, в) О 1s пики для ИГФ (б) и АГФ (в). (г, д) С 1s пики для ИГФ (г) и АГФ (д).

Из всего сказанного выше можно сделать вывод, что при анодировании происходит разрушение и окисление нескольких самых поверхностных слоев ГФ, так как не увеличивается общая площадь окисленного материала. Кислородсодержащие группы образуются на этих слоях. Но самое главное отличие этих двух поверхностей (ИГФ и АГФ) заключается в резком изменении адгезионной способности при нанесении полимерного покрытия. На необработанной ИГФ осаждаемая пленка практически не держится и в первых же экспериментах начинает разрушаться. Совсем другими свойствами обладает поверхность АГФ. Первые же капли раствора композитного материала, которые попадают на анодированную (разрыхленную) поверхность, легко впитываются. Только после нанесения значительного количества раствора на поверхности образца появляется жидкость. После высушивания такого электрода и помещения его в электрохимическую ячейку не замечено окрашивания электролита и разрушения или отслоения покрытия. В течение всего времени испытаний (проведения нескольких сотен циклов с разными скоростями, заряда-разряда) электрод был абсолютно стабилен, показывая устойчивое адсорбционное сцепление нанесенного полимера с поверхностью АГФ.
3.2. ЭС ПАни на двух поверхностях графитовой фольги
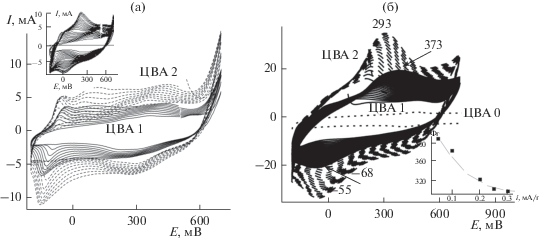
На рис. 6 приведены два сравнительных хода ЭС ПАни на двух поверхностях – ИГФ (рис. 6а, ЦВА 1) и АГФ (рис. 6а, ЦВА 2). Изначально были взяты две одинаковых пластины ИГФ размером 1 × 6 см. Одна из пластин была анодирована в течение 3 мин. Обе пластины в качестве рабочего электрода были погружены в электролит на глубину 1 см. Первые циклы ЭС ПАни показаны на рис. 6а. Из этого рисунка следует, что более активно ЭС начинается на поверхности АГФ (ЦВА 2). Количества полученного ПАни показаны на рис. 6б. Несмотря на то, что площади поверхности для обоих ГФ были рассчитаны методом BET как практически одинаковые, на АГФ ЭС проходит активнее, и за 30 циклов ЭС (рис. 6б, ЦВА 2) получено в два раза больше полимера, чем на ИГФ за 80 циклов (рис. 6б, ЦВА 1). Возможно, “взрыхленная” поверхность АГФ является более доступной для подхода мономера по сравнению с необходимостью проникновения внутрь слоев ИГФ, как это свидетельствует из фото СЭМ на рис. 2д, е. Особенность ЭС ПАни на обеих поверхностях заключается в том, что ЭС начинается практически с первого цикла в интервале потенциалов –0.2 + + 0.7 В. ЭС, начиная с бестокового потенциала 600 мВ (рис. 6а), характеризуется уже на первом цикле катодной ветки образованием катодного пика при 290 мВ и соответствующего ему на анодной ветви анодного пика при потенциале 360 мВ. Расположение этих пиков соответствует образованию пернигранилиновой пары задолго до начала электроокисления анилина на электродном материале. Такое начало ЭС наблюдается на обеих поверхностях. Начиная с третьего цикла, как можно рассмотреть на рис. 6а, появляются новые анодный и катодный пики –66 и –108 мВ соответственно. Такая пара пиков при ЭС ПАни в катодной области практически не встречается при классическом ЭС ПАни на других подложках-анодах (Pt, стеклоуглерод, другие аноды). Одним из объяснений такого начала ЭС может быть активное донорно-акцепторное взаимодействие анилина и образующихся на первых циклах димеров и олигомеров с поверхностью графлексовой фольги [16]. Указанная пара пиков растет в течение первых 5–6 циклов и далее начинает уменьшаться по интенсивности с параллельным началом роста стандартных пиков окисления-восстановления ПАни. Появление этих пиков на ЦВА свидетельствует о процессе окисления-восстановления форм лейкоэмеральдин-эмеральдин и начале стандартного электросинтеза ПАни [17–19]. Процесс перехода течения ЭС в стандартное русло показан на вкладке к рис. 6а. ЭС ПАни проходил практически одинаково на обеих поверхностях, но с разными скоростями. На АГФ ЭС проходит значительно быстрее (рис. 6б). Тем не менее, благодаря описанному началу ЭС, полностью отсутствует индукционный период, а интервал потенциалов ЭС сохраняется в очень благоприятной области для образования качественного ПАни. На рис. 7 приводятся ИК спектры двух ПАни, полученных на двух электродах: ИГФ (спектр 1) и АГФ (спектр 2). Очень активная полоса 853 см–1 в полимере 1 свидетельствует о сильной деформации кольца в области колебаний группы C–N–C [19, 20]. В ИК спектрах обоих полимеров в области 1400–1500 см–1 присутствуют слабые характерные для ПАни хиноидные и бензоидные пики соответствующей структуры ПАни. Однако, в ПАни на ИГФ (спектр 2) активная полоса 1215 см–1, а также поглощение в области 740 см–1 свидетельствует о присутствии иминной формы [21] азота, что отличает этот полимер от образца 1. В ПАни на АГФ более активна полоса 1296 см–1, что свидетельствует о аминной форме азота, т.е. полимер имеет более бензоидное строение. Возможно, не только строение поверхности, но и ее способность взаимодействовать с образующимся ПАни, улучшает проводящие свойства полимера, что также может определять и скорость его образования. Во время ЭС для обоих полимеров наблюдается активное смещение анодного пика в анодную область и соответствующее увеличение ΔЕА-К. Как следует из рис. 6б, для ПАни-АГФ ΔЕА-К почти на 100 мВ меньше, чем та же величина для ПАни-ИГФ. Значение катодного пика при этом в течение всего процесса ЭС остается практически постоянным. Из рис. 6б хорошо видно, что ЭС ПАни-АГФ (ЦВА 2) продолжается в течение всего времени ЭС. На ИГФ, как видно на ЦВА 1, ЭС практически прекратился или шел очень медленно уже к 30 циклу. Удалось провести зарядно-разрядное испытание для обоих электродов, после которого ПАни-ИГФ начал разрушаться, и дальнейшая работа с ним стала невозможной. Рассчитанные значения удельных емкостей (С Ф/г) составили 337 Ф/г для ПАни-АГФ и 185 Ф/г для ПАни-ИГФ при i = 0.2 мА/г. На вкладке к рис. 6б показаны значения снятой зависимости величины С от плотности тока для ПАни-АФГ. С увеличением плотности тока зависимость С/i уменьшается. Также этот электрод прекрасно циклировался в 1 М H2SO4, многократно вынимался из электролита и возвращался туда для дополнительных исследований. При этом потеря активности была минимальной, ПАни–АФГ вел себя очень устойчиво. Для того, чтобы выяснить, какую часть С составляет двойнослойная, а какую фарадеевская емкости, была снята ЦВА 0 ИГФ и АГФ в 1 М H2SO4. ЦВА обеих материалов практически не отличаются и Сг = 28 Ф/г. Пример такой ЦВА для АГФ (ЦВА 0) приведен на рис. 6б.

3.3. Приготовление и исследование электрода из АГФ, покрытой поли-4,4',4''-триc(N,N-дифенил-амино) трифениламином (ПТДТА) поливом из раствора
Раствор полученного полимера в МК наносился на подложку – ИГФ и АГФ. Сцепление наносимого покрытия с ИГФ было минимальным. Для работы такое изделие было непригодно, и поэтому все работы по исследованию композита ПТДАТА с подложкой были проведены с покрытием, посаженным на АГФ. В ИК-спектрах композита (рис. 8) полосы 730 и 870 см–1 указывают соответственно на деформацию кольца и C–H колебания выходящие из плоскости кольца (γ−(C−Η )−1,4 дизамещенного кольца) и аналогичные колебания хиноидного кольца [21–23]. Широкая полоса в районе 3260–3350 см–1 указывает на образовавшиеся на поверхности АГФ гидроксильные группы [24, 25]. Сама молекула ТДАТА плоская, и кольцо может подвергаться деформации при образовании каркаса полимера. Также пики 1370 и 1445 см–1 относятся к появлению связей N–Ar–Ar–N и С–N–С соответственно [26, 27], что может быть подтверждением образования полимера. Также образование полимерной цепи через фенильные кольца косвенно подтверждается наличием ряда пиков в районе 900–1200 см–1 [28]. В области 1500–1700 см–1 имеется ряд полос, которые относятся к С–С и С=С колебаниям хиноиднывх и бензоидных колец [24]. Из литературных данных известно об изучении различных функциональных материалов на основе трифениламина, таких как низкомолекулярные стекла, которые обладают проводящими/фотопроводящими свойствами. Авторы связывают это с наличием многочисленных волн окисления-восстановления такого полимера [29], причем практически все они являются обратимыми, как показывают ЦВА и наших композитных материалов.

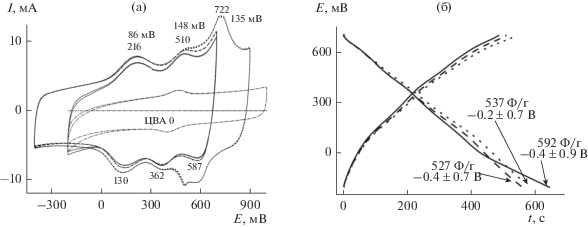
Электрохимическое циклирование композитного электрода АГФ-ПТДАТА было проведено в нескольких интервалах от –0.4 до +0.9 В. Во всех трех (–0.2 + 0.7; –0.4 + 0.7 и –0.4 + 0.9 В) исследованных интервалах (рис. 9а) электрод вел себя очень устойчиво, показывая три пары обратимых пиков с минимальным ΔА-К 85–148 мВ. При этом окислительно-восстановительные пороги, независимо от интервала циклирования сохраняли свои устойчивые положения. После каждого интервала циклирования были сняты зарядно-разрядные кривые (рис. 9б) и рассчитаны удельные электрохимические емкости (С Ф/г) для каждого случая. Расширение интервала потенциалов в катодную область практически не меняет величину С композитного электрода. Зато увеличение анодного потенциала от 0.7 до 0.9 В увеличивает значение С почти на 20%. Как видно из ЦВА (рис. 6а), такое расширение интервала циклирования позволяет полимеру пройти все стадии окислительных превращений. Во время 50 циклирований при $\text{v}$ = = 20 мВ/с полимер вел себя очень устойчиво, не меняя значения анодного и катодного потенциалов всех трех степеней окислительно-восстановительных превращений. Приведенная на рис. 9а ЦВА 0 чистого АГФ с С = 29 Ф/г показывает, что основной вклад в величину общего значения С композитного материала вносит полимерное покрытие ПТДАТА.
ЗАКЛЮЧЕНИЕ
Показано, что кратковременное анодное воздействие на поверхность графитовой фольги позволяет модифицировать ГФ и улучшить ее адсорбционную способность для закрепления на ней электрохимически выращенного или механически посаженного покрытия химически синтезированного полимера и получения нового функционального композитного материала. Этот композитный материал (с ПАни или ПТДАТА) показывает хорошую устойчивость при работе в протонной среде и высокие значения Суд (400 Ф/г для ПАни и 600 Ф/г для ПТДАТА). Полученные электрохимические характеристики важны для разработки перспективного органического катода на основе производных анилина и аминов. Также отметим, что проведение экспериментов на промышленно выпускаемых НПО Унихимтек графитовых фольгах-Графлекс показало аналогичные результаты.
Работа выполнена по темам госзадания 0089-2014-0036 и 0089-2014-0024 с использованием оборудования Центра коллективного пользования “Новые нефтехимические процессы, полимерные композиты и адгезивы”.
Список литературы
Huang X., Qi X., Boey F., Zhang H. // Chem. Soc. Rev. 2012. V. 41. P. 666.
Сорокина Н.Е., Никольская И.В., Ионов С.Г., Авдеев В.В. // Известия АН. Сер. хим. 2005. Т. 54. № 8. С. 1.
Savchenko D.V., Ionov S.G. // J. Phys. Chem. Solids. 2010. V. 71. P. 548.
Chung D.D.L. // J. Mater. Sci. 2016. V. 51. P. 554.
Chung D.D.L. // Jmepeg. 2000. V. 10. P. 161.
Shornikova O.N., Kogan E.V., Sorokina N.E., Avdeev V.V. // Russian J. of Physical Chemistry A. 2009. V. 83. № 6. P. 1022.
Шорникова О.Н., Коган Е.В., Сорокина Н.Е., Авдеев В.В. // ЖФХ. 2009. Т. 83. № 6. С. 1161.
Su Ch., He H., Xu L., Zhao K., Zheng Ch., Zhang Ch. // J. Mater. Chem. A. 2017. V. 5. P. 2701.
Savchenko D.V., Serdan A.A., Morozov V.A., Van Tendeloo G., Ionov S.G. // New carbon materials. 2012. V. 27. № 1. P. 12.
Gregg S.J., Sing K.S.W. // Adsorption, surface area and porosity. Academic Press. New York, 1982.
Trchová M., Stejska Ja. // Pure Appl. Chem. 2011. V. 83. № 10. P. 1803.
Kim M., Lee C., Jang J. // Adv. Funct. Mater. 2014. V. 24. P. 2489.
Yanga N., Zhaib J., Wanc M., Wanga D., Jiangc L. // Synth. Met. 2010. V. 160. P. 1617.
Wilamowska M., Kujawa M., Michalska M., Lipinska L., Lisowska-Oleksiaka A. // Synth. Met. 2016. V. 220. P. 334.
Goswami S., Maiti U.N., Maiti S., Nandy S., Mitra M.K., Chattopadhya K.K. // Carbon. 2011. V. 49. P. 2245.
Wang Y.G., Li H.Q., Xia Y.Y. // Adv. Mater. 2006. V. 18. P. 2619.
Kan J., Zhang Sh. // Synth. Met. 2004. V. 145. Is. 1. P. 37.
Liu H., Wang J.Y., Cui H.M., Shi R.J., Hu X.B. // Synth. Met. 2004. V. 145. Is. 1. P. 75.
Lindfors T., Kvarnstro C., Ivaska A. // J. Electroanal. Chem. 2002. V. 518. P. 131.
Kieffel Y., Travers P.J., Ermolieff A., Rouchon D. // J. Appl. Polym. Science. 2002. V. 86. P. 395.
Berrada K., Quillard S., Louarn G., Lefrant S. // Synth. Met. 1995. V. 69. P. 201.
Furukawa Y., Ueda F., Hyodo Y., Harada I., Nakajima T., Kawagoe T. // Macromol. 1988. V. 21. P. 1297.
Quillard S., Berrada K., Louarn G., Lefrant S., Lapkowski M., Pron A. // New J. Chem. 1995. V. 19. P. 365.
Kvarnstro C., Petr A., Damlin P., Lindfors T., Dunsch I.L. // J. Solid State Electrochem. 2002. V. 6. P. 505.
Pham T.A., Ashok N., Yeon K., Jeong T. // Synth. Met. 2010. V. 160. P. 2028.
Kvarnstr C., Petr A., Damlin P., Lindfors T., Ivaska A., Dunsch L. // J. Solid State Electrochem. 2002. V. 6. № 8. P. 505.
Adriana V., Szeghalmi M.E., Volker E., Michael S., Stephan A., Volker K., Gilbert N., Rainer S., Christoph L., Dirk L., Dietmar S., Z.Manfred, Jurgen P. // J. Am. Chem. Soc. 2004. V. 126. № 25. P. 7834.
Petr A., Kvarnstrom C., Dunsch L., Ivaska A. // Synth. Met. 2000. V. 108. P. 245.
Thelakkat M., Hagen J., Haarer D., Schmidt H.-W. // Synth. Met. 1999. V. 102. Is. 1–3. P. 1125.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов