

Физикохимия поверхности и защита материалов, 2019, T. 55, № 4, стр. 359-367
Фотоэлектрокаталитическое окисление муравьиной кислоты в видимой области спектра на пленках нанокристаллического диоксида титана, легированного висмутом
В. А. Гринберг 1, *, В. В. Емец 1, Н. А. Майорова 1, Д. А. Маслов 2, А. А. Аверин 1, С. Н. Поляков 2, 3, 4, И. С. Левин 2, М. В. Цодиков 2
1 Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский просп., 31, Россия
2 Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Ленинский просп., 29, Россия
3 Московский физико-технический институт (государственный университет)
141701 Московская обл., Долгопрудный, Институтский пер., 9, Россия
4 Институт сверхтвердых и новых углеродных материалов
142190 Троицк, ул. Центральная, 7а, Россия
* E-mail: vitgreen@mail.ru
Поступила в редакцию 25.07.2018
После доработки 02.02.2019
Принята к публикации 07.02.2019
Аннотация
На основе золь–гель синтеза разработан метод получения пленочных покрытий из легированного ионами висмута (Bi3+) диоксида титана со структурой анатаза. На поверхности фотоанода сформированы пленочные образцы, содержащие от 0.5 до 20 мас. % Bi. Показано, что легирование диоксида титана ионами висмута приводит к сдвигу поглощения в видимую область спектра электромагнитного излучения. Уровень поглощения зависит от концентрации висмута и достигает максимума для образцов, содержащих 0.5 и 1.0 мас. % Bi. На основании данных рентгенофазового анализа высказано предположение, что повышение содержания висмута до 20 мас. % приводит к разрушению кристаллических областей и аморфизации оксидов висмута и титана. Полученные покрытия исследованы в качестве катализаторов фотоэлектрокаталитического окисления муравьиной кислоты при освещении монохроматическим и видимым светом. Установлено, что наибольший каталитический эффект наблюдается на образцах, содержащих 1.0 мас. % висмута. Проведена оценка ширины запрещенной зоны по поглощению монохроматического излучения 464 нм и показано, что фотоэлектрокаталитическое окисление муравьиной кислоты в видимой области спектра, сопровождаемое адсорбцией формиат-иона на поверхности освещаемого фотоанода, вероятно, обусловлено снижением ширины запрещенной зоны легированного диоксида титана до 2.7 эВ.
ВВЕДЕНИЕ
Фудзишима и Хонда первыми использовали n‑TiO2 для фотоэлектрохимического разложения воды при освещении ультрафиолетовым светом [1]. Среди фотоэлектрохимических и фотоэлектрокаталитических процессов наиболее перспективны получение водорода из воды [2–9], деградация загрязнений окружающей среды [10–17], селективное превращение органических соединений [18, 19], восстановление диоксида углерода с целью получения топлив [20–26] и др. Реакции фотоэлектроокисления органических соединений на электродах из n-TiO2 (в модификации анатаза) исследуют уже более 30 лет [26–41]. Однако ширина запрещенной зоны анатаза (~3.2 эВ) обеспечивает его активность только в УФ области спектра, составляющей всего 3–5% от спектра солнечного света [37–40], что оставляет неиспользованной почти половину солнечной энергии. Повышение эффективности реакций фотокаталитического и фотоэлектрокаталитического окисления на n-TiO2 связывают, в первую очередь, с увеличением поглощения катализатором видимого света (λ = 400–780 нм) для более полного использования солнечной радиации, а также с уменьшением скорости рекомбинации фотогенерируемых пар электрон−дырка. Для решения этих задач в течение последних десятилетий разработаны легированные как металлами, так и неметаллами наноматериалы на основе диоксида титана для применения в различных областях, связанных с фотокатализом и фотоэлектрохимией [41–51]. В работе [40] была предпринята попытка провести фотоэлектрохимическое окисление метиленового синего на фотоаноде из легированного железом диоксида титана, поглощающего свет в области частот видимого спектра [43]. Эффективность преобразования энергии фотона в ток (IPCE) на таком электроде составила 0.5% (λ = 450 нм). Однако авторы другой работы не наблюдали фотоэлектрохимического окисления некоторых модельных систем (метанол и др.) на легированных железом фотоанодах, несмотря на заметный сдвиг поглощения в видимую область спектра [44]. Отсутствие фотоэлектрокаталитической активности в этом случае могло быть связано с усилением рекомбинационных процессов.
В последнее время было показано, что легирование n-TiO2 такими металлами как висмут, кадмий, хром, свинец также сдвигает область поглощения света полупроводником в видимую область спектра и увеличивает его фотокаталитическую активность в реакциях окисления органических соединений [45–48].
Предметом изучения в настоящей работе был выбран легированный ионами висмута диоксид титана в виде пленочного покрытия, синтезированный с использованием модифицированного золь–гель метода. Исследовано поглощение света фотоанодами с нанесенными пористыми пленками и их фотоэлектрокаталитическая активность в реакции фотоэлектроокисления муравьиной кислоты при освещении как монохроматическим светом с длиной волны 464 нм, так и видимым светом (400–780 нм).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
Аналитически чистые тетра-н-бутоксид титана (Ti(OBu)4) и пентагидрат нитрата висмута (Bi(NO3)3 · 5H2O) (Sigma Aldrich) были взяты в качестве исходных материалов без дополнительной обработки. Для приготовления растворов использовали деионизированную воду, этанол и ацетилацетон, который был очищен и обезвожен дистилляцией. Для изготовления фотоанодов использовали стекла марки ТСО 22–15 с электропроводящим покрытием из фтор – стабилизированного диоксида олова (F:SnO2; Solaronix; удельное сопротивление ≈15 Ом). Стекла были очищены выдержкой в растворе бихромата калия и серной кислоты, промыты дистиллированной водой и высушены при 100°С.
Приготовление фотоанодов
Образцы фотоанодов представляли собой поликристаллические покрытия из модифицированного висмутом TiO2, нанесенные на стекла со слоем F:SnO2, с одинаковой массовой долей покрытия в полученном фотоаноде, но с различным содержанием внесенных ионов Bi3+. Пленку формировали модифицированным золь–гель методом, включавшим приготовление Bi–Ti – содержащего золя и термообработку сформированного на его основе слоя. Приготовленные образцы обозначены как x% BiOx/TiO2, где x обозначает массовую долю Bi в нанесенном слое (мас. %).
Для приготовления золя к необходимому количеству бутилоксида титана добавляли эквимольное количество ацетилацетона для получения соединений состава Ti(OBu)4– x(acac)x, растворенных в образованном бутаноле (раствор 1). Нитрат висмута растворяли в смеси воды, этанола и ледяной уксусной кислоты (раствор 2). Вода и этанол были добавлены в количестве, необходимом для мольного соотношения [H2O]/[Ti] = 4 и [EtOH]/[H2O] = 0.3 (моль/моль). Количество уксусной кислоты варьировали в зависимости от количества внесенного нитрата висмута. Соотношение [Bi]/[AcOH] в растворе меняли от 0.001 до 0.053 (моль/моль), для образцов 0.5BiOx/TiO2 – 20BiOx/TiO2 соответственно. Раствор 2 добавляли по каплям и при интенсивном перемешивании к раствору 1, после чего наблюдалось помутнение полученой смеси без выделения осадка с последующим осветлением. Для интенсификации процесса растворы нагревали в колбе с обратным холодильником и кипятили в течение 1 ч до образования коллоидного раствора – золя. С повышением количества внесенного нитрата висмута наблюдалось изменение прозрачности получаемых золей (из которых в дальнейшем формировали пленочное покрытие) от почти бесцветного раствора при [Bi]/[Ti] = 0.004 (образец 0.5BiOx/TiO2) до молочноподобного коллоидного раствора при [Bi]/[Ti] = с 0.042 моль/моль (10BiOx/TiO2) и выше.
На втором этапе формировали пленочное покрытие каждого образца нанесением на стекло ТСО 22-15 соответствующего золя по каплям и при вращении стекла со скоростью 2000 об. мин–1 (spin-coating method). Скорость нанесения золя и его количество подбирали таким образом, чтобы раствор равномерно заполнял всю поверхность. После испарения растворителя на поверхности стекла формируется гель, который кристаллизуется до TiO2 в процессе термообработки. Термообработку образцов проводили в электрической печи при 500°С в течение 15 мин, с медленным нагреванием (10°С/мин) и медленным охлаждением. Процедуры нанесения и термообработки повторяли многократно до образования равномерного плотного покрытия с одинаковой массовой долей нанесенной пленки для всех образцов (0.1 мас. % от массы полученного фотоанода). Для удаления остаточных углеродсодержащих веществ образцы перед дальнейшими исследованиями подвергали дополнительному прокаливанию при 500°С в течение 5 ч. Таким образом были приготовлены образцы, содержащие 0.5; 1.0; 2.5; 5.0; 10 и 20 мас. % висмута, что соответствует 0.6; 0.13; 0.33; 0.67; 1.40 и 3.11 ат. %. Содержание висмута определяли методом атомно – абсорбционной спектрометрии.
Рентгеновская дифрактометрия
Съемку дифрактограмм проводили на дифрактометре Empyrean (Panalytical, Голландия), оснащенном высокочувствительным полупроводниковым детектором PIXel-3D. Для реализации геометрии Брэгга-Брентано использовали рентгенооптический модуль Bragg-BrentanoHD (многослойное параболоидное зеркало), осуществляющий монохроматизацию первичного пучка и обеспечивающий высокую интенсивность падающего на образец рентгеновского пучка. Кроме того, данный оптический модуль улучшает соотношение сигнал/шум на экспериментальных дифрактограммах и обеспечивает более низкое значение интенсивности возбуждаемого в образцах флуоресцентного излучения. Режим работы рентгеновского источника (отпаянная рентгеновская трубка, CuKα-излучение λ = 1.542 Å) при съемке образцов: U = 40 кВ, I = 40 мА, шаг сканирования 0.013 угл. град. Детектор работал в линейном (1D) режиме с угловым диапазоном 3.3 угл. град, определяемым входной апертурой детектора (14 × 14 мм2). Дифрактограммы регистрировались в угловом диапазоне от 15 до 90 град. Для обработки дифрактограмм использовали программу Jade9++ (Materials Data Inc., США).
Спектральные характеристики
Спектральные характеристики полученных пленок изучали в диапазоне 300–900 нм при комнатной температуре с использованием спектрометра Lambda35 “PerkinElmer”.
Фотоэлектроокисление
При проведении фотоэлектрохимических исследований использовали компьютеризированную установку, которая включала в себя фотоэлектрохимическую трехэлектродную ячейку PECC-2 (Zahner Elektrik, Германия), симулятор солнечного спектра 96 000 (Newport, США) мощностью 150 Вт и потенциостат IPC-Pro MF (ИФХЭ РАН, Россия). Рабочий электрод в ячейке представлял собой фотоанод из n-TiO2 или из n-TiO2, легированного висмутом, в виде пленочного покрытия с площадью поверхности 1 см2. Вспомогательным электродом служила платиновая проволока с площадью поверхности ≈3 см2. Серебряную проволоку использовали в качестве электрода сравнения, относительно которого приведены все потенциалы в данной работе. Освещение осуществляли с обратной стороны фотоанода, а мощность освещения на различных расстояниях от источника света определяли с помощью прибора Nova (OPHIR-SPIRICON Inc., Израиль). Фотоэлектрохимическое окисление муравьиной кислоты на приготовленных фотоанодах проводили либо монохроматическим светом (длина волны 464 нм) при мощности освещения от 1.5 до 10 мВт см–2, либо видимым светом при мощности 100 и 450 мВт см–2, при этом УФ область спектра отсекалась специальным стеклом.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Нанесенные пленки всех синтезированных образцов характеризовались высокой адгезией к поверхности стекла, покрытого слоем токопроводящего материала.
Полученные ранее данные атомно-силовой микроскопии показали, что покрытие, нанесенное из коллоидных органических золь–гель предшественников, состоит из сферических гранул размером 50–60 нм с достаточно плотной упаковкой [44].
На рис. 1 представлены дифрактограммы образцов висмут-содержащего диоксида титана с различной концентрацией висмута. В табл. 1 указаны параметры кристаллической структуры и усредненный размер микрокристаллитов.

Таблица 1.
Параметры кристаллической структуры образцов Bi–TiO2
| Образец, % Bi | Размер кристаллитов TiO2, Å | Период решетки а, Å | Период решетки с, Å | |
|---|---|---|---|---|
| мас. | атом. | |||
| 1.0 | 0.13 | 115 | 3.7782 | 9.4954 |
| 2.5 | 0.33 | 90 | 3.7725 | 9.4803 |
| 5 | 0.67 | 75 | 3.8077 | 9.4687 |
| 10 | 1.40 | 80 | 3.8120 | 9.5416 |
| 20 | 3.11 | 100 | 3.7714 | 9.4665 |
Как видно из рис. 1, на всех образцах четко прослеживается набор рефлексов от фазы анатаза (2Θ = 25.39–25.51; 37.08; 37.94; 38.70; 48.20; 54.10; 55.18–55.24; 62.34; 62.84–62.86; 69.04; 70.42–70.51; 75.35; 83.01 (карточка № 00-073-1764 базы данных ICDD_PDF-2). И лишь для образца, содержащего 0.33 ат. % (20 мас. %) Bi, в области угла 2Θ = 27.5 град наблюдается гало диффузного рассеяния, которое, по нашему предположению, связано, с началом образования фазы оксида висмута β-Bi2O3 (карточка № 00-077-0374 базы данных ICDD_PDF-2).
Ионные радиусы Ti4+ и Bi3+ существенно отличаются друг от друга, что исключает возможность образования твердых растворов замещения. Из данных табл. 1 видно уменьшение усредненного размера микрокристаллитов и небольшое возрастание величины периода кристаллической решетки фазы TiO2 при повышении содержания висмута до 10 мас. % (0.67 ат. %). Это, вероятно, указывает на возможность взаимодействия некоторого количества ионов висмута с атомами кислорода в микрокристаллитах оксида титана на границе раздела фаз, что приводит к незначительному искажению структуры. Подобное искажение структуры анатаза ионами железа Fe3+ при их взаимодействии с атомами поверхностного кислорода микрокристаллитов TiO2 было показано ранее при изучении структуры методами XAFS и мессбауэровской спектроскопии [52, 53]. Возрастание содержания висмута до 10 и 20 мас. %, по всей вероятности, приводит к микрорасслаиванию кристаллитов по фазовому составу и упорядочению структуры оксида висмута. На дифрактограмме образца с 20 мас. % Bi идентифицируется наиболее интенсивный рефлекс фазы Bi2O3 с угловым положением характерного для нее пика 2Θ = 27.5 град. Отсутствие рефлексов от оксида висмута при содержании висмута менее 10 мас. %, вероятно, объясняется высокой дисперсностью висмутсодержащих частиц.
Как было показано нами ранее, характеристики поглощения нанесенных покрытий из диоксида титана, легированного висмутом, характеризуются поглощением в области длин волн более 400 нм, в отличие от пленки диоксида титана, не содержащего легирующих добавок [44]. Оценка ширины запрещенной зоны по сдвигу поглощения в видимую область спектра показывает, что образцы, легированные висмутом, характеризуются снижением ширины запрещенной зоны до 2.7 эВ.
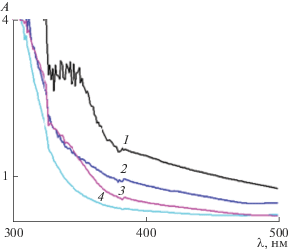
Аналогичный сдвиг поглощения в видимую область спектра для диоксида титана, содержащего добавки висмута, наблюдали в работе [46]. Зависимость степени поглощения от концентрации Bi приведена на рис 2. Видно, что максимальное поглощение наблюдается для пленочных покрытий, содержащих 1 мас. % Bi. Увеличение концентрации легирующей добавки сопровождается снижением поглощения образцом в области частот 300–500 нм.
Рис. 2.
Спектры поглощения пленок: (1) TiO2 + 1 мас. % Bi; (2) ТiО2 + 2.5 мас. % Bi; (3) ТiО2 + 5 мас. % Bi; (4) ТiО2 + 20 мас. % Bi.
Фотоэлектрохимические процессы окисления различных субстратов при освещении пленок нелегированного диоксида титана видимым светом были исследованы нами ранее [44]. При освещении фотоанода на поверхности пленки инициируется фотохимический процесс. TiO2 абсорбирует свет и генерирует пары электрон/дырка в соответствии с уравнением
(1)
${\text{Ti}}{{{\text{O}}}_{{\text{2}}}} + h\nu \to {\text{Ti}}{{{\text{O}}}_{{\text{2}}}}\left( {{\text{e}}_{{{\text{cb}}}}^{ - } + h_{{{\text{vb}}}}^{ + }} \right),$а фотогенерируемые дырки способны окислять широкий спектр органических соединений:
(2)
${\text{Ti}}{{{\text{O}}}_{{\text{2}}}}{\text{(}}{{h}^{{\text{ + }}}}{\text{)}} + {\text{S}} \to {\text{Ti}}{{{\text{O}}}_{2}} + {{{\text{S}}}^{ + }}.$Поглощаемое диоксидом титана излучение вызывает увеличение концентрации свободных носителей заряда в приповерхностном слое электрода, а также снижение поверхностного потенциального барьера (изгиб зон). Когда потенциал полупроводника положительнее потенциала плоских зон, то зоны загнуты вверх, и фотогенерированные дырки (h+) мигрируют к поверхности электрода, а электроны движутся к коллектору и – при замкнутой цепи – далее к катоду. Это предотвращает рекомбинацию фотогенерированных носителей. Низко расположенные поверхностные состояния являются сильными окислителями и способны окислять различные органические соединения [26–41].
На рис. 3а приведены вольтамперные характеристики пленочных фотоанодов из диоксида титана, легированного висмутом. Из рисунка видно, что уменьшение концентрации легирующего компонента приводит к сдвигу фототоков в отрицательную область потенциалов и их увеличению по сравнению с пленками, содержащими повышенную концентрацию висмута. Сдвиг к более отрицательным потенциалам отражает возможность анионов муравьиной кислоты окисляться фотогенерированными дырками и, таким образом, уменьшает их рекомбинацию. Следует отметить, что для пленок нелегированного диоксида титана, нанесенного из коллоидного раствора, мы не наблюдали фотоактивности в видимой области спектра, что согласуется с отсутствием поглощения в этой области для подобных пленок [44].
Рис. 3.
(а) Вольтамперные характеристики пленочных фотоанодов n-TiO2 + 1 мас. % Bi (1), n-TiO2 + 0.5 мас. % Bi (2), n-TiO2 + 2.5 мас. % Bi (3), n-TiO2 + 5 мас. % Bi (4), n-TiO2 + 10 мас. % Bi (5), и n-TiO2 + 20 мас. % Bi (6) в водных растворах 5 мл 0.1 М KOH + 350 мкл HCOOH (9.2 ммоль), рН 4. Скорость развертки потенциала 0.01 В с–1. (б, в) Транзиенты фотопотенциала и фототока на аноде с пленкой n-TiO2 + 1 мас. % Bi при Е = 0 В в водных растворах 5 мл 0.1 М KOH + 350 мкл HCOOH (9.2 ммоль), рН 4 в темновых условиях и при включении освещения. Условия освещения: ксеноновая лампа (150 Вт) с фильтром 464 нм; мощность освещения на фотоаноде – 10 мВт см–2.
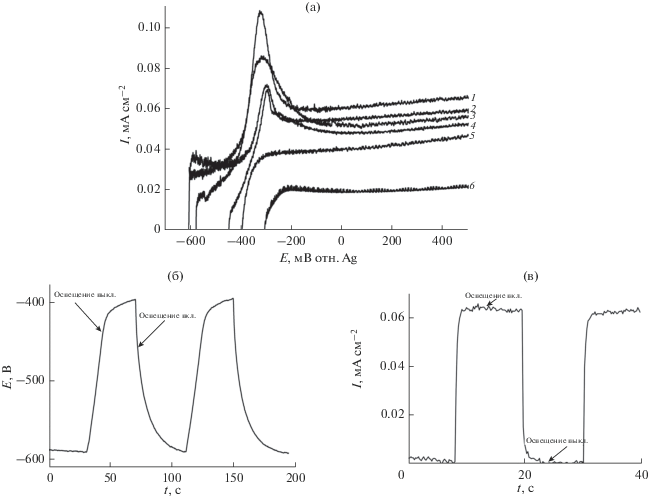
Наибольший сдвиг стационарного потенциала при освещении монохроматическим светом наблюдается для образца диоксида титана, легированного 1 мас. % Bi, что отражает наибольшее накопление положительного заряда в зоне пространственного заряда на поверхности изогнутой зоны. Снижение скорости фотоэлектрокаталитического окисления НСООН при увеличении концентрации легирующего компонента, по-видимому, связано со снижением степени поглощения (рис. 2) и возможным увеличением скорости рекомбинационных процессов, как и в случае легирования пленок диоксида титана железом [44].
На рис. 3б и 3в показаны транзиенты фотопотенциала и фототока при включении и выключении освещения для образца n-TiO2 + 1 мас. % Bi.
Как видно из рис. 3б, при освещении пленки монохроматическим светом с длиной волны 464 нм потенциал фотоанода смещается в отрицательную сторону на ~200 мВ, что связано с генерацией пары e−/h+ и обогащением зоны проводимости электронами, а валентной зоны – дырками. Прямоугольная форма транзиента тока (рис. 3в) указывает на быстрый отклик фотоанода на монохроматическое освещение. Также видно, что в темновых условиях токи отсутствуют.
В области потенциалов от –400 до –200 мВ наблюдается пик, имеющий адсорбционную природу. Фототок зависит от времени предадсорбции формиат-анионов на освещаемом фотоаноде при стационарном потенциале (рис. 4). За время циклирования в области потенциалов от стационарного до потенциала 500 мВ на фотоаноде не успевает накопиться необходимое количество положительных носителей, и адсорбционный пик не регистрируется на вольтамперной кривой уже на втором цикле (рис. 5).
Рис. 4.
Вольтамперные характеристики пленочного фотоанода n-TiO2 + 1 мас. % Bi в водных растворах 5 мл 0.1 М KOH + 350 мкл HCOOH (9.2 ммоль), рН 4 при различных временах выдержки при бестоковом потенциале, мин: (1) 5, (2) 3, (3) 1, (4) 0.5, (5) 0. Условия освещения: ксеноновая лампа (150 Вт) с фильтром 464 нм; мощность освещения на фотоаноде – 10 мВт см–2 .
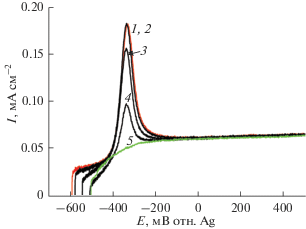
Рис. 5.
Циклические вольтамперограммы пленочного фотоанода n-TiO2 + 1 мас. % Bi в водных растворах 5 мл 0.1 М KOH + 350 мкл HCOOH (9.2 ммоль), рН 4: (1) первый цикл после выдержки при бестоковом потенциале в течение 0.5 мин, (2) второй цикл, без выдержки. Условия освещения: ксеноновая лампа (150 Вт) с фильтром 464 нм; мощность освещения на фотоаноде – 10 мВт см–2.

Предельный фототок наблюдается при потенциалах выше –200 мВ отн. Аg электрода. При более высоких потенциалах токи не зависят от потенциала и ограничиваются диффузией дырок к поверхности фотоанода, что отражается на вольтамперограмме в виде плато вплоть до потенциала 500 мВ [54, 55]. Следует подчеркнуть, что адсорбционный пик в области потенциалов –400–200 мВ наблюдается лишь в слабокислом растворе при рН 4.
Установлено, что в 0.1 М растворе КОН даже при содержании формиата калия 9.2 мМ (рН 14) мы не наблюдали фототоков окисления анионов муравьиной кислоты на пленке диоксида титана, легированного 1 мас. % Bi, что свидетельствует о сильной конкуренции формиат- и гидроксил- анионов за поверхность фотоанода. Аналогичное подавление минерализации НСООН при УФ облучении на пленках немодифицированного диоксида титана при рН > 6 было отмечено в работе [56].
Конкурентная адсорбция воды, неорганических анионов и муравьиной кислоты при окислении последней на фотоаноде из диоксида титана была отмечена в ряде работ [57–61, 62]. Было показано, что муравьиная кислота, растворенная в воде, хемосорбируется на поверхности TiO2 и окисляется путем прямого переноса генерируемой дырки из валентной зоны на молекулу кислоты [60, 61].
(3)
${\text{Ti}}{{{\text{O}}}_{{\text{2}}}} + h\nu \to {\text{Ti}}{{{\text{O}}}_{{\text{2}}}}\left( {{\text{e}}_{{{\text{cb}}}}^{ - } + h_{{{\text{vb}}}}^{ + }} \right),$(4)
${\text{HCOOH}} + h_{{\text{v}{\text{b}}}}^{ + } \to \quad\,{\text{HCO}}{{{\text{O}}}^{ \bullet }}\quad\, + \,{{{\text{H}}}^{ + }},$(5)
${\text{HCO}}{{{\text{O}}}^{\centerdot }} \to {\text{C}}{{{\text{O}}}_{{\text{2}}}} + {{{\text{H}}}^{ + }} + {{{\text{e}}}_{{({\text{С В }})}}}.$На рис. 6 представлены вольтамперограммы окисления муравьиной кислоты в зависимости от мощности освещения пленки при частоте монохроматического света 464 нм на образце, содержащем 1 мас. % Bi.
Рис. 6.
Вольтамперные характеристики пленочного фотоанода n-TiO2 + 1 мас. % Bi в водных растворах 5 мл 0.1 М KOH + 350 мкл HCOOH (9.2 ммоль), рН 4. Условия освещения: ксеноновая лампа (150 Вт) с фильтром 464 нм; мощность освещения на фотоаноде, мВт: (1) 10, (2) 4, (3) 1.5 мВт.
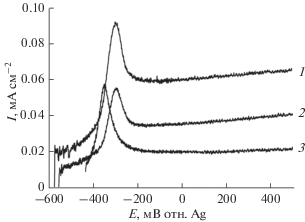
Как видно из рисунка, увеличение мощности освещения пленки n-TiO2 + 1 мас. % Bi приводит к сдвигу стационарного потенциала в отрицательную сторону, что, как было упомянуто выше, приводит к накоплению положительных зарядов в валентной зоне и электронов в зоне проводимости. Адсорбционные пики окисления анионов муравьиной кислоты при указанных мощностях освещения были получены при времени предадсорбции анионов 0.5 мин. Величина предельного фототока окисления НСООН на пленке (n-TiO2 + + 1.0 мас. % Bi) при мощности освещения 1.5 мВт см–2 составила 20 мкА см–2, что соответствует степени использования фотонов, т.е. фотоэлектрокаталитической активности (IPCE), равной 3.56%. IPCE вычисляли по формуле:
(6)
$\begin{gathered} {IPCE(\% )} = \\ {{ = \,\,1240{{J}_{{{\text{sc}}}}}({\text{м А с }}{{{\text{м }}}^{{--2}}})} \mathord{\left/ {\vphantom {{ = \,\,1240{{J}_{{{\text{sc}}}}}({\text{м А с }}{{{\text{м }}}^{{--2}}})} {\lambda ({\text{н м }})}}} \right. \kern-0em} {\lambda ({\text{н м }})}}P({\text{м В т с }}{{{\text{м }}}^{{--2}}}) \times 100, \\ \end{gathered} $Увеличение мощности освещения до 10 мВт см–2, согласно уравнению (4), приводит к закономерному снижению IPCE до 1.6%. Подчеркнем, что значения IPCE, полученные на диоксиде титана, модифицированном поли(три-s-триазином), в области частот видимого спектра (400–550 нм) составляют 0.1–0.2% при использовании НСООН (рН 3–4) в качестве акцептора дырок [63].
Вольтамперные характеристики образцов фотоанодов из легированного диоксида титана с различным содержанием Bi в реакции фотоэлектрокаталитического окисления муравьиной кислоты были исследованы также при освещении имитатором видимого света мощностью 100 мВт см–2. Из рис. 7 видно, что эти характеристики симбатны характеристикам, полученным при освещении монохроматическим светом, однако величина фототоков на порядок выше, что связано с увеличением количества генерируемых зарядов, ответственных за фотоэлектрокаталитическое окисление НСООН, вследствие увеличения мощности освещения. Пики токов в области потенциалов –400…–200 мВ также связаны с предварительной адсорбцией анионов муравьиной кислоты на пленочном фотоаноде при освещении.
Рис. 7.
Вольтамперные характеристики пленочных фотоанодов n-TiO2 + 1 мас. % Bi (1), n-TiO2 + 2.5 мас. % Bi (2), n‑TiO2 + 5 мас. % Bi (3), n-TiO2 + 10 мас. % Bi (4) и n-TiO2 + 20 мас. % Bi (5) в водных растворах 5 мл 0.1 М KOH + 350 мкл HCOOH (9.2 ммоль), рН 4. Скорость развертки потенциала 0.01 В с–1. Условия освещения: имитатор видимого света (ксеноновая лампа с УФ фильтром); мощность освещения на фотоаноде – 100 мВт см–2. Врезка: вольтамперные характеристики пленочного фотоанода n-TiO2 + 1% Bi при освещении имитатором видимого света (ксеноновая лампа с УФ фильтром); мощность освещения на фотоаноде, мВт см–2: (1) 100; (2) 450.

Полученные данные по фотоэлектрокаталитическому окислению НСООН на пленках n-TiO2 с различным содержанием Bi согласуются с результатами оптических измерений по поглощению пленкой света видимой области спектра [44, 46]. Проведенные исследования показали, что использование солнечной энергии и легирование n-TiO2 висмутом позволяют осуществлять на нем реакции фотокаталитического электроокисления муравьиной кислоты светом видимой области солнечного спектра, что несомненно представляет интерес с точки зрения очистки воды от органических загрязнений.
Из вольтамперных характеристик видно, что наибольший каталитический эффект наблюдается на образце n-TiO2, содержащем 1 мас. % Bi.
Из полученных результатов следует, что интенсивность фотоэлектрокаталитического окисления НСООН при освещении видимым светом фотоанодов из n-TiO2, легированного висмутом, в широкой области мощностей облучения зависит от рН среды и количества положительных носителей заряда в валентной зоне полупроводника, и не зависит от концентрации окисляемого субстрата в растворе.
Таким образом, синтез сложных оксидов титана с использованием алкоксо-метода позволяет получать наноразмерные оксиды со структурой анатаза как в виде дисперсных порошков, так и пленочных покрытий. Присутствие небольшой концентрации ионов висмута в структуре диоксида титана приводит к сдвигу поглощения в видимую область (по сравнению с нелегированным диоксидом титана) и проявлению фотоэлектрокаталитической активности в окислении НСООН, что, вероятно, связано со снижением ширины запрещенной зоны диоксида титана. Фотоэлектрокаталитическая активность зависит от содержания висмута и максимальна при его концентрации 1 мас. %. Дальнейшее повышение концентрации висмута в смешанном оксиде приводит к снижению активности, что может быть связано с различными факторами. Полученные результаты могут быть следствием размерного эффекта рентгеноаморфных висмут-содержащих частиц, распределенных на границе раздела микрокристаллитов оксида титана. Не исключено, что при небольшом содержании висмута наряду с высокодисперсными частицами оксидной фазы ионы Bi3+ могут взаимодействовать с кислородными атомами микрокристаллитов оксида титана на границе раздела фаз. В результате взаимодействия возможно изменение зарядового состояния атомов титана, что приводит к снижению ширины запрещенной зоны. Повышение содержания висмута более 1 мас. %, вероятно, приводит к увеличению микрофазового расслоения с упорядочением аморфизированной фазы оксида висмута и оксида титана. При содержании висмута 20 мас. % (3.3 ат. %) на дифрактограмме идентифицируется наиболее интенсивный рефлекс от фазы Bi2O3 (Θ – 27.5 град). Этот образец обладает минимальным уровнем поглощения в видимой области и практически неактивен в фотокаталитическом окислении муравьиной кислоты. С целью изучения влияния структуры полученных систем на фотоэлектрокаталитические свойства мы предполагаем исследовать тонкую структуру висмутсодержащих сложных оксидов методами XAFS и ПЭМ.
Спектры поглощения наноразмерных пленок получены на оборудовании ЦКП ФМИ ИФХЭ РАН.
Работа выполнена при финансовой поддержке Программы фундаментальных исследований Президиума РАН 1.8 П “Фундаментальные аспекты химии углеродной энергетики” и в рамках госзадания ИФХЭ РАН (Тема 47.23).
Список литературы
Fujishima A., Honda K. // Nature. 1972. V. 238. P. 37.
Ni M., Leung M.K.., Leung D.Y.C. // Internatioanl Journal of Hydrogen Energy. 2007. V. 32. № 13. P. 2305.
Ma Y., Wang X., Jia Y. et al. // Chemical Reviews. 2014. V. 114. № 19. P. 9987.
Li X., Yu J., Low J. et al. // Journal of Materials Chemistry A. 2015. V. 3. № 6. P. 2485.
Chen X., Shen S., Guo L. et al. // Chemical Reviews. 2010. V. 110. № 11. P. 6503.
Warren E.L., McKone J.R., Boettcher S.W. et al. // Chemical Reviews. 2010. V. 110. P. 6446.
Kudo A., Miseki Y. // Chemical Society Reviews. 2009. V. 38. № 1. P. 253.
Maeda K. // Journal of Photochemistry and Photobiology C: Photochemical Reviews. 2011. V. 12. № 4. P. 237.
Han F., Kambala V.S., Srinivasan M. et al. // Applied Catalysis A: General. 2009. V. 359. № 1–2. P. 25.
Antopoulou M., Evgenidou E., Lambropoulou D. et al. // Water Research. 2014. V. 53. P. 215.
Pelaez M., Nolan N.T., Pillai S.C. et al. // Applied Catalysis B: Environment. 2012. V. 125. P. 331.
Hoffmann M.R., Martin S.T., Choi W. et al. // Chemical Reviews. 1995. V. 95. P. 69
Akpan U.G., Hameed B.H. // Journal of Hazardous Materials. 2009. V. 170. № 2–3. P. 520.
Fujishima A., Rao T.N., Tryk D.A. // Journal of Photochemistry and Photobiology C: Photochemical Reviews. 2000. V. 1. № 1. P. 1.
Chen C., Ma W., Zhao J. // Chemical Society Reviews. 2010. V. 39. № 11. P. 4206.
Park H., Park Y., Kim W. et al. // Journal of Photochemistry and Photobiology C: Photochemical Reiews. 2013. V. 15. № 1. P. 1.
Shiraishi Y., Hirai T. // Journal of Photochemistry and Photobiology C: Photochemical Reiews. 2008. V. 9. № 4. P. 157.
Lincebigler A.L., Lu G., Yates J.T. // Chemical Reviews. 1995. V. 95. № 3. P. 735.
Xu Q., Yu J., Zhang J. et al. // Chemical Communications. 2015. V. 51. № 37. P. 7950.
Yu J., Low J. Xiao W. et al. // Journal of the American Chemistry Society. 2014. V. 136. № 25. P. 8839.
Li X., Wen J. Q., Low J. X. et al. // Science China Chemistry. 2014. V. 57. P. 70.
Fu J., Cao S., Yu J. et al. // Dalton Transactions. 2014. V. 43. № 24. P. 9158.
Li X., Liu H.L., Luo D.L. et al. // Chemical Engineering Journal. 2012. V. 180. P. 151.
Dhakshinamoorthy A., Navalon S., Corma A. et al. // Energy and Environment Science. 2012. V. 5. № 11. P. 9217.
Zhang Q.H., Han W.D., Hong Y.J. et al. // Catalysis Today. 2009. V. 148. № 3–4. P. 335.
Hemminger J.C., Carr R., Somorjai G.A. // Chemical Physics Letters. 1978. V. 57. № 1. P. 100.
Inoue T., Fujishima A., Konish S., Honda K. // Nature. 1979. V. 227. P. 637.
Hirano K., Bard A.J.Z. // Electrochemical Society. 1980. V. 127. № 5. P. 1056.
Reiche H., Bard A.J. // Journal of the American Chemical Society. 1979. V. 101. № 11. P. 3127.
Kanno T., Oguchi T., Sakuragi H. et al. // Tetrahedron Letters. 1980. V. 21. № 5. P. 467.
Taniguchi I., Nakashima K., Yamaguchi H., Yasukouchi K. // J. Electroanal. Chem. 1982. V. 134. P. 191.
Плесков Ю.В. // Электрохимия. 1981. Т. 17. С. 3.
Гринберг В.А., Джавришвили Т.В., Васильев Ю.Б. и др. // Электрохимия. 1984. Т. 20. Вып. 1. С. 121.
Гуревич Ю.Я., Плесков Ю.В. // Итоги науки и техники. Электрохимия. Т. 18. М.: ВИНИТИ, 1982. С. 3.
Mills A., Le Hunte S. // Journal of Photochemistry and Photobiology A: Chemistry. 1997. V. 108. № 1. P. 1.
Mills A., Lee S.K. // Journal of Photochemistry and Photobiology A: Chemistry. 2002. V. 152. № 1–3. P. 233.
Anpo M., Takeuchi M. // Journal of Catalysis. 2003. V. 216. P. 505.
Antoniadou M., Lianos P. // Journal of Nanoscience and Nanotechnology. 2010. V. 10. № 9. P. 6240.
Li L., Zhang S., Li G., Zhao H. // Analytica Chemica Acta. 2012. V. 754. P. 47.
Гринберг В.А., Емец В.В., Модестов А.Д. и др. // Электрохимия. 2017. Т. 53. № 2. С. 241. / Grinberg V.A., Emets V.V., Modestov A.D. et al. // Russian Journal of Electrochemistry. 2017. V. 53. № 2. P. 217.
Wen J., Li X., Liu W. et al. // Chinese Journal of Catalysis. 2015. V. 36. № 12. P. 2049.
Su R., Bechstein R., Kibsgaard J. et al. // Journal of Materials Chemistry. 2012. V. 22. № 45. P. 23755.
Tu Y.F., Huang S.Y., Sang J.P., et al. // Materials Research Bulletin. 2010. V. 45. № 2. P. 224.
Гринберг В.А., Емец В.В., Майорова Н.А. и др. // Физикохимия поверхности и защита материалов. 2018. Т. 54. С. 55.
. Baifu Xin, Liqiang Jing, Zhiyu Ren et al. // Journal of Physical Chemistry B. 2005. V. 109. P. 2805.
Sajjad S., Leghari S.A. K, Chen F. et al // Chemistry-A European Journal. 2010. V. 16. № 46. P. 13795.
Andronic L., Enesca A., Vladuta C., Duta A. // Chemical Engineering J. 2009. V. 152. P. 64.
Hajjaji A., Atyaoui A., Trabelsi K. et al. // American Journal of Analytical Chemistry. 2014. V. 5. № 8. P. 473.
Hoang S., Berglund S.P., Hahn N.T. et al. // Journal of American Chemical Society. 2012. V. 134. P. 3659.
Hoang S., Guo S.W., Hahn N.T. et al. // Nano Letters. 2011. V. 12. P. 26.
Wang G.M., Wang H.Y., Ling Y.C. et al. // Nano Letters. 2011. V. 11. P. 3026.
Maksimov Yu.V., Suzdalev I.P., Tsodikov M.V. et al. // Journal of Molecular Catalysis A: Chemical. 1996. V. 105. № 3. P. 167.
Kriventsov V.V., Kochubey D.I., Tsodikov M.V. et al. // Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Accociated Equement. 2001. V. 407. № 1–2. P. 331.
Jiang D., Zhao H., Zhang S. et al. // Journal of Physical Chemistry B. 2003. V. 107. № 46. P. 12774.
Zhang H., Zhao H., Zhang S. et al. // European Journal “ChemPhysChem”. 2008. V. 9. 117.
Kim D.H., Anderson M.A. // Journal of Photochemistry and Photobiology A: Chemistry. 1996. V. 94. № 2–3. P. 221.
Lana-Villarreal T., Goґmez R., Neumann-Spallart M. et al. // Journal of Physical Chemistry B. 2004. V. 108. P. 15172.
Lana-Villarreal T., M. Peґrez J. Goґmez R. // Comptes Rendus Chimie. 2006. V. 9. 806.
Gong X.-Q., Selloni A. and Vittadini A. // Journal of Physical Chemistry B. 2006. V. 110. P. 2804.
Montoya J.F., Peral J., Salvador P. // Journal of Physical Chemistry C. 2014. V. 118. № 26. P. 14266.
Montoya J.F., Atitar M.F., Bahnemann D.W. // Journal of Physical Chemistry C. 2014. V. 118. P. 14276.
Kim D.H., Anderson M.A. // Environmental Science and Technology. 1994. V. 28. № 3. P. 479.
Mitoraj D., Beranek R., Kisch H. // Photochemical and Photobiological Sciences. 2010. V. 9. № 1. P. 31.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов