

Физикохимия поверхности и защита материалов, 2019, T. 55, № 5, стр. 451-460
Влияние адсорбции на состояние равновесных шероховатых поверхностей границ раздела фаз
Е. С. Зайцева 1, Е. Е. Гвоздева 1, А. Б. Рабинович 1, Ю. К. Товбин 1, *
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
* E-mail: tovbinyk@mail.ru
Поступила в редакцию 26.11.2018
После доработки 12.04.2019
Принята к публикации 15.04.2019
Аннотация
Исследовано влияние адсорбции на характеристики равновесных шероховатых поверхностей границ раздела фаз пар–жидкость и пар–твердое тело около температуры плавления. Для расчета использована дискретная модель плотной фазы – модель решеточного газа, учитывающая прямые корреляции между всеми ближайшими компонентами системы в квазихимическом приближении. Модель для простоты предполагает соизмеримость размеров компонентов и учитывает колебания их частиц через эффективные вклады параметров латерального взаимодействия как для атомов адсорбента, так и молекул адсорбата. Рассчитаны равновесные профили адсорбента А и адсорбата В внутри переходной области (при условии неизменности объемной фазы адсорбента) для физической адсорбции и для хемосорбции. Рассмотрено влияние адсорбции на поверхностное натяжение плотной фазы. Построены оценки вероятности флуктуационных процессов формирования новой фазы, обусловленной шероховатостью поверхности чистого адсорбента А и при учете адсорбции на ней частиц В.
ВВЕДЕНИЕ
Во многих реальных многостадийных поверхностных процессах стадия адсорбции является неизбежной и/или необходимой стадией [1–10]. Монослойная граница раздела фаз является очень сильной идеализацией структуры поверхности. Более реальными являются представления о разной степени шероховатости поверхности. Существуют два типа процессов создания шероховатых поверхностей. В общем случае это относится к неравновесным процессам на границах раздела (осаждения или конденсации атомов, их растворения или десорбции, скола, деформаций сдвига, и т.д.) и равновесным процессам формирования шероховатости как результату теплового движения поверхностных атомов твердого тела.
В зависимости от соотношения энергии между атомами твердого тела и энергией взаимодействия с адсорбатом реализуется взаимосвязь между адсорбцией и шероховатостью. Если адсорбция слабо влияет на состояние поверхности, то в основном сохраняется степень шероховатости до начала адсорбции. В противном случае адсорбция начинает влиять на состояние степени шероховатости поверхности.
Целью работы является разработка атомистической модели адсорбции с учетом эффектов колебаний атомов адсорбента и адсорбата на открытой шероховатой поверхности и анализ изменения ее шероховатости за счет влияния температуры и адсорбции при равновесном состоянии системы. Модель ориентирована на широкий круг процессов, связанных с физической адсорбцией и хемосорбцией. Увеличение шероховатости увеличивает площадь поверхности по сравнению с гладкой поверхностью.
Данный вопрос исследовался в ранних работах (см. [12] и ссылки в ней). Однако он не получил широко распространения из-за сложного характера уравнений описывающих шероховатые поверхности. Недавно был предложен подход [13], позволяющий рассматривать шероховатые поверхности как переходную область между сосуществующими фазами, и использовать методы расчета поверхностного натяжения на границах раздела фаз [14–16] в равновесных условиях. В рамках такого подхода учет процесса адсорбции рассматривается как переход к системе, содержащей большее число компонентов, чем число компонентов в исходной расслаивающей системе, причем, возможность проникновения адсорбирующегося компонента в объемную плотную фазу исключается. Близкий подход был использован при адсорбции на неперестраивающуюся поверхность адсорбента [17, 18]. В этом случае шероховатая поверхность обладает широким диапазоном поверхностных центров с разной энергией связи. В отличие от старых подходов [12] ориентированных на один-два монослоя в переходной области, подход [13] позволяет рассматривать неограниченное число монослоев.
В случае равновесной адсорбции наличие адсорбата вызывает перестройку приповерхностных плотной фазы (жидкой или твердой) и промежуточная область представляет собой смесь из общего числа компонентов плотной фазы и адсорбата. Для жидкой плотной фазы такая смесь является очевидной, а для твердой фазы – нет.
Очевидно, что при низких температурах естественная шероховатость фазы невелика и ограничена малым числом монослоев, тогда как при увеличении температуры растет и число слоев переходной области. Существенное повышением температуры жидкой фазы может приводить к растворимости адсорбата, что будет менять число компонентов в ее объеме, поэтому наше внимание будет ограничено областью температур вблизи температуры плавления твердой фазы, когда важна только ее шероховатость. Здесь дополнительную роль играет фактор энергии связи между адсорбатом и адсорбентом: при их сопоставимости (в случае хемосорбции) адсорбция может существенно менять состояние поверхности.
Для расчета использована дискретная модель плотной фазы – модель решеточного газа, учитывающая прямые корреляции между всеми ближайшими компонентами системы в квазихимическом приближении [13–22] (модель многократно описана в литературе и здесь не дублируется). Молекулярная модель основана на модели решеточного газа [19–22]. Объем системы разбивается на отдельные элементарные ячейки со стороной в диаметр молекулы λ и объемом v0. Обсуждается граница раздела жидкость–пар для капли радиуса R. На границе ячейки объединяются в сферические монослои с номером q, отсчитываемым от крайнего монослоя жидкости, в которых они характеризуются числом связей с ближайшими соседями zqp(R) из монослоя p как функция от радиуса кривизны границы раздела фаз R [23]. Плоским границам отвечают большие значения радиуса кривизны.
Для простоты предполагается соизмеримость размеров компонентов и учитываются колебания их частиц через эффективные вклады параметров латерального взаимодействия как для атомов адсорбента, так и молекул адсорбата [24–26].
Описание модели подробно дано в работах [13, 17, 18], поэтому здесь приводятся основные уравнения для расчета профилей плотности $\theta _{q}^{A}$ для приповерхностной области чистого адсорбента А, и $\theta _{q}^{B}$ для адсорбированного газа В, и соответствующие им равновесные характеристики изучаемых систем. Формулы приводятся для общего случая искривленных границ, необходимых при анализе флуктуаций, хотя основные иллюстрации приведены для плоских границ.
УРАВНЕНИЯ МОДЕЛИ
Учет многослойности границы раздела фаз. Формирование поверхности адсорбента проводится в условиях равновесия между плотной фазой (жидкости или твердого) и паром вещества А при некоторой температуре Т. Концентрационный профиль А рассчитывается по уравнениям
(1)
$\begin{gathered} {{a}_{q}}{{P}^{A}} = {{\theta _{q}^{A}{{\Lambda }_{q}}(R)} \mathord{\left/ {\vphantom {{\theta _{q}^{A}{{\Lambda }_{q}}(R)} {\left( {1 - \theta _{q}^{A}} \right)}}} \right. \kern-0em} {\left( {1 - \theta _{q}^{A}} \right)}} \\ {{\Lambda }_{q}}(R) = \prod\limits_{p = q - 1}^{q + 1} {{{{\left[ {1 + t_{{qp}}^{{AA}}{{x}^{{AA}}}} \right]}}^{{{{z}_{{qp}}}(R)}}}} , \\ \end{gathered} $Адсорбция на сформированной шероховатой границе раздела фаз. Обозначим в переходной области границы через $\theta _{q}^{A}$ локальные плотности частиц адсорбента A и через $\theta _{q}^{B}$ локальные плотности молекул адсорбата В. В равновесном состоянии шероховатой поверхности эти локальные плотности получаются из решения следующей системы уравнений при заданной температуре T:
(1a)
$\begin{gathered} a_{q}^{i}{{P}^{i}} = {{\theta _{q}^{i}L_{q}^{i}} \mathord{\left/ {\vphantom {{\theta _{q}^{i}L_{q}^{i}} {\theta _{q}^{s}}}} \right. \kern-0em} {\theta _{q}^{s}}}, \\ \Lambda _{q}^{i} = \prod\limits_{p = q - 1}^{q + 1} {{{{\left[ {\sum\limits_{j = 1}^s {t_{{qp}}^{{ij}}} \exp \left\{ {\beta {{\varepsilon }_{{ij}}}} \right\}} \right]}}^{{{{z}_{{qp}}}(R)}}}} , \\ \end{gathered} $Уравнения на парные функции распределения в квазихимическом приближении выписываются в виде
(2)
$\hat {\theta }_{{qp}}^{{ij}}\hat {\theta }_{{qp}}^{{kl}} = \hat {\theta }_{{qp}}^{{il}}\hat {\theta }_{{qp}}^{{kj}},\,\,\,\,\hat {\theta }_{{qp}}^{{kj}}(r) = \theta _{{qp}}^{{kj}}\exp ( - \beta \varepsilon _{{qp}}^{{kj}}).$Нормировочные соотношения для парных функций компонентов имеют вид: $\sum\nolimits_{i = 1}^s {\theta _{{qp}}^{{ij}}} = \theta _{p}^{j}$ и $\sum\nolimits_{j = 1}^s {\theta _{{qp}}^{{ij}}} = \theta _{q}^{i},$ а для унарных функций они равны $\sum\nolimits_{i = 1}^s {\theta _{q}^{i}} = 1.$
Решение уравнений на концентрационный профиль (1) и (2) позволяет рассчитать все термодинамические характеристики системы.
Поверхностное натяжение границы. Поверхностное натяжение на границе раздела фаз определяется через концентрационный профиль молекул (или через плотность вакансий) [14–16]
(3)
$\sigma = \frac{1}{{{{\lambda }^{2}}{{f}_{{\rho }}}}}\left[ {\sum\limits_{1 \leqslant q \leqslant {{{\rho }}_{e}}} {{{f}_{q}}({{\pi }_{1}} - {{\pi }_{q}}) + \sum\limits_{{{{\rho }}_{e}} < q \leqslant \kappa } {{{f}_{q}}({{\pi }_{\kappa }} - {{\pi }_{q}})} } } \right],$(4)
$\beta {{\pi }_{q}}{{{v}}_{0}} = - \ln \theta _{q}^{V} - \sum\nolimits_p {{{z}_{{qp}}}ln{{\left( {{{\theta _{{qp}}^{{VV}}} \mathord{\left/ {\vphantom {{\theta _{{qp}}^{{VV}}} {\left( {\theta _{q}^{V}\theta _{p}^{V}} \right)}}} \right. \kern-0em} {\left( {\theta _{q}^{V}\theta _{p}^{V}} \right)}}} \right)} \mathord{\left/ {\vphantom {{\left( {{{\theta _{{qp}}^{{VV}}} \mathord{\left/ {\vphantom {{\theta _{{qp}}^{{VV}}} {\left( {\theta _{q}^{V}\theta _{p}^{V}} \right)}}} \right. \kern-0em} {\left( {\theta _{q}^{V}\theta _{p}^{V}} \right)}}} \right)} 2}} \right. \kern-0em} 2}} ,$ПАРАМЕТРЫ ЧИСЛЕННОГО АНАЛИЗА
Проведенные модельные расчеты иллюстрируют основные особенности поведения равновесных шероховатых поверхностей для случая физической адсорбции и для хемосорбции. На поверхностях жидкости рассматривается физическая адсорбция компонента В, а на поверхностях твердого тела моделируемого для простоты изотропной решеткой с малой шириной переходной области рассматриваются оба типа адсорбции. (Для более реальной модели твердого тела нужен учет присутствия других граней твердого тела.)
При физической адсорбции степень заполнения поверхности меняется в широких пределах. Приведенные выше уравнения соответствуют четырем типам равновесных ситуаций: адсорбция пара компонента А на твердом теле компонента А для условий (сосуществующие фазы), адсорбция жидкости компонента А на твердом теле компонента А (сосуществующие фазы), адсорбция пара В на твердом теле А и адсорбция жидкости В на твердом теле А. Для хемосорбции область изменения заполнения ограничена монослоем. Это отвечает обычным представлениям о ненасыщаемости связей для неспецифической физической адсорбции и специфичности связей при хемосорбции.
Напомним [21], что взаимное отталкивание хемосорбированных частиц описывается отрицательными параметрами латерального взаимодействия, а притяжение физически адсорбированных частиц – положительными. За основу энергетических соотношений выбрана величина εАА энергии взаимодействия компонентов адсорбента А. Для физической адсорбции были заданы: εАB = 0.015εAA, εBB = 0.14εAB; и для химической адсорбции: εАB = 0.15εAA, εBB = –0.33εAB. Величина εАА в обоих случаях адсорбции условно выбрана за единицу – это относит рассчитанные результаты к системам разного типа и дает относительные корреляции между ними.
Адсорбция газа рассматривается при сверхкритической температуре. В качестве условной меры для давления в данной работе введена величина ${\text{P}}_{{\text{n}}}^{{\text{B}}},$ при которой числовая плотность флюида в объеме равна 0.5. По отношению к ней рассматриваются разные варианты давлений рассмотренных систем.
РЕЗУЛЬТАТЫ РАСЧЕТА КОНЦЕНТРАЦИОННЫХ ПРОФИЛЕЙ
Физическая адсорбция. Для физической адсорбции рассматриваются четыре температуры, которым отвечают κ = 5 и 7 (вещество А в твердом состоянии) κ = 10 и 12 (вещество A в жидком состоянии), где κ – ширина переходной области плюс по монослою от каждой из сосуществующих фаз.
На рис. 1 представлены профили плотности в переходной области чистого вещества А (кривая 1) и плотности А (кривые 2, 3 и 4) в присутствии адсорбата В с профилями, описываемыми кривыми 5, 6 и 7, при давлении газа В, PB, приводящем к заполнению адсорбатом 10, 40 и 80% всей переходной области (монослои 1 < q < κ), при температурах, которым отвечают κ = 5 (рис. 1а) и 7 (рис. 1б) (вещество А в твердом состоянии) κ = 10 (рис. 1в) и 12 (рис. 1г) (вещество А в жидком состоянии). Отметим, что для 80% заполнения свободной части переходной области адсорбента А адсорбатом В, соответствует давление $P_{\kappa }^{{\text{B}}},$ где ${{P_{\kappa }^{{\text{B}}}} \mathord{\left/ {\vphantom {{P_{\kappa }^{{\text{B}}}} {P_{n}^{{\text{B}}}}}} \right. \kern-0em} {P_{n}^{{\text{B}}}}}$ = 6 (данное отношение несущественно меняется с варьированием температуры Т при используемых фиксированных εij).

Профили на рис. 1 показывают, что плотность В в переходной области (кривые 5–7) мало отличается от плотности В в газе, что обусловлено слабым взаимодействием адсорбата В с адсорбентом А. Заметное превышение плотности В в переходной области по сравнению с газом имеет место только при высоких температурах с κ = 10 и 12 (рис. 1в и 1г) и высоких давлениях, когда переходная область заполнена адсорбатом на 80% (кривая 7 на рис. 1в и 1г). Адсорбция газа несущественно меняет плотность вещества А в переходной области по сравнению с профилем чистого вещества А (кривая 1) при малых давлениях В (когда переходная область заполнена на 10% адсорбатом В) и низких температурах с κ = 5 и 7 (кривая 3 на рис. 1а и 1б).
При больших температурах и давлениях PB адсорбция В приводит к уменьшению шероховатости на поверхности вещества А: профили А (1–4) на рис. 1в и 1г с увеличением давления PB становятся более крутыми. Когда вещество А находится в жидком состоянии и его переходная область на 80% заполнена адсорбатом у поверхности А наблюдается монослой (q = 5 на кривой 4 на рис. 1в и 1г), обедненный содержанием А (плотность А в нем ниже, чем плотность А в объеме).
Кривые $\theta _{q}^{i}$ на рис. 1, представляющие собой доли от всего объема монослоя q, которые занимают частицы i, полностью согласуются с обычными представлениями о повышенной концентрации компонента В на поверхности адсорбента А. При расчете величины адсорбции требуется пересчитать плотность В в монослое q, к доле объема монослоя q, свободного от частиц А, (1 – $\theta _{q}^{A}$). В результате получаем величину $\theta _{q}^{B}$/(1 – $\theta _{q}^{A}$), представляющую собой реальную плотность в данном монослое q, которая входит в полную изотерму адсорбции, представленную ниже на рис. 4. (Такие же комментарии ниже относятся к рис. 2 и 3.)
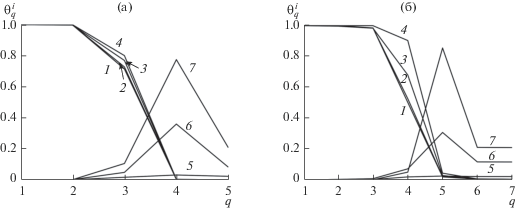
Рис. 3.
Профили плотности компонентов А и В в переходной области с учетом влияния колебаний при физической адсорбции (а) и хемосорбции (б).


Химическая адсорбция. Для хемосорбции рассматриваются две температуры, которым отвечают κ = 5 и 7 (вещество А в твердом состоянии), и три давления PB, при которых монослой на поверхности вещества А заполнен на 10, 50 и 100%. Так как взаимодействие между адсорбентом и адсорбатом сильное, то монослой заполняется при давлениях $P_{m}^{{\text{B}}}$ в несколько раз меньше, чем $P_{n}^{{\text{B}}}{\text{:}}$ ${{P_{m}^{{\text{B}}}} \mathord{\left/ {\vphantom {{P_{m}^{{\text{B}}}} {P_{n}^{{\text{B}}}}}} \right. \kern-0em} {P_{n}^{{\text{B}}}}}$ = 0.2 для рассматриваемых параметров (здесь также данное отношение слабо меняется с варьированием температуры Т при фиксированных εij).
На рис. 2 представлены профили плотности в переходной области чистого вещества А (кривая 1) и плотности А (кривые 2, 3 и 4) в присутствии адсорбата В с профилями, описываемыми кривыми 5, 6 и 7, при давлении газа В, PB, приводящем к заполнению адсорбатом 10, 50 и 100% монослоя на поверхности А, при температурах, которым отвечают κ = 5 (рис. 2а) и 7 (рис. 2б) (вещество А в твердом состоянии).
Профили на рис. 2 показывают, что плотность адсорбата В в переходной области (особенно в монослое q = 4 на рис. 2а и q = 5 на рис. 2б) намного превышает плотность газа В в объеме, что является следствием сильного взаимодействия εАВ. Также по сравнению с физической адсорбцией (рис. 1) при химической адсорбции адсорбат В глубже проникает в переходную область вещества А: имеется существенное содержание В в монослое q = 3 на рис. 1а и в монослое q = 4 на рис. 1б.
С увеличением содержания адсорбата в переходной области вещества А при хемосорбции также наблюдается тенденция к уменьшению шероховатости на поверхности А: плотность А на рис. 2 в центральном монослое (q = 3 на рис. 2а и q = 4 на рис. 2б) растет, но по сравнению с рис. 1 это выражено слабее, так как, во-первых, результаты по хемосорбции представлены для давлений PB в десятки раз меньше, чем при физической адсорбции, а во-вторых, проникновение адсорбата В в более глубокие монослои переходной области препятствуют тенденции А к уменьшению шероховатости своей поверхности.
Влияние колебаний на профили. Колебания учитывались через температурный вклад в энергию взаимодействия: $\varepsilon _{{ij}}^{*} = {{\varepsilon }_{{ij}}}\left( {{{1 - {{u}_{T}}kT} \mathord{\left/ {\vphantom {{1 - {{u}_{T}}kT} {{{\varepsilon }_{{ij}}}}}} \right. \kern-0em} {{{\varepsilon }_{{ij}}}}}} \right),$ ${{u}_{T}} = - 0.1$ [24, 25]. Учет колебаний в 3 раза понизил давление газа $P_{\kappa }^{{\text{B}}},$ при котором заполняется переходная область адсорбатом при физической адсорбции, и в 2 раза повысил давление газа $P_{m}^{{\text{B}}},$ при котором заполняется адсорбатом монослой на поверхности адсорбента А при хемосорбции.
На рис. 3 представлены профили плотности в переходной области чистого вещества А (кривая 1) и плотности А (кривые 2, 3 и 4) при физической (рис. 3а) и химической (рис. 3б) адсорбции В с профилем, описываемым кривыми 5, 6 и 7, при давлении газа В, PB, приводящем к заполнению адсорбатом 10, 50 и 100% всей переходной области в случае физической адсорбции (рис. 3а) и монослоя на поверхности А в случае хемосорбции (рис. 3б), при температуре с κ = 5 (вещество А в твердом состоянии).
Согласно рис. 3 учет влияния колебаний увеличил плотность вещества А (кривые 1–4) в переходной области в том числе и в отсутствии адсорбата В (кривая 1). Учет колебаний привел к росту взаимодействия εАВ, благодаря чему для физической адсорбции вид концентрационных профилей стал ближе к профилях при хемосорбции, в частности существенно повысилась разность плотности В в переходной области и в газе: появились пики на профилях 5–7 в монослое q = 4 на рис. 3а.
В случае хемосорбции (рис. 3б) учет колебаний привел к усилению эффекта сглаживания поверхности вещества А с ростом величины адсорбции В: профили 3 и 4 на рис. 3б характеризуются резким перепадом плотности в монослоях q = 3 и 4. Таким образом, учет влияния колебаний при физической адсорбции больше меняет профили адсорбата в переходной области, а при хемосорбции – профили адсорбента. В целом профили на рис. 3а и рис. 3б близки, но стоит заметить, что для хемосорбции они построены при давлениях PB в 4 раза меньше, чем для физической адсорбции.
Изотермы адсорбции. На рис. 4 представлены изотермы физической адсорбции (рис. 4а) и хемосорбции (рис. 4б) при температурах с κ = 5 (кривая 1) и 7 (кривая 2) (вещество А в твердом состоянии) и κ = 10 (кривая 3) и 12 (кривая 4) (вещество А в жидком состоянии). По оси ординат отложено давление В в газе, PB, нормированное на давление насыщения $P_{n}^{{\text{B}}}.$ По оси абсцисс отложены на рис. 4а величина адсорбции $\Gamma _{\kappa }^{{\text{B}}}$ в переходной области А, нормированная на емкость переходной области Мκ, а на рис. 4б величина адсорбции $\Gamma _{m}^{{\text{B}}}$ в монослое на поверхности А, нормированная на емкость монослоя на поверхности А Мm. Проблемы определения емкости монослоя на шероховатой поверхности разобраны в работах [17, 18], в которых показано, как прямой метод моделирования структуры поверхности соотносится с экспериментальными данными.
Рисунок 4а показывает, что с увеличением PB при физической адсорбции сначала быстро увеличивается количество адсорбата В в переходной области $\Gamma _{\kappa }^{{\text{B}}},$ пока не будет достигнуто $P_{n}^{{\text{B}}}.$ В данной области изотермы 1–4, построенные для разных температур, практически неотличимы. Затем, когда PB/$P_{n}^{{\text{B}}}$ > 1, с дальнейшим увеличением давления PB скорость роста величины адсорбции $\Gamma _{{\kappa }}^{{\text{B}}}$ замедляется. В этой области наблюдаются небольшие отклонения изотерм 1–4 друг от друга. В целом, требуются большие давления PB/$P_{n}^{{\text{B}}}$ = = 20–30, чтобы переходная область полностью заполнилась адсорбатом В $\Gamma _{\kappa }^{{\text{B}}}$/Мκ = 1.
Рисунок 4б показывает, что с увеличением PB при хемосорбции величина адсорбции в монослое $\Gamma _{m}^{{\text{B}}}$ растет практически линейно, пока не достигает значение емкости монослоя Мm. Монослой при заданных параметрах заполняется примерно при PB/$P_{n}^{{\text{B}}}$ = 0.2.
Положение разделяющей поверхности внутри переходной области при адсорбции зависит как функция степени заполнения поверхности адсорбатом. Отметим, что здесь положение эквимолекулярной поверхности ρe рассчитывается статистическим усреднением по всем конфигурациям шероховатой поверхности [13], поэтому изменения этой величины могут быть не дискретные.
На рис. 5 представлено, как с ростом давления PB изменяется положение разделяющей поверхности ρe (в качестве таковой выбрана эквимолекулярная поверхность) внутри переходной области А при физической (рис. 5а) и химической (рис. 5б) адсорбции при температурах с κ = 5 (кривая 1) и 7 (кривая 2) (вещество А в твердом состоянии) и κ = 10 (кривая 3) и 12 (кривая 4) (вещество А в жидком состоянии).
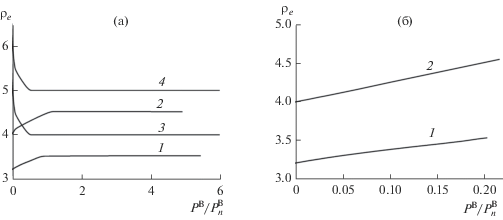
На рис. 5а кривые 1, 2, построенные для физической адсорбции на твердом теле, показывают, что с ростом давления PB реперная поверхность ρe смещается на один монослой к пару, пока не достигается давление $P_{n}^{{\text{B}}}.$ Затем реперная поверхность ρe практически не смещается. Последнее связано с замедлением скорости адсорбции после PB/$P_{n}^{{\text{B}}}$ = 1, наблюдаемое на рис. 4а. Кривые 3, 4, построенные для физической адсорбции на поверхности жидкости, показывают, что с ростом давления PB реперная поверхность ρe смещается на два монослоя к жидкости, достигая своего постоянного значения еще до давления $P_{n}^{{\text{B}}}.$
В случае хемосорбции на твердом теле (рис. 5б) реперная поверхность ρe смещается в пределах одного монослоя к пару практически линейным образом для обеих рассмотренных температур.
Поверхностное натяжение зависит от степени заполнения поверхности адсорбатом как функция его давления для обоих типов адсорбции (физической и химической) – здесь следует напомнить о разных диапазонах изменения рассматриваемого давления.
На рис. 6 представлено, как с ростом давления PB изменяется поверхностное натяжение σ, нормированное на поверхностное натяжение чистого вещества А σA, на разделяющей поверхности внутри переходной области при физической (рис. 6а) и химической (рис. 6б) адсорбции при температурах с κ = 5 (кривая 1) и 7 (кривая 2) (вещество А в твердом состоянии) и κ = 10 (кривая 3) и 12 (кривая 4) (вещество А в жидком состоянии).
Рис. 6.
Изменение поверхностного натяжения вещества А с ростом величины адсорбции на нем в результате физической адсорбции (а) и хемосорбции (б).
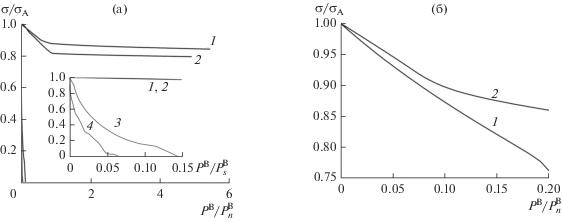
Рисунок 6а показывает, что при физической адсорбции на твердом теле (кривые 1 и 2) поверхностное натяжение убывает с увеличением PB. При PB/$P_{n}^{{\text{B}}}$ > 1 поверхностное натяжение продолжает убывать существенно медленнее, чем при PB/$P_{n}^{{\text{B}}}$ < 1, так как уменьшается скорость адсорбции (см. рис. 4а). При физической адсорбции на поверхности жидкости (кривые 3 и 4 на рис. 6а) поверхностное натяжение резко убывает и достигает нуля еще до того, как давление PB доходит до $P_{n}^{{\text{B}}}.$ Это является следствием резкого уменьшения неоднородности поверхности жидкости, наблюдаемое на рис. 1в и 1г, а именно уменьшения фактической ширины неоднородной области. С увеличением температуры получаем, что для заданного давления $P_{n}^{{\text{B}}}$ поверхностное натяжение отклоняется сильнее от значения σА.
Рисунок 6б показывает, что при хемосорбции поверхностное натяжение вещества А убывает с ростом PB. В отличие от физической адсорбции, поверхностное натяжение тем сильнее отклоняется от σA, чем ниже температура. В области давлений PB/$P_{n}^{{\text{B}}}$ < 0.2 поверхностное натяжение убывает быстрее при хемосорбции, чем при физической адсорбции.
ВЕРОЯТНОСТЬ ПОЯВЛЕНИЯ НОВОЙ ФАЗЫ
Задача описания динамики появления новой фазы должна решаться методами неравновесной термодинамики и/или кинетических теорий. Часто для упрощения решения такой задачи используют флуктуационный подход, когда для выбранного характерного масштаба времени вводят вероятность появления частицы новой фазы или зародыша вместо решения систем кинетических уравнений на изменение состояния изучаемой системы. Такой подход принят во многих приложениях [9, 14]. Расчет вероятности флуктуации появления новой фазы оценивается по формуле $\omega = K\exp \left( {{{ - {{R}_{{\min }}}} \mathord{\left/ {\vphantom {{ - {{R}_{{\min }}}} {{{R}_{B}}{{T}_{0}}}}} \right. \kern-0em} {{{R}_{B}}{{T}_{0}}}}} \right)$ [27, 28], где $K$ – константа скорости процесса роста фазы, соответствующая выбранной шкале времени, Rmin – минимальная работа образования зародыша. Для появления зародыша новой фазы, согласно [16], величина Rmin равна ${{R}_{{\min }}} = \sigma (R)A(R)$ (вместо такого же произведения с коэффициентом 1/3 [9, 14]), где функции $\sigma (R)$ и $A(R)$ относятся к размерным зависимостям поверхностного натяжения и площади поверхности трехмерной капли. Здесь T0 – температура состояния системы, из которой происходит флуктуационный переход.
Величины поверхностного натяжения, приведенные выше для макроскопических (плоских) границ, обладают закономерностями уменьшения $\sigma (R) \to 0$ при уменьшении размера капли до критического размера существования фазы R0 [16]. Этот факт приводит к тому, что если воспользоваться нормированными величинами ${\omega \mathord{\left/ {\vphantom {\omega K}} \right. \kern-0em} K},$ то они отражают размерные зависимости процесса флуктуационного появления новой фазы.
На рис. 7 показано, как с изменением радиуса капли R (величина R нормирована на межмолекулярное расстояние λ), меняется вероятность появления новой фазы ${\omega \mathord{\left/ {\vphantom {\omega K}} \right. \kern-0em} K}$ данного размера при двух температурах с κ = 10 (кривые 1 и 3) и 12 (2) в отсутствии адсорбата (1 и 2) и при хемосорбции компонента В (3) при низких давлениях PB/$P_{n}^{{\text{B}}}$ = 10–3.

Кривые для радиусов от R0 до 200λ для двух температур показывают, что вероятность образования новой фазы возрастает с уменьшением размера зародыша R (все кривые 1–3 имеют отрицательный наклон), с увеличением температуры (кривая 1 для более низкой температуры лежит ниже, чем кривая 2), и в результате адсорбции (кривая 3 лежит ниже кривой 1).
Качественный результат молекулярного описания зарождения шероховатой поверхности состоит в наличии скачка для значения радиуса зародыша, начиная с которого новое образование становится устойчивым. Здесь это величина равна R0/λ = 10. Данный результат согласуется ранее полученными значениями R0/λ ~ 10 для формирования равновесной фазы в объеме [16] и на плоской поверхности монослоя адсорбата [29].
ЗАКЛЮЧЕНИЕ
Для анализа влияние адсорбции на состояние равновесных шероховатых поверхностей границ раздела фаз использована дискретная модель плотной фазы – модель решеточного газа, учитывающая прямые корреляции между всеми ближайшими компонентами системы в квазихимическом приближении. Модель для простоты предполагает соизмеримость размеров компонентов и учитывает колебания их частиц через эффективные вклады параметров латерального взаимодействия, как для атомов адсорбента, так и молекул адсорбата.
Исследовано влияние физической и химической адсорбции на характеристики равновесных шероховатых поверхностей границ раздела фаз пар–жидкость и пар–твердое тело. Разработанные программы позволяют рассчитать поверхностное натяжение для всего температурного диапазона паро-жидкостных систем (от температуры точки плавления до критической температуры), и исследовать температурные зависимости адсорбции. Для твердых тел полученные результаты относятся к температурам около температуры плавления.
Рассчитаны равновесные профили адсорбента А и адсорбата В внутри переходной области (при условии неизменности объемной фазы адсорбента) для физической адсорбции и для хемосорбции. В случае хемосорбции всегда фиксировалось заметное превышение плотности В в переходной области по сравнению с газом и уменьшение шероховатости на поверхности вещества А под воздействием адсорбата В, а в случае физической адсорбции повышенная концентрация В в переходной области и сглаживание поверхности А наблюдались только при высоких температурах с κ = 10 и 12 и высоких давлениях, когда переходная область заполнена адсорбатом на 80%.
Рассмотрено влияние адсорбции на поверхностное натяжение плотной фазы. Получено, что поверхностное натяжение на границе шероховатой поверхности А убывает как с физической адсорбцией, так и с хемосорбцией.
Построены оценки вероятности флуктуационных процессов формирования новой фазы, обусловленной шероховатостью поверхности чистого адсорбента А и при учете адсорбции на ней частиц В. Они показали, что вероятность образования новой фазы возрастает с уменьшением размера зародыша R, с увеличением температуры, и в результате адсорбции.
Данный подход может быть обобщен на расплавы и сплавы многокомпонентных систем, а также на смеси адсорбатов.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (код проекта № 18-03-00030а).
Список литературы
Трепнелл Б. Хемосорбция. М.: изд-во иностр. лит. 1958. 326 с.
Киперман С.Л. Введение в кинетику гетерогенных каталитических реакций. М.: Наука, 1964. 607 с.
Волъкенштейн Ф.Ф. Физикохимия поверхности полупроводников. М.: Наука, 1973. 400 с.
Дельмон Б. Кинетика гетерогенных реакций / Пер. с фр. под ред. Болдырева В.В. М.: Мир, 1972. 554 с.
Розовский А.Я. Кинетика топохимических реакций. М.: Химия, 1974. 220 с.
Баре П. Кинетика гетерогенных процессов / Пер. с фр. под. ред. Болдырева В.В. М.: Мир, 1976. 399 с.
Киселев В.Ф., Крылов О.В. Адсорбционные процессы на поверхности полупроводников и диэлектриков. М.: Наука, 1978. 255 с.
Моррисон С. Химическая физика поверхности твердого тела / Пер. с англ. под ред. Волькенштейна Ф.Ф. М.: Мир, 1980. 488 с.
Адамсон А. Физическая химия поверхностей / Пер. с англ. под ред. Зорина 3.М., Мулера В.М. М.: Мир, 1979. 568 с.
Roberts M.W., McKee C.S. Chemistry of the metal-gas interface. Oxford: Clarendon Press, 1978.
Somorjai G.A. Chemistry in two- dimension surface. N.Y., Ithaca: Cornell Univ. Press, 1981.
Даннинг В. // Межфазовая граница газ–твердое тело. Под ред. Флада Э. М.: Мир, 1970. С. 230. [Benson G.G., Yun K.S. // The solid – gas interface / Edited by E. Alison Flood, Dekker Marcel, Inc., N.Y. 1967.]
Товбин Ю.К. Зайцева Е.С., Рабинович А.Б. // ЖФХ 2018 Т. 92. № 3. С. 474.
Оно С., Кондо С. Молекулярная теория поверхностного натяжения. М.: Изд-во иностр. лит., 1963. (Ono S., Kondo S. Molecular theory of surface tension in liquids. Berlin–Gottinhen–Heidelberg: Springer–Verlag, 1960.)
Роулинсон Дж., Уидом Б. Молекулярная теория капиллярности. М.: Мир, 1986. (Rowlinson J.S., Widom B. Molecular Theory of Capillarity. Oxford: ClarendonPress, 1982.)
Товбин Ю.К. Малые системы и основы термодинамики. М.: Физматлит, 2018. 404 с. (Tovbin Yu.K. Small systems and fundamentals of thermodynamics. Boca Raton, Fl.: CRC Press, 2019.)
Зайцева Е.С., Товбин Ю.К. // ЖФХ. 2018. Т. 92. № 5. С. 816.
Зайцева Е.С., Товбин Ю.К. // Физикохимия поверхности и защита материалов. 2018. Т. 54. № 4. С. 365.
Хилл Т. Статистическая механика. М.: Изд-во иностр. лит., 1960. (Hill T.L. Statistical Mechanics. Principles and Selected Applications. N.Y.: McGraw–Hill Book Comp.Inc., 1956.)
Гиршфельдер Дж., Кертисс Ч., Берд Р. Молекулярная теория газов и жидкостей. М.: ИЛ, 1961. 930 с.
Товбин Ю.К. Теория физико-химических процессов на границе газ – твердое тело. М.: Наука, 1990. (Tovbin Yu.K. Theory of physical chemistry processes at a gas–solid surface processes. Boca Raton, Fl.: CRC Press, 1991.)
Товбин Ю.К. Молекулярная теория адсорбции в пористых телах. М.: Наука, 2012. 624 с. (Tovbin Yu.K. Molecular Theory of Adsorption in Porous Solids. Boca Raton, Fl.: CRC Press, 2017.)
Товбин Ю.К., Рабинович А.Б. // Изв. Акад. наук. Сер. химич. 2009. № 11. С. 2127.
Товбин Ю.К., Рабинович А.Б., Гвоздева Е.Е. // ЖФХ. 2014. Т. 88. № 10. С. 1624.
Товбин Ю.К., Рабинович А.Б., Гвоздева Е.Е. // Физикохимия поверхности и защита материалов. 2015. Т. 51. № 1. С. 8.
Товбин Ю.К. Зайцева Е.С., Рабинович А.Б. // ЖФХ. 2016. Т. 90. № 1. С. 138.
Ландау Л.Д., Лившиц Е.М. Теоретическая физика. Т. 5. Статистическая физика. М.: Наука, 1964. 568 с.
Товбин Ю.К., Еремич Д.В., Комаров В.Н., Гвоздева Е.Е. // Химическая физика. 2007. Т. 26. № 9. С. 98.
Товбин Ю.К., Зайцева Е.С., Рабинович А.Б. // ЖФХ. 2016. Т. 90. № 6. С. 917.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов