

Физикохимия поверхности и защита материалов, 2019, T. 55, № 4, стр. 346-349
Особенности сольватации галоидных ионов в протонных и апротонных средах
Р. Н. Куклин 1, *, В. В. Емец 1
1 ФГБУН Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский проспект, 31, Россия
* E-mail: rnkuklin@mail.ru
Поступила в редакцию 14.01.2019
После доработки 04.02.2019
Принята к публикации 07.02.2019
Аннотация
Рассмотрены особенности взаимодействия галоидных анионов с молекулами апротонного и протонного растворителя в ближней координационной сфере и их влияние на сольватацию в растворах. Если энергия сольватации в апротонном растворе представлена в основном электрической составляющей, то в протонных средах энергия сольватации содержит наряду с электрической химическую составляющую, обусловленную водородной связью. Предложена микроскопическая модель взаимодействия аниона с молекулой протонного растворителя, имитирующая динамику атома водорода в полях протонного донора и акцептора, позволяющая объяснить механизм формирования химической составляющей энергии сольватации.
ВВЕДЕНИЕ
Ионы в растворе, взаимодействуя с ближайшими молекулами растворителя, организуют сложные образования – сольватные структуры, которые определяют электрохимические и термодинамические свойства электролита. Природа их формирования сложна, соответственно существуют различные подходы интерпретации механизмов их образования: физические, химические, квантово-механические, молекулярной динамики, компьютерные симуляции, отражающие участие различных степеней свободы среды в процессе сольватации [1, 2].
Физические модели, претендующие на универсальность подхода в описании явления сольватации, оказались успешными при оценке вклада поляризационных степеней свободы раствора в энергию сольватации [3]. Первоначальная идея Борна получила дальнейшее развитие в работах [4, 5], использующих континуальные модели поляризуемости сред с пространственной дисперсией с детализацией описания форм-фактора заряда источника. Модели оказались успешными при интерпретации сольватации в средах с минимальным влиянием химической составляющей – в апротонных растворителях. Однако в протонных растворах важные моменты, обусловленные водородными связями, оказываются за рамками теории.
Проблема разделения химической и электрической составляющих энергии сольватации является актуальной в теории сольватационных явлений. Очевидно, что невозможно объяснить многообразие взаимодействий в ближних координационных сферах ионов только лишь электростатическими моделями. Так при взаимодействии анионов с молекулами протонных растворителей имеют место частичный перенос как электронной, так и протонной плотности. Как выход из положения были предложены некоторые решения в рамках электрохимической феноменологии [3, 6]. Однако существует ощущение необходимости моделей, более детально отражающих химические механизмы.
Современные вычислительные средства позволяют постановку как квантово-химических расчетов конкретных структур фактически из первых принципов, так их имитацию средствами молекулярной динамики или методами компьютерных симуляций [7], но большой объем вычислений утрачивает наглядность и общность получаемых результатов.
Существует, однако прецедент, где удается рассмотрение парциальных вкладов физических и химических факторов в сольватационные процессы. Это случай сольватации галоидных анионов в протонных растворителях.
Настоящая работа посвящена исследованию особенностей взаимодействий щелочно-галоидных ионов с молекулами апротонного (диметилформамида (ДМФА)) и протонного (формамида (ФА)) растворителей. В протонной среде оказывается существенным учет фактора водородной связи. Предложена модель, имитирующая взаимодействие галоидного аниона с молекулой протонного растворителя по механизму водородной связи. Ее использование допускает раздельное рассмотрение динамики электронных и протонных степеней свободы в растворе и оценку их вкладов в процесс сольватации щелочно-галоидных ионов в полярных протонных растворах.
Таблица 1.
Характеристики валентных связей галогенидов водородом [13]
| Связь | Длина связи, Å | Энергия валентного колебания ћω1, эВ |
|---|---|---|
| Cl–H | 1.275 | 3.7 |
| Br–H | 1.413 | 3.4 |
| I–H | 1.604 | 2.86 |
СОЛЬВАТАЦИЯ ИОНОВ В ПОЛЯРНОМ АПРОТОННОМ РАСТВОРИТЕЛЕ
Полярный апротонный растворитель (например, ДМФА) является конденсированной фазой молекул, электроны которых локализованы в пределах своих определенных атомно-молекулярных структур. Молекулы полярных апротонных растворителей представляют собой многоатомные органические соединения, в структуру которых включены электроотрицательные атомы (N, O, P, S, галогены (Ha)), заполненные электронные оболочки которых содержат наряду с электронами, вовлеченными в ковалентные связи, электронные пары, не принимающие участия во внутри молекулярных связях. Волновые функции таких электронов локализованы на корах указанных атомов, а их электронные уровни являются наивысшими из заполненных состояний молекулы. Они задают характер взаимодействия конкретного компонента раствора с его ближним окружением в конденсате. Соотношение электроотрицательностей атомов в структуре определяет направленность и величину дипольных моментов как отдельных связей, так и общего значения дипольного момента в молекуле [9].
Ближний порядок в среде поддерживается межмолекулярными диполь-дипольными взаимодействиями. Введенные в объем раствора щелочно-галоидные ионы с заполненными электронными оболочками как источники электрического заряда поляризуют растворитель. Энергия сольватации в апротонной среде представляет собой результат взаимодействия ионного заряда с полем наведенной им в среде поляризации.
ОСОБЕННОСТЬ СОЛЬВАТАЦИИ АНИОНА В ПОЛЯРНОМ ПРОТОННОМ РАСТВОРИТЕЛЕ
В случае протонного растворителя (например, ФА) электроотрицательный атом молекулы (азот) кроме свободных электронных пар содержит ковалентно связанный атом водорода. Рассмотренный выше электрический механизм сольватации по-прежнему имеет место, но общая ситуация изменяется в сравнении с первым случаем. Открывается дополнительная возможность межмолекулярного взаимодействия – механизм водородной связи.
При достаточно близком расстоянии между галоидным анионом и молекулой растворителя можно рассмотреть детализованную эволюцию протона в полях акцептора и донора. Процесс локального установления равновесных распределений электронной и протонной плотностей идентичен элементарному акту реакции переноса протона p+ от донора (молекулярного иона ФА – mI–) на акцептор (анион галогенида Ha–) [9]:
(1)
$\begin{gathered} {\text{H}}{{{\text{a}}}^{{\text{--}}}} + {{{\text{p}}}^{{\text{ + }}}}{\text{m}}{{{\text{I}}}^{{\text{--}}}} \to \\ \to \,\,{\text{H}}{{{\text{a}}}^{{\text{--}}}} + {{{\text{p}}}^{{\text{ + }}}} + {\text{m}}{{{\text{I}}}^{{\text{--}}}} \to {\text{H}}{{{\text{a}}}^{{\text{--}}}}{{{\text{p}}}^{{\text{ + }}}} + {\text{m}}{{{\text{I}}}^{{\text{--}}}}{\text{.}} \\ \end{gathered} $Состояния электронной системы и протона в процессе (1) описываются уравнением Шредингера, полная потенциальная энергия протона и электронов которого включает потенциальную энергию электрон-электронного, электронно-ядерного и ядерно-ядерного взаимодействий. Согласно теории Борна–Оппенгеймера [10] существует возможность разделения электронных и протонных эволюций вследствие большого различия масс электрона и протона. В результате оказывается возможным характеризовать общее состояние системы поверхностью потенциальной энергии взаимодействующих частиц в координатах положений протона относительно донора и акцептора реакции (1) [9, 10]. Цепь положений протона между указанными аттракторами на поверхности потенциальной энергии взаимодействующих частиц, отвечающих минимуму энергии, имеет вид ассиметричной потенциальной ямы с двумя минимумами, разделенными максимумом (активированным комплексом) рис. 1. Нижайший минимум соответствует состоянию протона, ковалентно связанного с электроотрицательным атомом молекулы растворителя. Другой (метастабильный) минимум отвечает состоянию протона в потенциальном поле аниона галогенида. Эффект водородного связывания состоит в относительном понижении энергии протона в двуямном потенциале в сравнении с его состоянием в молекуле растворителя, достаточно удаленной от аниона. Стимулирующими факторами процесса являются приближенное соответствие энергетических уровней основных состояний протона в каждой из потенциальных ям и достаточная для него проницаемость разделяющего их потенциального барьера.
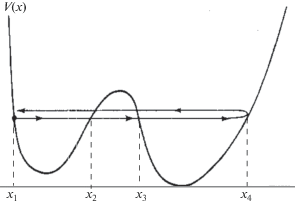
Состояние квантовой частицы p+ в двуямном потенциале предполагает частичное заполнение обеих ям протонной плотностью заряда с соответствующим понижением энергии основного состояния – формальным аналогом одноэлектронной межатомной связи [11]. Рассматриваемая система является асимметричным аналогом молекулярного иона водорода (Н2)–, с тем различием, что в данном случае положительный заряд – протон совершает осциллирующее движение относительно молекулярных коров донора и акцептора. Взаимодействие молекул по электронному и протонному механизмам протекает практически независимо, поэтому можно ожидать приближенно их аддитивного вклада в энергетику короткодействующих взаимодействий как между молекулами растворителя, так и в процесс сольватации анионов в протонном растворе.
МОДЕЛЬ ХИМИЧЕСКОЙ СОСТАВЛЯЮЩЕЙ ЭНЕРГИИ СОЛЬВАТАЦИИ
Механизм водородной связи и его вклад в энергию сольватации можно интерпретировать моделью осциллятора в поле двойной ассиметричной потенциальной ямы. Рассмотрим квазиклассическую динамику протона в поле поверхности потенциальной энергии процесса (1). Эффективное поле представляет собой потенциальную энергию, изображенную на рис. 1.
Энергия основного состояния квазиклассической частицы в поле асимметричного осциллятора V(x), дается величиной смещения уровня ΔE
где E1= hω1, E2= hω2, ω1, ω2 частоты осцилляций в разделенных минимумах. Недиагональный матричный элемент H12 гамильтониана равен [12](3)
${{H}_{{12}}} = {{\left( {h{{\omega }_{1}} \times h{{\omega }_{2}}} \right)}^{{\frac{1}{2}}}}{{e}^{{ - {\theta }}}}.$Фактор туннельной проницаемости барьера протоном дается выражением интеграла от функции действия, взятым между точками поворота x2, x3 траектории движения протона, μ – масса протона
(4)
$\theta = \int\limits_{{{x}_{2}}}^{{{x}_{3}}} {dx{{{\left( {2\mu \left[ {V\left( x \right) - E} \right]} \right)}}^{{\frac{1}{2}}}}} .$В результате подстановок значений E1, E2, H12формула (2) принимает согласно [12] следующий вид
(5)
$\Delta E = 2h{{\frac{{{{\omega }_{1}} \times {{\omega }_{2}}}}{{{{\omega }_{1}} - \omega }}}_{2}}{{e}^{{ - 2{\theta }}}}.$Искомый вклад водородной связи в энергию сольватации аниона можно рассчитать, воспользовавшись параметрами валентных связей, приведенных в табл. 1.
Валентное колебание протона связи N–H в молекуле ФА согласно [14, 15] характеризуется интервалом энергий ћω2 = 3.8–4.3 эВ при длине связи 0.99 Å.
Расчет по формуле (5) дает следующие значения величин предэкспоненциальных факторов формулы (5): 26.6, 16.4, 8.5, быстро уменьшающиеся при переходе от аниона Cl– к I–. На рис. 2 для двух галоидных анионов с разными ионными радиусами представлены аппроксимации двуямных потенциалов электронными термами связей: Ha––p+, p+–mI–. Показатели степеней формулы (5) определяются величинами площадей треугольников, образуемых пересечениями линий 1, 2, ΔE1; 1, 3, ΔE2. Из рис. 2 можно заметить, что показатель степени не критично уменьшается при переходе от иона с меньшим радиусом к иону с большим радиусом. Таким образом, проницаемость барьера для протона не слишком сильно изменяется при переходе от одного вида галоидных ионов к другому. Отсюда можно заключить, что энергия взаимодействия галоидных анионов с молекулой растворителя, обязанная водородной связи, возрастает в следующем порядке Cl– > Br– > I–.
Рис. 2.
Аппроксимация двуямного потенциала электронными термами связей для двух галоидных анионов с разными ионными радиусами.

Современные экспериментальные методы, использующие технику синхротронного излучения. позволяют исследовать локальную структуру растворов. В работах [16, 17] на основе анализа тонкой структуры K-края дальних спектров рентгеновского поглощения была получена информация о ближайшем окружении анионов Br–, I–. В растворах щелочно-галоидных электролитов для серии протонных и апротонных растворителей было установлено, что для галоидных анионов в протонных растворителях координационная сфера охвата взаимодействием молекул растворителя значительно шире, чем в апротонном. В протонном растворителе только первая координационная сфера содержит 6 молекул на расстоянии ~3.2 Å, но упорядочивающее влияние анионов в растворе распространяется значительно дальше. В протонных растворах наблюдается корреляция координационного числа с акцепторным числом Гутмана AN .
В апротонных растворах лишь ближайшее окружение аниона координирует молекулы (4–8 молекул ДМФА) первой координационной сферы растворителя на расстоянии ~3.6 Å. Кроме того, апротонные растворители подразделяются на две группы сольватных структур с большей и меньшей степенью зависимости химической связи аниона с метильными группами, содержащимися в молекуле растворителя.
Рассмотренные модели процессов сольватации анионов в протонных и апротонных растворителях показывает, что водородные связи вносят существенные изменения в баланс сил, формирующих структуру растворов электролитов, и играют важную роль в энергетике процессов сольватации ионов.
ЗАКЛЮЧЕНИЕ
Рассмотрена модель взаимодействия галоидных анионов с молекулами полярного растворителя, основанная на описании эволюции атома водорода в полях протонного донора и акцептора. Ее использование для интерпретации сольватационных процессов в протонных растворителях позволяет выделить химическую составляющую энергетического эффекта анионов. В частности, позволяет объяснить характер зависимостей величин энергий сольватации в ряду галоидных анионов, а также их увеличение, например, при переходе от апротонного растворителя диметилформамида к протонному формамиду.
Работа выполнена при финансовой поддержке Гранта РФФИ № 16-3-01078.
Список литературы
Reichardt C. Solvents and Solvent Effect in Organic Chemistry. VCH. Weinheim. 1988.
Чуев Г.Н., Базилевский М.В. // Успехи химии. 2003. Т. 72. С. 827.
Проблемы химии растворов. Ионная сольватация. Ред. Крестов Г.А. М.: Наука, 1987.
Воротынцев М.А., Корнышев А.А. Электростатика сред с пространственной дисперсией. М.: Наука, 1993.
Рубашкин А.А., Конев Д.В., Воротынцев М.А. // Международный научно-исследовательский журн. 2016. № 5(47). Часть 5. С. 112.
Кришталик Л.И. // Электрохимия. 2008. Т. 44. С. 48.
Haibo Yu., Mazzanti C.L., Whitfield T.W. et al. // J. Amer. Chem. Soc. 2010. V. 132. P. 10847.
Куклин Р.Н. Устойчивость адсорбционных фаз. Размерные критерии наноэлектрохимии. 2015. М.: ГЕОС.
Эйринг Г., Лин С.Г., Лин С.М. Основы химической кинетики. М.: Мир, 1983.
Борн М., Хуан Кунь. Динамическая теория кристаллических решеток. М.: ИЛ, 1958.
Куклин Р.Н. // Физикохимия поверхности и защита материалов. 2014. Т. 50. С. 354.
Miller W.H. // J. Phys. Chem. 1979. V. 83. P. 960.
Краткий справочник физико-химических величин. Ред. Равдель А.А., Мищенко К.П. Л.: Химия, 1974.
Tukachev N.V., Bataev V.A., Godunov I.A. // Comp. and Theor. Chem. 2017. V. 1113. P. 82.
Гордон Ф., Форд Р. Спутник химика. М.: Мир, 1976.
Tanida H., Sacane H., Watanabe I. // J. Chem. Soc. Dalton Trans. 1994. P. 2321.
Tanida H., Watanabe I. // Bull. Chem. Soc. of Jpn. 2000. V. 73. P. 2747.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов