

Физикохимия поверхности и защита материалов, 2019, T. 55, № 5, стр. 479-487
Исследование сорбции катионов цезия сорбентом на основе фосфата титана
М. В. Маслова 1, *, В. И. Иваненко 1, Л. Г. Герасимова 1, Н. Л. Вилкова 1
1 Институт химии и технологии редких элементов и минерального сырья
им. И.В. Тананаева ФИЦ КНЦ РАН
Апатиты, Мурманская обл., Россия
* E-mail: maslova@chemy.kolasc.net.ru
Поступила в редакцию 17.10.2018
После доработки 23.03.2019
Принята к публикации 15.04.2019
Аннотация
Изучено влияние температуры и концентрации цезия во внешнем растворе на сорбционную способность фосфата титана, полученного при гетерогенном взаимодействии кристаллической соли (NH4)2TiO(SO4)2 ⋅ H2O с фосфорной кислотой. Показано, что сорбция на фосфате титана наилучшим образом описывается изотермой Ленгмюра. При описании кинетики сорбции растворов необходимо учитывать как возможность диффузионного, так и адсорбционного лимитирования скорости процесса. Применение моделей химической кинетики показало, что вклад в общую скорость процесса также вносит стадия химического взаимодействия ионов металлов с функциональными группами сорбента.
ВВЕДЕНИЕ
Неизбежным следствием эксплуатации любого объекта, где добываются, используются или перерабатываются радиоактивные вещества, является образование радиоактивных отходов (РАО). Проблема обращения с РАО и их переработка вызывает повышенный интерес в связи с потенциальной опасностью для экосферы. Важность указанной проблемы подтверждается постоянным ростом числа публикаций в этом направлении за последнее десятилетие [1–3]. Особо токсичными являются долгоживущие актиниды, а также стронций-90 и цезий-137, образующиеся в радиохимических процессах. Высокая миграционная способность радионуклидов ведет к быстрому распространению их в природных водных средах [4, 5]. Сорбционные свойства материалов в значительной мере определяются природой матрицы сорбента и его функциональных групп. Помимо селективности сорбционные материалы должны обеспечивать высокую скорость извлечения веществ. При выборе сорбента необходимо учитывать его устойчивость в водных средах (химическую, механическую, радиохимическую), а также такие факторы, как простота получения сорбента, доступность и стоимость используемых для синтеза материалов. Кроме того, следует учитывать возможность дальнейшей переработки или длительного хранения сорбционного материала.
В последнее время для переработки ЖРО все большее применение находят неорганические сорбционные материалы, в частности фосфаты титана [6–11]. Они характеризуются высокими значениями обменной емкости, хорошей кинетикой обмена, радиационной стабильностью и совместимостью с матрицами для захоронения.
Наибольший интерес представляют фосфаты титана, имеющие в своем составе дигидрофосфатные группы. Среди обширного класса фосфатов титана на сегодняшний день известно всего 2 соединения, имеющего в своем составе только дигидрофосфатные группы (TiHP) [13–17 ]: Ti2O3(H2PO4) ⋅ 2H2O и TiO(OH)(H2PO4) ⋅ 2H2O. Как правило, синтез таких материалов достаточно сложен, это длительный и многоступенчатый процесс, требующий жестких условий ведения процесса, большого расхода реагентов, высоких температур, использование автоклавного оборудования и органических темплатов. Внимание исследователей в основном сосредоточено на поиске доступных методов синтеза, свойства новых сорбентов изучены недостаточно. В работе [12] показано, что сорбенты на основе дигидрофосфата титана обладают высокой ионообменной емкостью и хорошей кинетикой по отношению к катионам цветных, а наличие сильнокислотных обменных центров обеспечивает их успешную работу при низких значениях рН. Данные по изучению сорбционной способности TiHP по отношению к катионам-аналогам радионуклидов отсутствуют как в отечественной, так и зарубежной литературе.
В данной работе представлены результаты исследований сорбционной способности TiHP, полученного из кристаллического прекурсора – аммоний титанил сульфата (NH4)2TiO(SO4)2 ⋅ Н2О, являющегося продуктом переработки титансодержащего сырья и техногенных отходов. Гетерогенное взаимодействие твердого прекурсора с разбавленной фосфорной кислотой позволяет значительно сократить время синтеза и получать фосфат титана заданного структурного типа в одну стадию. Для обоснования возможности применения нового сорбента требуются знания механизмов его функционирования для выбора конкретных условий его эксплуатации. В работе представлены данные по ионообменным равновесиям с применением различных моделей и изучены кинетические свойства сорбента.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез фосфата титана
Для получения сорбента заданное количество титановой соли (NH4)2TiO(SO4)2 ⋅ H2O порционно загружали в нагретую до 60°С 30% фосфорную кислоту при мольном отношении TiO2 : P2O5 = 1 : 1.5 и выдерживали при перемешивании в течение 4 часов. Полученную суспензию фильтровали, для удаления аммонийного иона, присутствующего в структуре сорбента, осадок промывали раствором 0.1 N HCl и водой, сушили при 60°С. Для перевода сорбента в солевую форму, навеску обрабатывали 0.1 М раствором Na2CO3. На основании данных химического, термического и рентгенофазового анализов исходный образец сорбента может быть представлен, как аморфное соединение состава (мас. %): TiO2 – 29.60, P2O5 – 39.40, Na2O – 5.69, H2O – 26.78.
Методы анализа
Элементный состав фосфата титана определяли с использованием плазменного атомно-эмиссионного спектрометра ICPE-9000 фирмы Shimadzu после разложения осадка смесью кислот HF–HNO3–HCl. Для получения информации по составу продукта и типам функциональных групп использовали 31P ЯМР анализ, выполненный на Varian/Chemagnetics InfinityPlus CMX-360 спектрометре и рентгено-фазовый анализ с использованием, дифрактометра Siemens D 5000 с монохромным CuKα-излучением (λ = 1.5418 Å). Удельную поверхность образцов определяли методом низкотемпературной адсорбции азота с использованием анализатора поверхности Micrometrics ASAP 2000. Распределение пор по размерам рассчитывали по BJH методу. Концентрацию цезия в фильтрате после сорбционных экспериментов определяли методом атомно-адсорбционной спектрометрии на спектрометре AAS 300 Perkin-Elmer. Для определения среднего размера частиц сорбента проводили ситовой анализ, для работы использовали фракцию сорбента с размером частиц 0.1 мм.
Эксперименты по сорбции
Экспериментальное изучение процесса сорбции катионов цезия из хлоридных растворов проводили в статических условиях с концентрациями от 1 до 15 ммоль л–1 при температурах от 25 до 65°С. Сорбент в солевой форме в количестве 1 г заливали 200 мл раствора заданной концентрации и выдерживался при термостатировании до состояния равновесия при постоянном перемешивании. Количество сорбированного иона металла определяли по разности концентраций ионов цезия в растворе над сорбентом до и после сорбции. Величину сорбции (ммоль г–1) рассчитывали по формуле:
где Сисх и Сравн – исходная и равновесная концентрации металла в растворе, ммоль л–1, V – объем раствора, мл, m – навеска сорбента, г.Полученные результаты обрабатывали с использованием изотермы Ленгмюра и Фрейндлиха.
Кинетику сорбции цезия изучали при температуре 25, 45 и 65°С методом ограниченного объема из водных растворов солей хлорида цезия. Концентрация цезия в растворе составляла 10 и 1 ммоль л–1, объем раствора, контактирующего с сорбентом – 200 мл, навеска сорбента – 1 г. Сорбцию проводили при рН 7.0, исходные растворы термостатировали при заданной температуре с точностью ±1° в течение 1 ч, затем вводили сорбент. Процесс сорбции вели при интенсивном перемешивании со скоростью вращения мешалки 300 об. мин–1. Методом отдельных навесок через определенные интервалы времени устанавливали характер изменения концентрации раствора, находящегося в контакте с сорбентом до установления равновесия. Концентрацию сорбата во всех точках объема раствора и непосредственно у поверхности сорбента при данных скоростях считали постоянной, т.е. рассматривали диффузию из хорошо перемешиваемого раствора.
F – степень достижения равновесия в системе рассчитывали как F = Сt /Сe, где Сt – количество сорбированного вещества в момент времени t, ммоль г–1; Сe – количество сорбированного вещества в состоянии равновесия, ммоль г–1.
Для моделирования кинетики сорбции катионов цезия на фосфате титана использовали диффузионные модели Морриса–Вебера и Бойда, модели псевдо-первого порядка Лагергрена, псевдо-второго порядка Хо и Маккея, модель Еловича.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
На основании данных по элементному составу, РФА и 31Р ЯМР исследований синтезированный материал соответствует химическому соединению TiO(OH)(H2PO4) ⋅ 2H2O. Измеренные текстурные свойства образца позволяют его характеризовать как мезопористый материал с общим объемом пор 0.26 см3 г–1.
Сорбционную способность кислого фосфата титана оценивали, анализируя полученные изотермы сорбции. Как видно из представленных результатов (рис. 1а), TiHP обладает хорошей сорбционной способностью по отношению к катиону цезия. На всех изотермах отмечается резкий подъем в области малых равновесных концентраций катиона металла и наблюдается линейная зависимость количества сорбированных ионов цезия от равновесной концентрации в растворе, которая подчиняется уравнению Генри (рис. 1б).
Рис. 1.
Изотермы сорбции катионов цезия при различных температурах (а) и изотерма Генри при 25°С (б).

Количество поглощенного катиона цезия закономерно увеличивается по мере возрастания его содержания в исходном растворе до 1.5 г л–1, а затем остается неизменным. Судя по показателю степени сорбции (рис. 2), исследуемый ионит обеспечивает практически полную очистку от ионов металлов в растворах с исходной концентрацией до 100 мг л–1. С повышением температуры процесса сорбционная способность TiHP увеличивается.

Для получения количественных характеристик процесса сорбции экспериментальные данные обрабатывали с помощью уравнения изотермы Ленгмюра (2) и Фрейндлиха (3)
(2)
$\frac{1}{A} = \frac{1}{{{{A}_{{\max }}}}} + \left( {\frac{1}{{K{{A}_{{\max }}}}}} \right)\left( {\frac{1}{{{{C}_{p}}}}} \right),$Модель Ленгмюра предполагает эквипотенциальную поверхность адсорбента, т.е. энергетическую эквивалентность адсорбционных центров. При этом каждый адсорбционный центр взаимодействует только с одной молекулой адсорбата, в результате чего образуется мономолекулярный слой [18]. Для модели Фрейндлиха характерно экспоненциальное распределение адсорбционных центров по энергиям, предполагающее увеличение концентрации адсорбата на поверхности сорбента с увеличением концентрации адсорбтива в растворе [19]. Предложенные уравнения изотерм лианезировались в координатах (1/Cр)– (1/А) для модели Ленгмюра; lg Cp–lg A для модели Фрейндлиха. Анализ прямых позволил рассчитать параметры уравнений изотерм (табл. 1).
Таблица 1.
Сорбционные параметры уравнений и коэффициенты корреляции (R) для моделей Ленгмюра и Фрейндлиха
| Изотерма | Температура, К | ||
|---|---|---|---|
| Ленгмюра | 298 | 318 | 338 |
| Аmax, ммоль г–1 | 1.38 | 1.21 | 0.91 |
| K, л ммоль–1 | 7.7 | 19.2 | 114.6 |
| R2 | 0.999 | 0.995 | 0.986 |
| Фрейндлиха | |||
| Kf, мг г–1 | 1.102 | 1.293 | 1.456 |
| n | 1.70 | 1.79 | 2.07 |
| R2 | 0.972 | 0.978 | 0.988 |
Согласно полученным результатам, сорбция цезия на фосфате титана лучше описывается моделью Лэнгмюра. Анализ значений Аmax изотермы Ленгмюра показал, что полученные значения максимальной сорбции близки к экспериментальным значениям. С повышением температуры константа сорбционного равновесия повышается более чем в 15 раз. Такое сильное взаимодействие катионов цезия с сорбционными центрами может быть обусловлено преобладанием химических процессов при сорбции.
Значение константы n в уравнении Фрейндлиха больше 1, что свидетельствует о благоприятных условиях, связанных с уменьшением энергия связи сорбент–сорбат по мере заполнения поверхности. Значение константы Kf подтверждает легкость перехода сорбируемых катионов цезия из раствора в фазу сорбента.
Движущей силой процесса сорбции из водных растворов является градиент химического потенциала сорбата. По достижении равенства химических потенциалов последнего в объеме раствора и в сорбенте наступает химическое равновесие. Лимитирующее влияние на скорость сорбции оказывают подвод сорбируемого вещества к зерну сорбента (внешний массоперенос) и перемещение его молекул внутри зерна пористого сорбента (внутренняя диффузия).
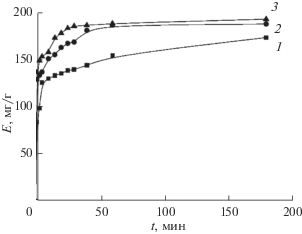
Кинетические кривые процесса сорбции катиона цезия в зависимости от концентрации внешнего раствора и температуры представлены на рис. 3 и рис. 4.
Рис. 3.
Кинетика сорбции цезия в зависимости от концентрации исходного раствора. Исходная концентрация катионов цезия в растворе (моль л–1): 1 – 10–2; 2 – 10–3.

Рис. 4.
Кинетика сорбции цезия в зависимости от температуры исходного раствора. Исходная концентрация цезия 10–3 моль л–1. Температура раствора, °С: 1 – 25; 2 – 45; 3 – 65.

Согласно полученным результатам, при малых исходных концентрациях цезия в растворе (10–3 моль л–1) равновесие в системе ион металла– сорбент достигается за несколько минут и не зависит от температуры раствора. При исходной концентрации цезия 10–2 моль л–1 равновесие достигается за 50–60 мин в зависимости от температуры раствора. Такая высокая скорость достижения равновесия обусловлена текстурными свойствами синтезированного материала: развитой удельной поверхностью и наличием широких мезопор.
В разбавленных растворах скорость процесса может лимитироваться пленочной диффузией. Согласно уравнению пленочной диффузии (4) проведен анализ кинетических данных в координатах –ln(1 – F) – t .
(4)
$--\ln (1--F) = {{3(Dct)} \mathord{\left/ {\vphantom {{3(Dct)} {(r\delta m)}}} \right. \kern-0em} {(r\delta m)}},$Полученные зависимости представляют собой прямые линии, при этом степень достижения равновесия приближается к F = 1 за 10 мин протекания процесса сорбции. Тангенс угла наклона прямой к оси абсцисс, определяет кажущуюся константу скорости внешней диффузии γ, мин–1:
(5)
$\gamma = {{3(Dc)} \mathord{\left/ {\vphantom {{3(Dc)} {(r\delta m)}}} \right. \kern-0em} {(r\delta m)}}.$Полученные зависимости были обработаны с использованием уравнения первого порядка, что позволило вычислить скорость реакции обмена во внешнедиффузионной области (табл. 2).
Таблица 2.
Кинетические параметры ионного обмена Cs+ на фосфате титана в зависимости от температуры и концентрации цезия в исходном растворе
| Исходная концентрация катионов Cs+ во внешнем растворе 10–3 моль/л | ||||||||
|---|---|---|---|---|---|---|---|---|
| Внешнедиффузионная область | ||||||||
| Т, °С | γ, мин–1 | R2 | Ea, кДж моль–1 | |||||
| 25 | 1.73 | 0.997 | 19.22 | |||||
| 45 | 2.61 | 0.999 | ||||||
| 65 | 3.78 | 0.999 | ||||||
| Исходная концентрация катионов Cs+ во внешнем растворе 10–2 моль/л | ||||||||
| Внешнедиффузионная область | Внутридиффузионная область | |||||||
| Т, °С | γ, мин–1 | R2 | Ea, кДж моль–1 | D × 10–7, см2 с–1 | B, мин–1 | R2 | Ea, кДж моль–1 | t1/2, мин |
| 25 | 0.45 | 0.999 | 14.59 | 7.02 | 0.713 | 0.999 | 33.97 | 7.1 |
| 45 | 1.11 | 0.999 | 7.36 | 0.604 | 0.999 | 6.8 | ||
| 65 | 1.18 | 0.997 | 7.86 | 0.502 | 0.999 | 6.3 | ||
Зависимость константы скорости от температуры описывается уравнением Аррениуса [20]:
(6)
$k = A\exp ({{ - {{E}_{{{\text{акт}}}}}} \mathord{\left/ {\vphantom {{ - {{E}_{{{\text{акт}}}}}} {RT}}} \right. \kern-0em} {RT}}),$(7)
$\lg k = \lg A - {{{{E}_{{{\text{акт}}}}}} \mathord{\left/ {\vphantom {{{{E}_{{{\text{акт}}}}}} {2.3RT}}} \right. \kern-0em} {2.3RT}},$При высоких концентрациях цезия в исходных растворах кинетические зависимости имеют сложный вид (рис. 5). Вид кинетических кривых свидетельствует о нескольких периодах сорбции. На первом участке за небольшой промежуток времени скорость извлечения катионов цезия наивысшая. Реальные ионообменные процессы редко могут быть описаны в рамках представлений о единственной лимитирующей стадии. Для таких систем правильное описание кинетики может быть получено путем учета влияния нескольких стадий.
Рис. 5.
Кинетика сорбции цезия в зависимости от температуры исходного раствора. Исходная концентрация цезия 10–2 моль л–1. Температура раствора, °С: 1 – 25; 2 – 45; 3 – 65.
Стадия внутренней диффузии – это перенос ионов внутри частицы сорбента от ионообменных центров к ее поверхности и наоборот. Синтезированный фосфат титана относится к аморфным материалам, частицы которого представляют собой агломераты в которых пространство между частицами заполнено жидким раствором. Это поровое пространство играет важную роль в ионообменных процессах, поскольку по нему осуществляется почти весь транспорт ионов вглубь частицы. Следовательно, структура пор сорбента оказывает существенное влияние на скорость протекания диффузионных процессов.
При описании кинетики сорбции с учетом внутридиффузионного механизма использовали модели гелиевой диффузии из ограниченного объема в тело, имеющее форму шара. Диффузионное уравнение Бойда (8) целесообразно использовать для описания случаев внутридиффузионного лимитирования кинетики сорбции и расчета эффективных коэффициентов диффузии [21].
(8)
$F = 1 - \frac{6}{{{{\pi }^{2}}}}\sum\limits_{n = 1}^\infty {\left( {{1 \mathord{\left/ {\vphantom {1 {{{n}^{2}}}}} \right. \kern-0em} {{{n}^{2}}}}} \right)\exp \left( {{{ - Dt{{\pi }^{2}}{{n}^{2}}} \mathord{\left/ {\vphantom {{ - Dt{{\pi }^{2}}{{n}^{2}}} {{{r}^{2}}}}} \right. \kern-0em} {{{r}^{2}}}}} \right),} $Расчет эффективных коэффициентов диффузии проводили по формуле:
(9)
$D = {{(Bt \cdot {{r}^{2}})} \mathord{\left/ {\vphantom {{(Bt \cdot {{r}^{2}})} {(t{{\pi }^{2}})}}} \right. \kern-0em} {(t{{\pi }^{2}})}}.$Значение Bt определяли по формуле [22]:
Уравнение модели Морриса–Вебера [23], описывающее внутрипористую диффузию, имеет следующий вид:
где kid – константа скорости диффузии, мг г–1 мин–1/2; С – параметр, связанный с толщиной пограничного слоя, мг г–1.Для первичного разграничения внутри- и внешнедиффузионного лимитирования сорбции в рамках диффузионных моделей Бойда и Морриса–Вебера проведен анализ кинетических данных в координатах – ln(1 – F) – t и F – t1/2 соответственно. При выбранной исходной концентрации цезия во внешнем растворе 10–2 моль л–1 для всех исследуемых температур наблюдается нелинейная зависимость –ln(1 – F) – t изменения адсорбции от времени (рис. 6).
Рис. 6.
Зависимость –ln(1 – F) от t для катионов цезия при исходной концентрации стронция в растворе 10–2 моль л–1. Температура раствора, °С: 1 – 25; 2 – 45; 3 – 65.
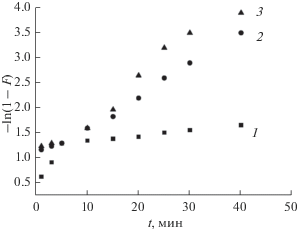
Зависимости F – t1/2 в координатах уравнения Морриса–Вебера для сорбции цезия также не являются линейными (R2 = 0.956–0.964), что говорит о смешанно-диффузионном механизме кинетики адсорбции, когда процесс сорбции сопровождается диффузией в пленке и диффузией в зерне. С повышением температуры продолжительность периода сорбции, протекающего по внешнедиффузионному механизму, увеличивается от 5 мин (25°С) до 15 мин (45 и 65°С). Дегидратация катиона цезия при повышенной температуре позволяет занимать сорбционные центры на поверхности сорбента без существенных стерических затруднений. Уменьшение концентрации поглощаемых ионов во внешнем растворе приводит к увеличению вклада внешней диффузии в кинетику ионного обмена вследствие роста соотношения концентраций ионов в ионите и растворе. Построение экспериментальных данных в координатах –ln(1 – F) от t может быть адекватно описано линейными зависимостями при малых временах контакта фаз, но при высокой степени завершенности процесса F > 0.7 для всех выбранных температур. Кинетические параметры внешнедиффузионного механизма представлены в табл. 2.
Определив экспериментально степени достижения равновесия F для данного времени t, и, рассчитав Bt по уравнению (10) были построены зависимости Bt = f(t). Графики (рис. 5) представляют собой прямую линию, что подтверждает гелевую диффузию ионного обмена на фосфате титана. По тангенсу угла наклона В рассчитали скорость реакции ионного обмена (табл. 1). Рассчитанные эффективные коэффициенты диффузии представляют собой кинетические коэффициенты, учитывающие диффузионные особенности транспорта молекул в сорбционной системе. Очевидно, что с повышением температуры диффузия в зерне увеличивается (табл. 1). Коэффициент диффузии цезия в воде при 25°C рассчитывали по формуле [24]:
(12)
$D = {{(RT\lambda )} \mathord{\left/ {\vphantom {{(RT\lambda )} {({{F}^{2}}z)}}} \right. \kern-0em} {({{F}^{2}}z)}},$Рассчитанный коэффициент диффузии цезия равен 2.06 × 10–5 см2 с–1. Найденные приближенные значения D составляют 7.02 × 10–7–7.86 × × 10–7 см2 с–1, что свидетельствует о том, что в зерне сорбента присутствуют тормозящие диффузию эффекты. С использованием эффективных коэффициентов диффузии цезия в ионите рассчитаны кажущиеся энергии активации процесса сорбции по уравнению, подобному уравнению Аррениуса [25]:
(13)
$D = {{D}_{{\text{o}}}}\exp ({{ - {{E}_{{{\text{акт}}}}}} \mathord{\left/ {\vphantom {{ - {{E}_{{{\text{акт}}}}}} {RT}}} \right. \kern-0em} {RT}}),$По тангенсу угла наклона прямой построенной в координатах D от 1/Т рассчитали энергию активации, которая составила 33.9 ± 1.54 кДж моль–1, коэффициент детерминации 0.992.
Численные значения времени полуобмена, рассчитанные по формуле t1/2 = 0.03r2/D сведены в табл. 2. Очевидно, что синтезированный фосфат титана обладает высоким сродством к катионам цезия. Рассчитанное время полуобмена не превышает 7 мин, такая высокая скорость извлечения цезия во многом определяется мезопористой структурой материала.
Для выявления вклада химического взаимодействия в общую скорость процесса использовали кинетические модели псевдо-первого, псевдо-второго порядка и модель Еловича. Кинетическое уравнение псевдо-первого порядка Лагергрена можно представить в виде: dq1/dt = k1(qe – qt) [26] или в линейной форме:
(14)
$\lg ({{q}_{e}} - {{q}_{t}}) = \lg {{q}_{e}} - {{{{k}_{{\text{1}}}}t} \mathord{\left/ {\vphantom {{{{k}_{{\text{1}}}}t} {2.3}}} \right. \kern-0em} {2.3}},$Уравнение псевдо-второго порядка Хо и Маккея [28] широко используется для описания кинетических закономерностей адсорбции. В интегральной форме оно может быть представлено следующим образом:
(15)
${t \mathord{\left/ {\vphantom {t {{{q}_{t}}}}} \right. \kern-0em} {{{q}_{t}}}} = {1 \mathord{\left/ {\vphantom {1 {\left( {{{k}_{2}}q_{e}^{2}} \right)}}} \right. \kern-0em} {\left( {{{k}_{2}}q_{e}^{2}} \right)}} + {t \mathord{\left/ {\vphantom {t {{{q}_{e}}}}} \right. \kern-0em} {{{q}_{e}}}},$Экспоненциальная модель Еловича описывает случаи гетерогенной хемосорбции на твердых поверхностях. Уравнение Еловича, упрощенное Ченом и Клейтоном [29], имеет следующий вид:
(16)
${{q}_{t}}~ = ({1 \mathord{\left/ {\vphantom {1 \beta }} \right. \kern-0em} \beta })\ln (\alpha \beta ) + ({1 \mathord{\left/ {\vphantom {1 \beta }} \right. \kern-0em} \beta })\ln (t),$Уравнение псевдо-первого порядка адекватно описывает закономерности сорбции когда значительное влияние на процесс оказывает явление пленочной диффузии. Уравнение псевдо-второго порядка позволяет учитывать не только взаимодействия сорбат–сорбент, но и межмолекулярные взаимодействия адсорбируемых веществ. Для всех выбранных концентраций цезия в исходном растворе кинетика описывается уравнением псевдо-второго порядка (коэффициенты детерминации составляют >0.999). Для низких концентраций цезия в исходном растворе, когда кинетика сорбции лимитируется внешнедиффузионными торможениями, применимость модели псевдо-второго порядка может быть связана с процессами обмена на поверхности частиц сорбента. Модель Еловича учитывает вклад в кинетику извлечения вещества как процессов сорбции, так и явление десорбции, приобретающей значительное влияние при приближении к равновесному состоянию. Для модели Еловича линейной зависимости удалось добиться только на начальных участках кривых (при малых степенях завершенности процесса). Это может быть объяснено высокой скоростью кинетики сорбционного процесса. Рассчитанные значения α на два порядка выше, чем β, что свидетельствует о необратимости процесса сорбции. Высокое химическое сродство катионов цезия к фосфатным группам определяет перспективность использования фосфата титана для концентрирования радионуклидов и их последующей иммобилизации в фосфатных матрицах. Значения констант и величин достоверности аппроксимации кинетических моделей адсорбции цезия представлены в табл. 3.
Таблица 3.
Кинетические параметры сорбции цезия на фосфате титана при различной исходной концентрации цезия в растворе
| Cs+ × 10–2 моль/л | Cs+ × 10–3 моль/л | ||||||
|---|---|---|---|---|---|---|---|
| Модель | Параметры | Температура процесса, К | |||||
| 298 | 318 | 338 | 298 | 318 | 338 | ||
| qexp, мг г–1 | 176.8 | 187.2 | 190.4 | 25.2 | 25.4 | 25.6 | |
| Псевдо-первого порядка | k1, мин–1 | 0.032 | 0.039 | 0.079 | 0.76 | 0.29 | 0.22 |
| qe, мг г–1 | 74.5 | 68.5 | 62.3 | 30.5 | 24.4 | 21.6 | |
| R2 | 0.946 | 0.968 | 0,947 | 0.933 | 0.958 | 0.978 | |
| Псевдо-второго порядка | k2, г мг–1 мин–1 | 1.8 × 10–3 | 2.5 × 10–3 | 3.7 × 10–3 | 0.22 | 0.37 | 0.42 |
| qe, мг г–1 | 166.1 | 188.3 | 191.9 | 25.2 | 25.5 | 25.6 | |
| R2 | 0,998 | 0,999 | 0,999 | 0.999 | 0.999 | 0.999 | |
| Еловича | α, мг г–1 мин–1 | 7.62 | 15.1 | 70.3 | 5.18 | 4.32 | 4.25 |
| β, г мг–1 | 0.062 | 0.064 | 0.078 | 0.016 | 0.015 | 0.077 | |
| R2 | 0.881 | 0.905 | 0.829 | 0.824 | 0.867 | 0.919 | |
ЗАКЛЮЧЕНИЕ
Изучено влияние температуры и концентрации цезия во внешнем растворе на сорбционную способность фосфата титана, полученного при гетерогенном взаимодействии кристаллической соли (NH4)2TiO(SO4)2 ⋅ H2O с фосфорной кислотой. Данные о синтезе фосфатов титана из кристаллических титановых солей отсутствуют как в отечественной так и зарубежной литературе.
Полученные экспериментальные данные обработаны с применением моделей Ленгмюра и Фрейндлиха. Показано, что сорбция на фосфате титана наилучшим образом описывается изотермой Ленгмюра. С повышением температуры константа сорбционного равновесия повышается более чем в 15 раз. Такое сильное взаимодействие катионов цезия с сорбционными центрами может быть обусловлено преобладанием химических процессов при сорбции.
Исследованы закономерности кинетики сорбции ионов цезия из модельных растворов. В результате обработки кинетических данных показано, что механизм сорбционного процесса имеет сложный характер, включающий обмен на поверхности сорбента, диффузию ионов в объем и гетерогенную ионообменную реакцию с образованием новой фазы. Благодаря наличию мезопор обеспечивается высокая скорость массопереноса при сорбции на исследуемом материале, причем равновесие в системе сорбат–сорбент устанавливается от 10 до 50 мин в зависимости от концентрация цезия во внешнем растворе. В скорость сорбционного процесса вносят вклад как диффузионное лимитирование (внутри- и внешнедиффузионное), так и скорость химической стадии сорбции. При сорбции цезия на фосфате титана необходимо учитывать полимолекулярные взаимодействия в системе сорбат–раствор–сорбент, что показано применением моделей псевдопервого и псевдовторого порядка. В связи с быстрой кинетикой процесса применение модели Еловича затруднено.
В целом фосфат титана характеризуются большим сродством к катионам цезия и может быть рекомендован для концентрирования из водных сред.
Исследования выполнены при финансовой поддержке Российского научного фонда (РНФ) в рамках научного проекта № 17-19-01522.
Список литературы
Полуэктов П.П., Суханов Л.П., Матюнин Ю.И. // Российский химический журн. 2005. Т. 49. № 4. С. 123.
Мясоедов Б.Ф. // Вопросы радиационной безопасности. 1997. № 1. С. 3.
Мясоедов Б.Ф. // Российский химический журнал. 2005. Т. 49. № 2. С. 64.
Попова Н.Н., Тананаев И.Г., Ровный С.И. и др. // Успехи химии. 2003. Т. 72. № 2. С. 115.
Новиков А.П., Калмыков С.Н., Ткачев В.В. // Российский химический журнал. 2005. Т. 49. № 2. С. 119.
Milonjić S., Bispo I., Fedoroff M., Loss-Neskovic C. et al. // J. Radioanalytical and Nuclear Chemistry. 2002. V. 252. № 3. P. 497.
Horwit E.P. // J. Inorganic Nuclear Chemistry. 1966. V. 28. № 6/7. P. 1469.
Долматов Ю.Д., Булавина З.Н., Долматов М.Ю. // Радиохимия. 1972. Т. 14. № 4. С. 526.
Локшин Э.П., Иваненко В.И., Авсарагов Х.Б. // Атомная энергия. 2002. Т. 92. № 2. С. 118.
Ortiz-Oliveros H.B., Flores-Espinosa R.M., Ordonez-Regil E. et al. // Chemical Engineering Journal. 2014. V. 236. P. 398.
Kapnisti M., Hatzidimitriou A.G., Noli F. et al. // J. Radioanalytical and Nuclear Chemistry. 2014. V. 302. P. 679.
Trublet M., Maslova M., Rusanova D. et al. // RSC Advances. 2017. V. 7. P. 1989.
Bereznitski Y., Jaroniec M., Bortun A. et al. // Colloid and Interface Science. 1997. V. 191. P. 442.
Takahashi H., Oi T., Hosoe M. // J. Materials Chemistry. 2002. V. 12. P. 2513.
Korosi L., Papp S., Dekany I. // Chemistry of Materials. 2010. V. 22. P. 4356.
Li Y., Whittingham M. // Solid State Ionics. 1993. V. 63–65. P. 391–395.
Trublet M., Maslova M., Rusanova D. et al. // Materials Chemistry and Physics. 2016. V. 183. P. 467.
Langmuir I. // J. American Chemical Society. 1916. V. 38. P. 2221.
Freundlich H.M.F. // J. Physical Chemistry Society. 1906. V. 40. P. 1361.
Кемпбел Дж. Современная общая химия. М.: Мир, 1975. Т. 2. 450 с.
Неудачина Л.К., Петрова Ю.С., Засухин А.С. и др. // Аналитика и контроль. 2011. Т. 15. № 1. С. 88.
Знаменский Ю.П. // Журн. физической химии. 1993. Т. 67. № 9. С. 1924.
Piplai T., Kumar A., Alappat B.J. // Water Environment Research. 2018. V. 90. № 5. P. 409.
Элькинд К.М., Трунова И.Г. // Труды Нижегородского государственного технического университета им. Р.Е. Алексеева. 2013. № 4(97). С. 272.
Кокотов Ю.А., Пасечник В.А. Равновесие и кинетика ионного обмена. Л.: Химия, 1979. 336 с.
Saiers J.E., Hornberger G.M., Liang L. // Water Resources Research. 1994. V. 30. P. 2499.
Ho Y.S., Ng J.C.Y., McKay G. // Separation and purification methods. 2000. № 2(29). P. 189.
Ho Y.-S. // J. Hazardous Materials B. 2006. V. 136. P. 681.
Kołodyńska D., Gęca M., Skwarek E., Goncharuk O. // Nanoscale Research Letters. 2018. V. 13. P. 1.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов