

Физикохимия поверхности и защита материалов, 2019, T. 55, № 4, стр. 350-358
ТЕРМОИНИЦИИРУЕМЫЕ ПРОЦЕССЫ В АКТИВИРОВАННЫХ МЕТАЛЛОПОЛИМЕРНЫХ КОМПОЗИТАХ, ПОЛУЧЕННЫХ ПЛАСТИЧЕСКИМ ДЕФОРМИРОВАНИЕМ СМЕСЕЙ НА ОСНОВЕ АЛЮМИНИЯ ПОД ВЫСОКИМ ДАВЛЕНИЕМ
В. А. Жорин 1, *, М. Р. Киселев 2, В. А. Котенев 2
1 Учреждение Российской академии наук Институт химической физики
им. Н.Н. Семенова РАН
119991 Москва, ул. Косыгина, 4, Россия
2 Учреждение Российской академии наук Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119991 Москва, Ленинский пр., 31, Россия
* E-mail: vzhorin@mail.ru
Поступила в редакцию 14.01.2019
После доработки 16.01.2019
Принята к публикации 07.02.2019
Аннотация
Для металл-полимерного композита, полученного методом интенсивного пластического деформирования смесей алюминиевый порошок-полимер, исследовались пассивирующие свойства различных полимерных матриц, а также процессы их термического разложения в присутствии наполнителя – активных частиц алюминия, а также процессы последующей депассивации и окисления алюминия, когда полимерная фаза уже разложилась. Для этого смеси различных полимеров с 50 и 80 мас. % алюминия подвергали пластическому деформированию под давлениями 1 и 4 ГПа, а затем исследовали термогравиметрическим методом в температурном диапазоне 20–800°С в воздушной среде и в азоте. В диапазоне 20–450°С регистрировали снижение массы образов, связанное с разложением полимера, а в диапазоне 450–800°С увеличение массы, связанное с окислением и азотированием алюминия. В низкотемпературном диапазоне активность кислорода была ниже активности азота, а в высокотемпературном – выше.
ВВЕДЕНИЕ
Полимер-матричные композиционные материалы с органическими и неорганическими наполнителями широко используется в самых разных областях науки и техники [1, 2]. Помимо широко распространенных конструкционных материалов, такие композиты также используются в качестве функциональных наноматериалов и нанослоев. Особый интерес в последнее время вызывает использование, в качестве неорганической подсистемы металл-полимерного композита, активных металлических наночастиц, что позволяет получать энергонасыщенные композиционые материалы с выражеными свойствами активных металлических и металл-оксидных наночастиц [2], в комбинации с контролируемыми защитными и/или активирующими свойствами контактирующей с наночастицами полимерной матрицы [2, 3].
Одним из наиболее эффективных металлов, используемых в качестве энергонасыщенных композиционных материалов и компонентов твердых топлив, является алюминий [4, 5]. В настоящее время проводятся широкие исследования процесса горения как индивидуальных порошков алюминия разной дисперсности [6], так и смесей алюминия с разными компонентами с целью активирования порошкоа алюминия для интенсификации процессами горения [7]. В качестве добавок используют как порошкообразные металлы, так и органические соединения [8–10]. Так, в работе [11] исследовали влияние нитроцеллюлозы, олеиновой и стеариновой кислоты на процесс окисления нанодисперсных порошков алюминия. В работе [12] установили, что скорость окисления наночастиц алюминия с покрытием из олеиновой кислоты выше, чем с оксидным или фторполимерным покрытием, нанесенным на поверхность. В [13] было установлено, что скорость горения алюминия в нанокомпозитах алюминий-октаген тем выше, чем больше размер кристаллов октогена на частицах металла.
Эффективным методом активирования порошкообразных металлов является формирование свежевскрытых поверхностей частиц порошков, свободных от поверхностных оксидных фаз, например в ходе механического разрушения или в ходе осаждения и конденсации на подложке в глубоком вакууме [14, 15]. Одним из методов механического создания свежевскрытых поверхностей в твердых телах различной химической природы является пластическое деформирование под высоким давлением на аппаратуре высокого давления типа наковален Бриджмена [14, 16, 17]. При деформировании происходит не только уменьшение размеров индивидуальных частиц, сопровождаемое образованием свежевскрытых поверхностей, но и формирование развитой межфазной границы.
Известно, что свежевскрытая поверхность алюминия обладает высокой химической активностью, но на воздухе быстро пассивируется, покрываясь оксидной пленкой. При этом в случае ультрадисперсных порошков с большой свободной поверхностью [4–6] получаемые порошки настолько активны, что сгорают при контакте с воздухом. Даже при высоких степенях вакуумирования при комнатной температуре алюминий чрезвычайно быстро взаимодействует с окислителем (кислородом) остаточного вакуума [15].
Поэтому активную поверхность активированного алюминиевого порошка необходимо защищать от воздействия воздушной атмосферы. Для этого алюминий подвергают пассивации, окисляя при высоких степенях вакуумирования [15], когда экзотермические эффекты выделения тепла невысоки.
Между тем, для пасссивации свежевскрытую поверхность порошкообразного алюминия можно формировать непосредственно в объеме полимерной матрицы при пластическом деформировании смесей металл-полимер, когда формируется плотный межфазный контакт. При этом, очевидно, высокая активность алюминия проявится главным образом только во время деформирования. После завершения обработки под давлением а также при последующем нагревании деформированных смесей в этом случае полимерная фаза будет существенно ограничивать доступ кислорода к активной поверхности алюминиевых частиц. Очевидно, полученный металл-полимерный композит будет консервировать химически активный алюминий, но при превышении температуры выше предельной температуры плавления/деградации полимерной фазы, будет окисляться с достаточно высокой эффективностью экзотермических процессов.
Однако в строении различных полимеров присутствуют химически активные функциональные группы (спиртовые, кислотные, амидные, Cl, F), активность которых может возрасти под действием высокого давления. Это может оказать влияние на структуру и пассивирующие свойства слоев образующихся на поверхности алюминиевых частиц, которые в свою очередь могут оказать влияние на процесс окисления алюминия. Так, в работах [18–20] исследовали смеси алюминия с различными полимерами (полипропилен, поливинилхлорид, тефлон) после пластического деформирования под давлением 1 ГПа. При нагревании деформированных смесей в диапазоне 20–250°С в них протекают экзотермические процессы, связанные с взаимодействием полимера со свежевскрытой поверхность алюминия на металлических частицах. Энтальпия этих процессов зависела от соотношения компонентов и достигала максимальных значений при 40–60 мас. % алюминия. Кроме того, при нагревании деформированных смесей органическая кислота – алюминий и полиэтилен – алюминий регистрировали интенсивные экзотермические процессы [21, 22], а в работах [23–25] было установлено, что в деформированных смесях полиэтилен – оксиды металлов энтальпия плавления полиэтилена возрастает в 2–8 раз.
В данной работе для активированного металл-полимерного композита, полученного методом интенсивного пластического деформирования смесей алюминиевый порошок – полимер, исследовались пассивирующие свойства различных полимерных матриц, а также процессы их термического разложения в присутствии наполнителя – активных частиц алюминия, а также процессы последующей депассивации и окисления алюминия, когда полимерная фаза уже разложилась.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве объектов исследования был выбран порошкообразный алюминий марки АСД-6 с размером частиц 5–10 мкм и порошкообразные полимеры с размером частиц 10–30 мкм: полиэтилен низкой плотности (ПЭНП), полиэтилен высокой плотности (ПЭВП), полипропилен (ПП), полистирол (ПС), поливинилхлорид (ПВХ), поливинилфторид (ПВФ), поливиниловый спирт (ПВС), микрокристаллическая целлюлоза (МКЦ), поликарбонат (ПК), полисульфон (п-сульфон), поликетон (п-кетон), полифенилацетилен (ПФАЦ), полифениленоксид (ПФО), полиимид (п-имид), полиакриламид (ПАА). Смеси исходных компонентов готовили в ступке. Деформирование исходных компонентов и металлополимерных смесей проводили под давлением 1 и 4 ГПа при комнатной температуре на наковальнях из каленой стали ХВГ с диаметром рабочих поверхностей 20 мм. Для анализа выбирали краевую зону образцов шириной 3 мм. Толщина образцов в этой зоне составляла 80–100 мкм. Угол поворота наковален (величина пластической деформации) составлял 600 град. Изменения массы образцов при нагревании изучали на приборе Q500 TA INSTRUMENT при скорости нагревания 20 град мин–1 в диапазоне 25−900°С в воздушной среде и в азоте марки “нулевой”. Массу образцов варьировали от 3 до 8 мг.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Исходный алюминий представлял собой серый порошок, цвет которого определялся оксидной пленкой на поверхности металлических частиц. Количество оксида алюминия на поверхности алюминиевых частиц в зависимости от способа получения и условий хранения может варьироваться от 3 до 13%. После деформирования образцы представляли собой монолитные диски с ярким, характерным металлическим блеском. В исходном порошкообразном алюминии в температурном диапазоне 30–420°С происходило снижение массы (“–м”) на 0.3%, а в образцах обработанных под давлением “–м” составило 0.1% [1, 2]; эффект снижения “–м” скорее всего связан с компактированием порошкообразного алюминия при пластическом деформировании. Дальнейшее повышение температуры до 800°С приводило к увеличению массы образцов (“+м”) на 1%, связанному с окислением алюминия.
В ряду выбранных полимеров присутствуют как аморфные, так и аморфно-кристаллические полимеры, различающиеся своими характерными температурами; так для ПЭНП Тпл = 110°С, а для ПК Тпл = 260°С; для ПС Тс = 85°С, а для ПАА Тс = 240°С; степени полимеризации полимеров варьируются в пределах 102–106. После деформирования полимерные образцы представляли собой прозрачные слабо окрашенные диски. При пластическом деформировании в полимерах протекает деструкция, приводящая к снижению молекулярной массы, а в аморфно-кристаллических полимерах разрушается кристаллическая структура. Все выбранные полимеры полностью разлагаются при нагревании, и этот процесс не зависит от обработки под давлением; начало процесса разложения для разных полимеров лежит в диапазоне температур от 240 до 320°С. В исходных металлополимерных смесях разного состава присутствие частиц алюминия не влияло на процесс разложения полимера.
В предварительных экспериментах со смесями разного состава было установлено, что изменения с полимерами, происходящие при температурах до 400°С, удобнее исследовать в смесях полимер – 50 мас. % Al, а за поведением алюминия при окислении (температуры выше 450°С) удобнее следить в смесях полимер – 80 мас. % Al. В ультрадисперсных порошках алюминия, пассивированных органическими компонентами, содержание органики также бывает близко к 20 мас. %.
Образцы смесей полимер-50% Al после деформирования под давлением 1 ГПа были черного цвета и источали резкий неприятный запах, что свидетельствует о выделении летучих продуктов, которые, скорее всего, образовались при химических превращениях уже на стадии деформирования под давлением.
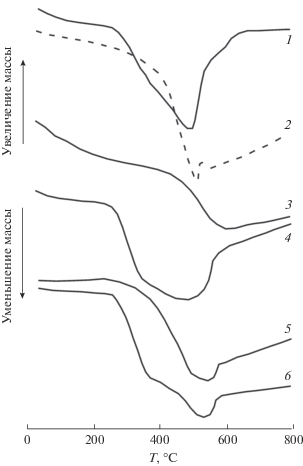
На рис. 1 приведены зависимости изменения массы для некоторых смесевых образцов при нагревании в воздушной среде, которые свидетельствуют о том, что в деформированных смесях “–м”, связанное с разложением полимера, начинается уже при 25–30°С и заканчивается при 450–550°С. Снижение массы было всегда меньше 50%, что указывает на образование термостойких продуктов. При дальнейшем повышении температуры в образцах происходило увеличение массы, связанное с взаимодействием алюминия с компонентами воздушной среды – кислородом и азотом воздуха; при этом максимальное значение “+m” не превышало 14%.
Рис. 1.
Изменение массы смесей полимер-50% Al, деформированных под давлением 1 ГПа при нагревании: ПВФ (1), п-кетона (2), МКЦ (3), п-сульфона (4), ПЭВП (5), ПВС (6).
В табл. 1 приведены значения “–m” для смесей полимер-50% Al после деформирования под давлением 1 ГПа, которые можно разделить на две группы. В первую группу можно отнести смеси, для которых снижение массы варьируется в пределах 15–23% (расчет проведен на полную массу образцов); в гетероцепных полимерах, входящих в эту группу, нет боковых заместителей, хотя они значительно различаются строением цепи. Во вторую группы можно отнести смеси, в которых снижение массы выражено значительно сильнее и лежит в пределах 40–45% (расчет проведен на полную массу образцов); в этом случае все полимеры являются линейной цепочкой, построенной из атомов углерода, с большим содержанием различных боковых групп (–СH3, –C–OH, –C–CONH2).
Таблица 1.
Снижение массы (–м, %), конверсия полимерной фазы при отмывке растворителем (КI, %), приращение массы (+м, %) в смесях алюминий-50 мас. % полимера после деформирования под давлением 1 ГПа
| Полимер | –м, % | KI, % | +м, % |
|---|---|---|---|
| ПФО | 18 | 97 | 5.3 |
| ПК | 20 | 80 | 6.6 |
| п-Кетон | 20 | 3.7 | |
| п-Имид | 20 | 2.0 | |
| ПФАЦ | 22 | 60 | 2.0 |
| п-Сульфон | 23 | 80 | 0.4 |
| МКЦ | 28 | 93 | 14.0 |
| ПВФ | 15 | 11.0 | |
| ПС | 20 | 87 | 12.2 |
| ПЭВП | 40 | 12.2 | |
| ПЭНП | 42 | 10.5 | |
| ПВХ | 45 | 43 | 11.0 |
| ПП | 45 | 12.0 | |
| ПВС | 45 | 70 | 10.0 |
Малые значения “–м” означают, что в смесях образовалось много термостойких продуктов, а большие “–м” то, что термостойких продуктов образовалось мало. Таким образом, химическая активность полимеров первой группы в процессах, приводящих к образованию термостойких продуктов, выше, чем для полимеров второй группы. Как видно из данных таблицы в первую группу попали ПВФ и ПС, хотя по строению они должны были бы находиться во второй группе. Возможно, это связано с тем, что ПВФ содержит в своем строении высокоактивный фтор, а в ПС при обработке под высоким давлением образуются сшитые структуры [8].
В линейных полимерах (ПП, ПВС, ПВХ, ПАА), содержащих одинаковое количество, но разных заместителей (–СН3, –СОН, –Cl, –С–СОNН2) величины “–м” совпадали. В ряду ПП-ПЭВП-ПЭНП в качестве боковой выступает –СН3– группа. При этом количество разветвлений в ПП составляет n = 50 на 100 атомов углерода основной цепи; аналогичный параметр в ПЭВП и ПЭНП составляет n = 0.3–0.6 и n = 1.5–2.5, соответственно. Не смотря на это различие величины “–м” для этих полимеров также совпадают.
Таким образом, можно сказать, что боковые группы в исследованных линейных полимерах не оказывают влияния на процессы, приводящие к образованию термостойких продуктов при нагревании деформированных металлополимерных смесей.
В табл. 1 приведены значения “+м” для смесей полимер-50 мас. % Al. Полученные результаты можно разделить на две группы. В первую группу входят все гетероцепные полимеры; в этой группе величины “+м” варьируются от 0.4 до 6.6% (средняя величина составляет 3.2%). Во вторую группу входят все линейные полимеры, для которых значения “+м” варьируются от 10 до 14% (средняя величина “+м” в этой группе составляет 11.6%). Минимальное значение “+м” = 0.4% было установлено в смеси с поли-сульфоном, а максимальное “+м” = 14% было достигнуто в смеси с МКЦ.
При нагревании деформированных смесей в них протекают два последовательных процесса: снижение массы, связанное с термическим разложением полимера, и увеличение массы, связанное с окислением алюминия. Процесс окисления интенсивно протекает в диапазоне 500–580°С и этот диапазон не зависит от полимера смеси. В то же время процесс разложения линейных полимеров, в основном, заканчивается при 450°С, а гетероцепных полимеров – при температурах выше 600°С. Таким образом, различие в величинах “+м”, скорее всего, связано не с природой полимерных компонентов, а с их термической стабильностью.
Образование термостойких продуктов в металлополимерных смесях возможно как при деформировании, так и при последующем нагревании деформированных смесей. О протекании химического взаимодействия между полимером и металлом уже во время деформирования свидетельствует почернение образцов и газовыделение. Представляет интерес вопрос о том, когда происходит химическое превращение в полимерной фазе – при деформировании смесей или при их нагревании.
Некоторые из выбранных полимеров в обычных условиях растворяются в простых растворителях (вода, ацетон, хлороформ и т.д.). Обработка этих полимеров под высоким давлением не приводила к изменению их растворимости; исключением является ПС, в котором при деформировании под высоким давлением образуются сшитые структуры. В этой связи, полимерную фазу в некоторых деформированных смесях отмывали растворителями, определяли количество нерастворимых продуктов и рассчитывали конверсию (КI,% = количество нерастворимых продуктов относили к количеству полимера в исходной смеси). Оказалось (табл. 1), что конверсии для большинства смесей были 60% и выше. Таким образом, большая часть полимера в деформированных металлополимерных смесях не растворялась. Это может быть связано с образованием сшитых структур, привитием полимерной цепи к свежевскрытой поверхности металлических частиц, образованием элементоорганических соединений, карбонизацией.
Растворение полимеров в деформированных металлополимерных смесях сопровождалось интенсивным газовыделением, которое продолжалось в течение 15–20 мин. Наиболее интенсивное газовыделение наблюдали в смесях с ПС, ПАА и ПВС, а в смесях с ПФАЦ газовыделения вообще не было. Таким образом, интенсивность газовыделения при растворении полимерной фазы не связана с химической активностью полимеров. Для смеси ПАА-50% Αl методом ГПХ был проведен анализ газов, выделяющихся при растворении. Оказалось, что в газовой смеси содержатся водород и метан. Возможно, что газовыделение связано с взаимодействием растворителей с атомами алюминия на свежевскрытых поверхностях металлических частиц.
Дальнейшие термогравиметрические измерения проводили на смесях содержавших 80 мас. % Al. После деформирования такие образцы представляли собой монолитные диски с характерным ярким металлическим блеском. Зависимости изменений массы при нагревании деформированных смесей полимер + 80% Al состояли из двух характерных участков (рис. 2). В диапазоне от комнатной температуры до 500°С происходило снижение массы, связанное с разложением полимера, начинавшееся при 280–290°С (исключением является ПФАЦ, у которого разложение начиналось при 430°С). В смеси на основе ПВФ потери массы зарегистрировать не удалось – полимер полностью реагировал с алюминием с образованием термостойких продуктов; в случае смеси с ПАА полимер разлагался полностью, то есть вообще не реагировал с алюминием; в остальных смесях снижение массы было всегда меньше количества полимера находившегося в смеси. При температурах выше 530–540°С регистрировали возрастание массы, связанное с взаимодействием алюминия с воздушной средой, в которой проводили измерения.
Рис. 2.
Изменение массы смесей полимер-80% Al, деформированных под давлением 1 ГПа при нагревании в воздушной среде – ПВФ (1), МКЦ (2), ПВС (3), ПФАЦ (4), ПЭВП (6) и в азоте – ПЭВП (5).

При увеличении содержания алюминия в смесях от 50 до 80 мас. % в температурном диапазоне до 500°С регистрировали общее снижение численных значений “–м”, связанное с уменьшением количества полимера в смесях, но, при этом возрастало относительное содержание термостойких продуктов.
В смесях с гетероцепными полимерами после обработки под давлением 1 ГПа максимальное значение “–м”) составило 2.2% (п-сульфон), а минимальное значение “–м” = 0.8% регистрировали в смесях с ПК. Средняя величина “–м” для гетероцепных полимеров после деформирования под давлением 1 ГПа составила 1.68%. Если в смесях с 50 мас. % Al до 60% полимера участвовало в образовании термостабильных продуктов, то в смесях с 80 мас. % Al это значение возрастало до 96%. Увеличение давления обработки до 4 ГПа приводило к увеличению среднего значения “–м” до 3.0% (исключение ПФАЦ). Такой эффект может быть связан с тем, что при более высоком давлении в смесях образуется большое количество летучих продуктов. Таким образом, давление деформирования может существенно изменять состав образующихся продуктов.
В линейных полимерах после деформирования под давлением 1 ГПа минимальное снижение массы составило 7.6% (ПВС), а максимальное – 20% зарегистрировали в смеси с ПАА (полимер полностью разлагался); среднее значение “–м” составляет 12.4%. Увеличение давления обработки до 4 ГПа приводило к снижению “–м” во всех смесях, то есть к увеличению количества термостойких продуктов; величина эффекта снижения была минимальной для смеси с ПВС – 1.2 раза, а максимальной для смеси с ПЭНП – 2.5 раза; среднее значение “–м” = 8%. Это может быть связано с тем, что при увеличении давления обработки усиливаются химические процессы при деформировании, больше полимера расходуется на образование термостойких продуктов, и, поэтому, уменьшаются массовые потери.
В табл. 2 приведены данные об увеличении массы в смесях полимер-80 мас. % Al аосле деформирования под давлением 1 и 4 ГПа. В ряду смесей с гетероцепными полимерами, обработанными под давлением 1 ГПа, величина “+м” варьировалась от 4% в п-имиде до 18% в ПК; в этом ряду среднее значение “+м” = 11.3%. Увеличение давления деформирования до 4 ГПа для всех смесей приводило к снижению “+м”; среднее значение снижалось до 6.9%.
Таблица 2.
Снижение массы (–м, %), конверсия полимерной фазы при нагревании (KII, %) и приращение массы (+м, %) для разных полимеров в смесях с 80% алюминия после деформирования под давлением (Р, ГПа)
| Полимер Р, Гпа C, % | Среда –м, % +м, % | P, ГПа | –м, % | K II, % | +м, % |
|---|---|---|---|---|---|
| ПК | Воздух | 1 | 0.8 | 96 | 18.0 |
| 4 | 3.7 | 81 | 12.5 | ||
| Азот | 1 | 2.7 | 86 | 3.6 | |
| 4 | 7.1 | 64 | 1.3 | ||
| п-Имид | Воздух | 1 | 1.0 | 95 | 4.0 |
| 4 | 3.9 | 80 | 1.5 | ||
| ПФО | Воздух | 1 | 2.0 | 90 | 13.0 |
| 4 | 5.4 | 73 | 6.0 | ||
| п-Сульфон | Воздух | 1 | 2.2 | 89 | 8.0 |
| 4 | 1.8 | 91 | 5.5 | ||
| ПФАЦ | Воздух | 1 | 2.0 | 90 | 8.6 |
| 4 | 1.1 | 94 | 3.0 | ||
| п-Кетон | Воздух | 1 | 1.8 | 91 | 14.0 |
| 4 | 2.3 | 88 | 10.0 | ||
| МКЦ | Воздух | 1 | 2.2 | 89 | 13.8 |
| 4 | 2.8 | 86 | 10.0 | ||
| Азот | 1 | 4.5 | 77 | 2.3 | |
| 4 | 3.1 | 84 | 1.3 | ||
| ПС | Воздух | 1 | 3.9 | 80 | 32.0 |
| 4 | 2.3 | 88 | 9.6 | ||
| Азот | 1 | 5.4 | 73 | 6.8 | |
| 4 | 3.5 | 82 | 5.4 | ||
| ПВФ | Воздух | 1 | 0 | 100 | 27.0 |
| 4 | 0.7 | 96 | 18.0 | ||
| Азот | 1 | 4.5 | 78 | 8.4 | |
| 4 | 5.6 | 72 | 2.6 | ||
| ПВС | Воздух | 1 | 7.6 | 62 | 24.0 |
| 4 | 6.2 | 69 | 16.8 | ||
| Азот | 1 | 8.8 | 56 | 3.1 | |
| 4 | 6.0 | 70 | 3.2 | ||
| ПП | Воздух | 1 | 10.0 | 50 | 27.0 |
| 4 | 5.4 | 73 | 34.0 | ||
| Азот | 1 | 14.2 | 25 | 5.0 | |
| 4 | 16.9 | 16 | 10.4 | ||
| ПЭНП | Воздух | 1 | 15.6 | 22 | 17.8 |
| 4 | 6.5 | 69 | 30.2 | ||
| Азот | 1 | 4.3 | 78 | 8.4 | |
| 4 | 5.4 | 72 | 2.6 | ||
| ПЭВП | Воздух | 1 | 15.0 | 25 | 13.0 |
| 4 | 11.0 | 45 | 24.0 | ||
| ПВХ | Воздух | 1 | 14.7 | 26 | 8.4 |
| 4 | 6.8 | 66 | 7.6 | ||
| ПАА | Воздух | 1 | 20.0 | 0 | 6.4 |
| 4 | 20.0 | 0 | 7.9 |
В смесях с линейными полимерами, деформированными под давлением 1 ГПа, величина “+м” варьировалась в широких пределах – от 6.4% в ПАА до 32% в ПС. В смесях с ПС, ПВФ, ПВС, ПП, содержащих одинаковое число значительно различающихся по химическим свойствам боковых групп, значения “+м” совпадали. В ряду ПП, ПЭНП, ПЭВП величина “+м” снижается по мере уменьшения числа CH3–групп, приходящихся на 100 атомов углерода основной цепи.
При увеличении давления деформирования до 4 ГПа величина “+м” в смесях изменялась разнонаправлено: для ПС, ПВФ, ПВС, ПВХ “+м” снижалась, а для ПП, ПЭНП, ПЭВП, ПАА – увеличивалась. В ряду ПС-ПВХ-ПАА значения “+м” различались мало (9.6–7.6–7.9%), а для ПВФ и ПВС практически совпадали (18 и 17%). В ряду ПП-ПЭНП-ПЭВП величины “+м” снижались, как и после обработки под давлением 1 ГПа, но если после обработки под давлением 1 ГПа снижении составило 2.2 раза, то после обработки под давлением 4 ГПа снижение составило 1.36 раза.
В работах [26, 27] было установлено, что при горении на воздухе ультрадисперсные порошки алюминия взаимодействуют не только с кислородом, но и с азотом. Представляло интерес провести термогравиметрическое исследование некоторых деформированных смесей в атмосфере азота. Зависимость изменения массы при нагревании в деформированной смеси ПЭВП-80% Al приведена на рис. 2. Видно, что температура начала разложения полимера смещается в высокотемпературную область, а также изменяется характер зависимости, описывающей процесс возрастания массы в высокотемпературном диапазоне.
При измерениях в азоте исключается взаимодействие материала образца с кислородом воздуха и при этом не происходит окисление алюминия. В табл. 2 приведены данные о поведении “–м” в смесях полимер-80% Al в азотной среде после деформирования под давлением 1 ГПа, которые свидетельствуют о возрастании “–м” по сравнению с воздушной средой (исключение составил ПЭНП, у которого “–м” в азоте снижалась в 3.6 раза по сравнению с воздушной средой). Увеличение давления деформирования до 4 ГПа приводило к уменьшению “–м” (увеличивалось количество термостойких продуктов) в смеси с МКЦ в 1.45 раза, в ПС – в 1.5 раза, а в ПВС – в 1.46 раза. В то же время в некоторых смесях регистрировали увеличение “–м” – так в смесях с ПК увеличение составило 2.6 раза, в смесях с ПВФ – 1.3 раза, с ПП – 1.2 раза, а в ПЭНП – 1.25 раза.
Во всех металлополимерных смесях, исследования которых проводили в азоте, значения “+м” были меньше, чем при измерениях в воздушной среде: в ПВФ в 3.1 раза, а в ПВС в 1.4 раза. В то же время в ПП зарегистрировали 2.1 кратное увеличение “+м”.
Представляло интерес сравнить активности азота и кислорода на разных стадиях нагревания деформированных смесей в воздушной среде, когда возможно взаимодействие, как с кислородом воздуха (окисление), так и с азотом (азотирование). В случае азота его активность можно оценить из данных табл. 2 по количеству образовавшихся термостойких продуктов при измерениях в азоте. В воздухе содержится 70% азота, поэтому количество термостойких продуктов, которое образуется при взаимодействии с азом при измерениях в воздухе, буде составлять 70%, от количества термостойких продуктов, полученного при измерениях в азоте. Количество термостойких продуктов, образующихся за счет взаимодействия с кислородом воздуха, можно получит как разницу между общим количеством термостойких продуктов образующихся при измерениях в воздушной среде и рассчитанного количества термостойких продуктов, образующихся при взаимодействии с азотом. Результаты такой обработки для смесей, деформированных под давлением 1 и 4 ГПа, представлены в табл. 3.
Таблица 3.
Количество термостойких продуктов, образующихся в температурном диапазоне разложения полимера (диапазон –м, %), и количество новых продуктов, образующихся в высокотемпературном диапазоне (диапазон +м, %) при взаимодействии с азотом и кислородом после деформирования под давлением 1 и 4 ГПа
| Полимер | Давление ГПа | Количество термостойких продуктов в диапазоне –м, % | Количество новых продуктов в диапазоне +м, % | ||
|---|---|---|---|---|---|
| азот | кислород | азот | кислород | ||
| ПВФ | 1 | 11.0 | 9.0 | 5.9 | 21.1 |
| 4 | 10.1 | 9.2 | 1.8 | 16.2 | |
| ПК | 1 | 12.1 | 7.1 | 2.5 | 15.5 |
| 4 | 9.0 | 7.3 | 0.9 | 11.6 | |
| ПС | 1 | 10.2 | 5.9 | 4.8 | 32.0. |
| 4 | 11.5 | 6.2 | 3.8 | 5.8 | |
| МКЦ | 1 | 11.0 | 6.8 | 7.0 | 6.8 |
| 4 | 11.8 | 5.4 | 0.9 | 1.4 | |
| ПВС | 1 | 7.8 | 4.6 | 2.2 | 21.8 |
| 4 | 9.8 | 8.0 | 2.2 | 14.6 | |
| ПП | 1 | 3.6 | 6.4 | 3.5 | 23.5 |
| 4 | 2.3 | 12.3 | 7.3 | 26.7 | |
| ПЭНП | 1 | 11.0 | 10.1 | 5.9 | 11.9 |
| 4 | 3.8 | 1.8 | 28.4 | ||
После обработки под давлением 1 ГПа и при измерениях в азоте количество термостойких продуктов в большинстве смесей различались мало, а среднее значение составило 9.5% (табл. 3). При измерениях в воздушной среде количество термостойких продуктов, связанное с взаимодействием с кислородом, варьировалось от 4.6 до 10%, а среднее значение составило 6.6%. Увеличение давления деформирования до 4 ГПа приводило лишь к небольшим изменениям, как для азота, так и для кислорода. Среднее количество термостойких продуктов образовавшихся при взаимодействии с азотом составило 9.2%, а с кислородом – 7.4%. Таким образом, количество термостойких продуктов образующихся при взаимодействии с азотом несколько больше, чем при взаимодействии с кислородом. Это свидетельствует о том, что активность азота в химических процессах, протекающих в низкотемпературном диапазоне, выше, чем активность кислорода.
В высокотемпературном диапазоне после деформирования под давлением 1 ГПа и при измерениях в азоте среднее количество образующихся продуктов составило 4.5%, а среднее количество продуктов при окислении составило 19.7%. Увеличение давления деформирования до 4 ГПа приводило к снижению количества образующихся продуктов, как при азотировании, так и при окислении – среднее значение для азотирования снижалось до 2.72%, а для окисления до 16.1%. Полученные результаты свидетельствуют о том, что в высокотемпературном диапазоне активность кислорода значительно выше активности азота.
ЗАКЛЮЧЕНИЕ
В смесях гетероцепных полимеров с 50 мас. % алюминия после деформирования под давлением 1 ГПа 80–97% полимерной фазы перестает растворяться. В смесях с линейными полимерами доля нерастворимой фракции варьировалась от 36 до 43%. Эти результаты свидетельствуют о том, что уже во время деформирования в присутствии алюминия в исследуемых полимерах протекают химические процессы. При нагревании деформированных смесей полимерная фаза разлагается, и при этом образуются термостабильные продукты, количество которых в смесях с гетероцепными полимерами достигает 30%, а в смесях с линейными – 10% от общей массы образцов. Таким образом, смеси с гетероцепными полимерами химически более активны как при деформировании, так и при последующем нагревании.
В смесях гетероцепных полимеров содержавших 80 мас. % алюминия после деформирования под давлением 1 ГПа до 95% полимера участвовало в образование термостойких продуктов, а в смесях ПЭНП, ПЭВП, ПВХ этот процент не превышал 26%.
Наибольшие приращения массы в высокотемпературном диапазоне были зарегистрированы в смесях с ПС – 32%, ПП и ПВФ – 27%. В этих смесях увеличение давления обработки до 4 ГПа приводило к снижению “+м”. В то же время в смесях с ПП и ПЭНП величина “+м” возрастала до 34 и 30%, соответственно. Таким образом, максимальное значения “+м” в деформированных смесях при Т = 800°С не превышало 34%.
Измерения в азоте показали, что азот и кислород взаимодействую с материалом деформированных образцов. В температурном диапазоне разложения полимера активность азота выше, чем активность кислорода. В высокотемпературном диапазоне химическая активность кислорода значительно превышает активность азота.
Список литературы
Полимерные смеси – под ред. Пола Д., Ньюмена С. М.: Мир, 1981.
Hybrid Nanocomposites for Nanotechnology. Ed. Mehrani L. Springer Science + Business Media, LLC 2009.
Oxide Thin Films, Multilayers, and Nanocomposites. Ed. Mele P., Endo T., Arisawa S., Li C., Tsuchiya T. Springer Science + Business Media, 2015.
Иванов В.Г., Гаврилюк О.В. // Физика горения и взрыва. 1999. Т. 35. № 6. С. 53–60.
Ильин А.П., Громов А.А., Яблуновский Г.В. // Физика горения и взрыва. 2001. Т. 37. № 4. С. 58–62.
Сандарам Д., Янг В., Зарко В.Е. // Физика горения и взрыва. 2015. Т. 51. № 2. С. 37–63.
Громов А.А., Попенко Е.М., Ильин А.П. // Химическая физика. 2005. Т. 24. № 4. С. 69–83.
Dreizin E.L., Shosin Yu.L., Mudryy R.S., Hoffman V.K. // Combustion and Flame. 2002. V. 130. № 4. P. 381–387.
Брейтлер А.Л., Мальцев В.М., Попов Е.И. // Физика горения и взрыва 1990. Т. 26. № 1. С. 97–104.
Шевченко В.Г., Кононенко В.И., Булатов М.Л., Латош И.Н., Чупова И.А. // Физика горения и взрыва 1998. Т. 34. № 1. С. 45–49.
Громов А.А., Ильин А.П., Бозе-Бат У., Тайпель У. // Физика горения и взрыва. 2006. Т. 42. № 2. С. 61–69.
Lewis W.K., Rumchik C.G., Smith M.J., Fernando K.A.S., Crouse C.A., Spowart J.E., Guliants E.A., Bunker C.E. // J. Appl. Phys. 2013. V. 113. P. 044907. https://doi.org/10.1063/ 1.4790159
Жигач А.Н., Лейпунский И.О., Пивкина А.Н., Муравьев И.В., Моногаров К.А., Кусков М.Л., Афанасенкова Е.С., Березкина Н.Г., Пшечков П.А., Брагин А.А. // Физика горения и взрыва. 2015. Т. 51. № 1. С. 117–124.
Котенев В.А., Жорин В.А., Киселев М.Р., Высоцкий В.В., Аверин А.А., Ролдугин В.И., Цивадзе А.Ю. // Физикохимия поверхности и защита материалов. 2015. V. 51. № 5. С. 512–516.
Котенев В.А. // Физикохимия поверхности и защита материалов. 2000. Т. 36. № 5. С. 451–461.
Zhorin, V.A., Kiselev, M.R., and Kotenev, V.A., Prot. Met. Phys. Chem. Surf., 2018, vol. 54 no. 5, pp. 853–858.
Zhorin, V.A., Kiselev, M.R., and Kotenev, V.A., Prot. Met. Phys. Chem. Surf., 2017, vol. 53, no. 6, pp. 1075–1081.
Жорин В.А., Киселев М.Р., Ролдугин В.И. // Журн. прикл. химии. 2012. Т. 85. № 4. С. 571–575.
Жорин В.А., Киселев М.Р., Ролдугин В.И. // Журн. прикл. химии. 2013. Т. 86. № 1. С. 18–23.
Жорин В.А., Киселев М.Р., Ролдугин В.И. // Физикохимия поверхности и защита материалов. 2014. Т. 50. № 3. С. 331–336.
Жорин В.А., Киселев М.Р., Ролдугин В.И. // Журн. физической химии. 2017. Т. 91. № 11. С. 1–9.
Жорин В.А., Киселев М.Р., Грачев А.В., Ладыгина Т.А. // Физика горения и взрыва. 2018. Т. 54. № 1. С. 1–13.
Жорин В.А., Киселев М.Р. // Пластические массы. 2017. № 3–4. С. 6–11.
Zhorin V.A., Kiselev M.R., Kotenev V.A. // Protection of metals and physical chemistry of surfaces. 2017. V. 53. № 5. P. 819–825.
Жорин В.А., Киселев М.Р., Котенев В.А. // Физикохимия поверхности и защита материалов. 2017. Т. 53. № 4. С. 418–425.
Ильин А.П., Яблуновский Г.В., Громов А.А. // Физика горения и взрыва. 1996. Т. 32. № 2. С. 108–110.
Ильин А.П., Толбанова Л.О. // Физика горения и взрыва. 2007. Т. 43. № 4. С. 59–64.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов