

Физикохимия поверхности и защита материалов, 2019, T. 55, № 6, стр. 641-648
Коррозионные и физико-механические свойства сплава Cr–P–W полученного электроосаждением из водно-диметилформамидных электролитов
А. В. Тележкина 1, В. В. Кузнецов 1, 3, *, Е. А. Филатова 1, Н. Е. Некрасова 1, **, В. В. Жуликов 2, В. А. Колесников 1
1 Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, Миусская пл., 9, Россия
2 Институт физической химии и электрохимии им. А.Н. Фрумкина
119071 Москва, Ленинский проспект, 31, корп. 4, Россия
3 Национальный исследовательский ядерный университет “МИФИ”
115409 Москва, Каширское шоссе, 31, Россия
* E-mail: Vitkuzn1@mail.ru
** E-mail: Madjerre@mail.ru
Поступила в редакцию 02.04.2019
После доработки 04.04.2019
Принята к публикации 10.04.2019
Аннотация
Защитные покрытия Cr–P–W были получены электроосаждением из водно-диметилформамидных растворов на основе хлорида хрома(III), в которые вводили фосфинат и вольфрамат натрия. Полученные сплавы содержали до 8 мас. % фосфора, 1.5 мас. % вольфрама, а также некоторое количество углерода. Полученные покрытия обладали удовлетворительной коррозионной стойкостью в коррозивных средах, содержащих хлорид-ионы. Количество сквозных пор в покрытиях становилось пренебрежимо малым при толщине покрытия более 15 мкм, что позволило обеспечить достаточную защитную способность разработанных покрытий.
ВВЕДЕНИЕ
Защитные покрытия, наносимые методами гальванотехники, представляют существенный интерес для противокоррозионной защиты. Хромовые покрытия характеризуются удачным сочетанием физико-механических и защитных свойств. Однако их существенным недостатком является нестойкость в коррозивных средах в присутствии хлорид-ионов.
По этой причине задача повышения коррозионной стойкости и защитной способности хромовых покрытий является актуальной. С этой целью в их состав дополнительно вводят другие элементы, как металлы (Ni [1, 2], Co [3, 4], Mo [5], W [6, 7]), так и неметаллы (P [8, 9], B [10], C [6, 7, 11, 12]). Однако из растворов на основе оксида хрома(VI), традиционно применяемых в технологии хромирования, электроосаждение сплавов хрома затруднено [13]. Это заставляет обратиться к растворам на основе Cr(III), применение которых позволяет существенно расширить количество сплавов хрома, получаемых электролизом [1–12]. Из электролитов “трехвалентного” хромирования возможно также получение композиционных покрытий на основе хромовой матрицы [14].
Особенности процесса хромирования из растворов на основе Cr(III) связаны с образованием в них устойчивых и кинетически инертных координационных соединений трехвалентного хрома [15–19]. Химический состав координационных соединений в прикатодном слое определяет как закономерности электрокристаллизации хрома, так и элементный состав образующихся покрытий. В работах было показано [9, 20] что введение фосфината натрия в растворы на основе сульфата хрома(III) позволяет получить Cr–P покрытия, обладающие более высокой, по сравнению с хромовыми, коррозионной стойкостью в присутствии хлорид-ионов. Электроосаждение сплавов хром-фосфор, по-видимому, происходит из комплексов Cr(III) с фосфинат-ионами [15]. Электролитические покрытия Cr–P представляют собой либо нанокристаллические твердые растворы фосфора в хромовой матрице [21], либо рентгеноаморфные материалы [22, 23], что благоприятно с точки зрения противокоррозионной защиты [24, 25]. В [15] показано, что соосаждение хрома с кобальтом приводит к образованию более однородной структуры нанокристаллического хромового покрытия.
Введение тугоплавких металлов в состав хромовых покрытий также приводит к повышению их коррозионной стойкости. Известно, что даже небольшое (до 1%) количество вольфрама (или молибдена) в составе защитных хромовых покрытий вызывает снижение скорости общей коррозии в растворах, содержащих хлорид-ионы, приблизительно на порядок [5, 6]. Высказано мнение [26], что при воздействии коррозионной среды на поверхности сплавов происходит формирование тонкого (~20 нм) слоя, обогащенного соединениями тугоплавкого металла. Образование этого барьерного слоя приводит к существенному торможению коррозионного процесса.
Для дальнейшего повышения коррозионной стойкости и защитной способности хромовых покрытий представлялось целесообразным одновременное введение в их состав фосфора и вольфрама. Целью данной работы являлись: 1) получение Cr–P–W покрытий из растворов, содержащих соединения Cr(III) и 2) определение коррозионной стойкости и защитной способности полученных покрытий в ряде коррозионных сред, в том числе, и в присутствии хлорид-ионов.
МЕТОДИКИ ИССЛЕДОВАНИЯ
Для электроосаждения сплавов хрома c фосфором и вольфрамом были использованы водно-диметилформамидные растворы. Выбор водно-органических сред был продиктован следующими соображениями. Во-первых, применение этих электролитов позволяет получать покрытия, содержащие 1–2 мас. % вольфрама, что невозможно из водных растворов [7]. Во-вторых, из этих растворов возможно электроосаждение слоев хрома значительной толщины (до 800 мкм [27]). Это позволяет легко получить компактное покрытие требуемой толщины путем изменения времени электролиза. В-третьих, очистка промывных и сточных вод от диметилформамида может быть легко проведена электрохимическими методами: он легко подвергается полной деструкции на нерастворимом аноде.
Раствор, используемый для электроосаждения Cr–P–W покрытий, содержал (моль дм–3): CrCl3 ⋅ 6H2O – 1, Na2WO4 ⋅ 2H2O – 0.05. Объемное соотношение вода: ДМФА в электролите составило 1 : 1. Методика приготовления электролита описана в [7]. В растворы дополнительно вводили фосфинат натрия в концентрациях 0.01–0.1 моль/л.
Нанесение Cr–P–W покрытий проводили в гальваностатических условиях (i = 0.22–0.25 A см–2) в ячейке без разделения катодного и анодного пространств. Выделения токсичного хлора при электролизе не происходило, поскольку на инертном Pt/Ti-аноде окислялись молекулы диметилформамида. Этот процесс приводил к уменьшению содержания ДМФА в электролите. Электрохимическая стабильность раствора без корректировки по диметилформамиду составляла 65–70 А ч дм–3. Это свидетельствует о том, что из разработанного раствора возможно осаждение качественных покрытий в достаточно широком интервале концентраций ДФМА.
При электролизе на инертном аноде, по-видимому, происходило также окисление фосфинат-ионов. По этой причине при проведении экспериментов, особенно в области низких концентраций фосфината натрия, всегда использовали свежеприготовленные растворы. В случае практического применения разработанных электролитов следует периодически проводить определение содержания ионов ${{{\text{H}}}_{{\text{2}}}}{\text{PO}}_{{\text{2}}}^{ - }$ в электролите и осуществлять необходимую корректировку раствора.
Водно-органические электролиты, использовавшиеся для осаждения Cr–P–W-покрытий, обладали низкой макрорассеивающей способностью. По этой причине было необходимо оптимизировать первичное распределение тока, что было осуществлено с помощью экранов из непроводящего материала и отвлекающих катодов.
Химический состав полученных покрытий был определен по данным рентгеновского микроанализатора, которым был оснащен сканирующий электронный микроскоп JSM-6490LV фирмы JEOL. Данные об относительном содержании элементов были получены, по крайней мере, в десяти точках на поверхности образца. Диаметр участка поверхности, с которого получали аналитическую информацию составлял ~1 мкм. Результаты определений подвергались статистической обработке.
Фазовый состав полученных покрытий был определен на рентгеновском дифрактометре STOE STADI P (“STOE&Cie GmbH”, Германия) с CuKα-излучением. Прибор был оснащен устройством для подавления флуоресцентного излучения.
Состав поверхностных слоев покрытий, а также степени окисления элементов в них были определены методом рентгеновской фотоэлектронной микроскопии. В работе использовали спектрометр НВ100 (Vacuum Generators, GB) с АlKα-излучением (энергия излучения 1486.6 эВ, мощность 200 Вт). Энергия пропускания анализатора составляла 50 эВ. Камеру вакуумировали до остаточного давления менее 10–8 мм рт. ст. Погрешность определения положения максимума пика составила не более 0.3 эВ.
Коррозионно-электрохимическое поведение осадков Cr–P–W определяли в стандартной трехэлектродной ячейке с использованием цифрового потенциостата IPC–Pro (Вольта, Россия). Потенциал рабочего электрода задавали относительно насыщенного хлорид-серебряного электрода сравнения, вспомогательным электродом служила сетка платинированного титана. Перед измерением исследуемые покрытия наносили на поверхность стеклоуглерода (СУ), средняя толщина нанесенных покрытий составила 5 мкм. Поляризационные характеристики были получены в потенциодинамических условиях при скорости развертки потенциала 0.167 мВ с–1. Анодные поляризационные кривые (АПК) были сняты в деаэрированных аргоном в течение 30 мин растворах, чтобы исключить влияние тока растворенного кислорода (~10–5 А см–2). Снятие АПК проводили из катодной области от потенциала на 50 мВ отрицательнее потенциала свободной коррозии при развертке потенциала в анодном направлении со скоростью 0.167 мВ с–1.
Данные о скорости коррозии Cr–P–W-покрытий, полученные электрохимическим методом, были сопоставлены с результатами гравиметрии по убыли массы Cr–P–W/СУ образцов в результате их длительной (7 дней) выдержки в исследуемых коррозивных средах.
Пористость полученных покрытий, осажденных на низкоуглеродистую сталь (0.2 ат. % C), определяли методом Уокера [28]; на поверхность образца накладывали фильтровальную бумагу, пропитанную раствором, содержащим 3 г дм–3 K3[Fe(CN)6] и 20 г дм–3 NaCl в течение 5 мин. О наличии сквозных пор в покрытии судили по появлению синих точек на фильтровальной бумаге. Дополнительно для оценки пористости нанесенных покрытий был измерен стационарный потенциал стальных образцов с медным подслоем и нанесенными Cr–P–W-покрытиями различной толщины в 0.1 М растворе хлороводородной кислоты.
Ускоренные коррозионные испытания проводили в камере солевого тумана в течение 30 суток; в исследовании определялось время появления первых очагов коррозии.
Для оценки возможности применения Cr–P–W-покрытий важно знать не только их коррозионно-электрохимические и защитные характеристики, но и физико-механические свойства. В данной работе была измерена микротвердость покрытий. Измерения были проведены методом вдавливания алмазной пирамидки Виккерса на микротвердомере ПМТ-3. Чтобы минимизировать влияние стальной основы, толщина нанесенных покрытий была не менее 15 мкм, нагрузка – 50 г.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
После электролиза на катоде было зафиксировано образование светло-серого покрытия хорошего качества. В составе катодного осадка зафиксированы хром, вольфрам, фосфор и некоторое количество углерода. Включение углерода, главным образом в виде карбидов, характерно для электролитов на основе соединений Cr(III) [11, 12, 29], содержащих органические вещества. Хром является основным компонентов полученных сплавов, его содержащие составляет 85–89 ат. %.
На рис. 1 представлены зависимости относительного содержания этих элементов от концентрации фосфината натрия в электролите (i = 0.22 А см–2). При увеличении концентрации фосфината натрия в растворе от 0.01 до 0.10 моль дм–3 содержание фосфора в формирующихся на катоде покрытиях возрастает, а количество углерода снижается. Возможно, что это связано с тем, что фосфинат-ионы, участвующие в реакции восстановления соединений хрома, адсорбируются в приэлектродном слое, вытесняя оттуда молекулы диметилформамида, что и приводит к наблюдаемым изменениям в содержаниях неметаллов в покрытии. Содержание вольфрама в катодном осадке постоянно и практически не зависит от концентрации фосфината натрия в растворе. Азот в составе катодного осадка зафиксирован не был.
Рис. 1.
Содержание фосфора, углерода и вольфрама в Cr–P–W-покрытиях, полученных в гальваностатических условиях при i = 0.22 А см–2 по данным рентгеновского микроанализа.
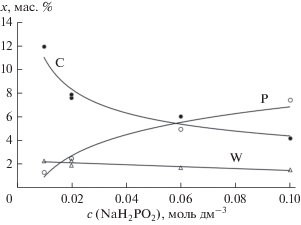
Для дальнейших исследований были выбраны покрытия, полученные при концентрации фосфината натрия в электролите, равной 0.10 моль дм–3, поскольку они содержали наибольшее количество фосфора. Это позволяло надеяться на их высокую коррозионную стойкость. Однако исследование морфологии полученных катодных осадков показало (рис. 2), что увеличение содержания фосфора в них вызывало появление трещин. Вероятно, появление трещин связано с образованием фосфидов хрома в катодном осадке, что приводит к возникновению внутренних напряжений и, соответственно, к растрескиванию.
Рис. 2.
Морфология поверхности Cr–P–W-покрытий (i = 0.22 А см–2, 10 мкм). Концентрация фосфината (гипофосфита) натрия в растворе (моль/л): (а) – 0, (б) – 0.02, (в) – 0.10.

Наличие сквозных трещин должно приводить к резкому снижению защитной способности покрытий. По этой причине особое значение приобретают определение оптимальной толщины Cr–P–W-покрытий и решение вопроса о целесообразности применения медного подслоя при защите стальных изделий от коррозии.
Покрытия Cr–P–W, полученные электроосаждением, были ренгеноаморфными: при толщине покрытий 10 мкм на дифрактограммах присутствовали лишь рефлексы, соответствующие медной подложке. Получение рентгеноаморфных покрытий, характеризующихся повышенной коррозионной стойкостью, благоприятно с точки зрения противокоррозионной защиты.
Включение фосфора и вольфрама в состав образующихся при электролизе покрытий было подтверждено результатами рентгеновской фотоэлектронной спектроскопии (РФЭС). Согласно данным РФЭС, в поверхностном слое покрытий после их экспозиции на воздухе были зафиксированы хром, вольфрам и фосфор.
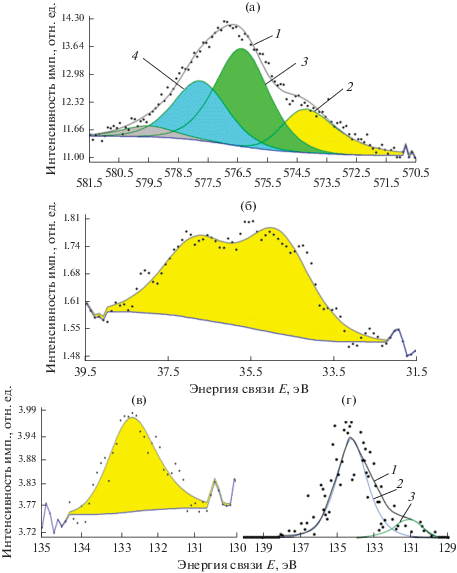
В составе поверхностной пленки, образовавшейся в результате поверхностного окисления сплава в атмосфере воздуха лаборатории, хром находился как в металлическом, так и в окисленном (Cr+3) состояниях (рис. 3а). Окисленные формы хрома были представлены оксидом и гидратированными формами оксида хрома(III). В поверхностном слое покрытий вольфрам был обнаружен лишь в окисленном состоянии (WO3) (рис. 3б). Соединения фосфора также входили в состав пассивной пленки, сформировавшейся на поверхности покрытия (рис. 3в). Фосфор, зафиксированный на поверхности сплава Cr–P–W после экспозиции образца с нанесенным покрытием в воздухе лаборатории находится в степени окисления +5, что, по-видимому, связано с окислением его соединений на поверхности образца. Этот процесс приводит к формированию защитной пленки. После травления на глубину ~20 нм большая часть фосфора по-прежнему находится в степени окисления +5 (рис. 3г), однако детальное рассмотрение спектра РФЭС высокого разрешения позволяет выделить “плечо” в области более низких энергий связи (~131 эВ), что может соответствовать неокисленным формам фосфора (P0 или фосфидам [30]). Очевидно, что в процессе электроосаждения при сильно отрицательных потенциалах катода (<–1.0 В, с. в. э.) в состав катодного осадка включаются атомы фосфора, находящиеся в нулевой или более низких степенях окисления. Однако при воздействии кислорода воздуха происходит их поверхностное окисление до фосфатов.
Рис. 3.
Спектры РФЭС Cr 2p (a), W 4f (б) и P 2p (в, г) для Cr–P–W-покрытий после их экспозиции в воздухе лаборатории (3 дня). (а, б, в) – на поверхности образца, (г) – после ионного травления на глубину ∼10 нм. Цифрами обозначены: на подрисунке а: 1 – экспериментальный спектр, 2 – Cr, 3 – Cr2O3, 4 – Cr(OH)3; на подрисунке г: 1 – экспериментальный спектр, 2 – P(+5), 3 – фосфиды.
Бестоковый потенциал сплавов Cr–P–W, полученных электролизом, в 0.5 М H2SO4 равен ~0.01 В (рис. 4а), что близко к равновесному потенциалу водородной реакции и значительно положительнее потенциала свободной коррозии металлургического хрома (~ –0.5 В). Диаграмма Эванса представлена на рис. 4а. На анодной ветви поляризационной кривой отсутствует область потенциалов активного растворения. По этой причине определение плотности тока коррозии экстраполяцией тафелевых участков на катодной и анодной ветвях поляризационной кривой невозможно.
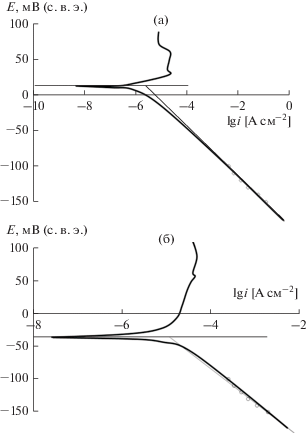
Значение lg iкор, в принципе, может быть получено аппроксимацией прямолинейного участка, зафиксированного на катодном скане диаграммы Эванса, на потенциал коррозии [31]. Однако надежно тафелевый участок на катодной кривой наблюдается лишь при потенциалах отрицательнее потенциала свободной коррозии более, чем на 70 мВ, когда токами обратного направления можно пренебречь. В этих условиях на поверхности электрода происходит интенсивное выделение водорода, что снижает точность вольтамперометрических измерений. По этой причине величину логарифма тока коррозии электролитических осадков Cr–P–W, найденную экстраполяцией тафелевого участка на потенциал свободной коррозии (lg iкор ≈ –5.6) следует рассматривать как ориентировочную. Более надежные результаты определения тока коррозия могут быть получены методами гравиметрии (–6.0) и анализа коррозионной среды (–5.7). Скорость коррозии Cr–P–W покрытий существенно меньше по сравнению с Cr–C покрытиями, полученными электроосаждением из водно-диметилформамидных растворов [7] и сопоставима с покрытиями, имитирующими сплав стеллит [32]. В последнем случае снижение скорости коррозии было вызвано образованием на поверхности сплава, подвергаемого воздействию коррозионной среды, тонкой (~10 нм) пленки, состоящий из оксидов хрома и вольфрама. Возможно, что аналогичное явление имеет место и для Cr–P–W покрытий.
Пассивное состояние Cr–P–W сплавов при потенциале свободной коррозии сохраняется и при переходе от растворов серной кислоты к растворам хлороводородной кислоты с той же активностью ионов водородов, при этом Eкор лишь незначительно смещается в сторону более отрицательных потенциалов (до –0.04 В). Методы гравиметрии и анализа коррозионной среды показали, что ток коррозии увеличился лишь в ~5 раз по сравнению с раствором серной кислоты. Следовательно, активирующее действие хлорид-ионов, хотя и проявляется, относительно невелико.
В небуферируемых растворах хлорида натрия потенциал коррозии Cr–P–W покрытий равен ~ –0.2 В. Значение тока коррозии невозможно определить электрохимическим методом, что связано с сильными осцилляциями тока, наблюдаемыми на вольтамперных кривых, вблизи потенциала коррозии [33]. Расчет скорости коррозии на основе анализа коррозионной среды привел к значению логарифма тока коррозии –6.0. Это значение ожидаемо ниже скорости коррозии в хлороводородной кислоте и сопоставимо с результатами, полученными для электролитических сплавов хрома, осажденных из растворов на основе соединений Cr(III). Следует отметить, что скорости коррозии электролитических осадков Cr–P–W существенно ниже по сравнению с Cr–C и имеют один порядок величины по сравнению с Cr–C–W.
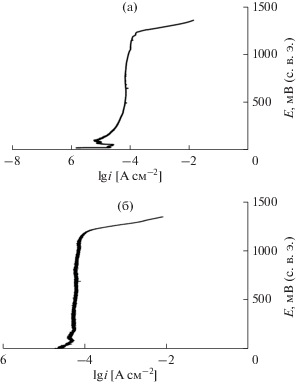
На АПК электролитически осажденных Cr–P–W сплавов в 0.5 М серной кислоте (рис. 5а) наблюдается протяженная область потенциалов пассивного состояния (до ∼1.2 В). Плотность тока пассивации в этом интервале потенциалов составляет ~10–5–10–4 А см–2. Интересно, что подобная область зафиксирована и на анодных кривых в 1.0 М HCl (рис. 5б). По сравнению с растворами серной кислоты можно отметить лишь незначительное уменьшение протяженности области потенциалов пассивного состояния (до ~1.1 В), плотности тока растворения сплава, находящегося в пассивном состоянии, немного меньше 10–4 А см–2. На основании этого можно сделать вывод о том, что активирующее действие хлорид-ионов на анодное растворение Cr–P–W-покрытий не столь велико, чтобы вызвать нарушение пассивного состояния сплава при анодной поляризации. Аналогичная область потенциалов пассивного состояния наблюдается и в 0.5 М растворе хлорида натрия.
Рис. 5.
Анодные поляризационные кривые Cr–P–W/СУ электрода в 0.5 М H2SO4 (а) и 1.0 М HCl (б). Скорость развертки потенциала 0.167 мВ с–1. Кривые получены при развертке потенциала из катодной области в анодную сторону.
Наличие сквозных пор в хромовом покрытии при их нанесении на сталь недопустимо: в случае их возникновения будет происходить коррозия стальной основы под пассивным хромовым покрытием. Результаты СЭМ показывают наличие трещин в электролитических осадках Cr–P–W (рис. 2). Однако микроскопическое исследование шлифов нанесенных покрытий показало, что при толщине покрытия 10 мкм трещины, в основном, не являются сквозными (рис. 6). К такому же выводу привела и оценка пористости покрытия с помощью реактива Уокера: для стальных образцов с толщиной покрытия 15 и 20 мкм не было обнаружено синих точек, соответствующих непокрытой стальной основе. Ускоренные коррозионные испытания, проведенные в камере солевого тумана, показали, что для стальных образцов с нанесенным Cr–P–W покрытием толщиной 15 мкм первые очаги коррозии фиксируются на 25-й день выдержки. Этот показатель существенно лучше по сравнению с Cr–W-покрытиями: для них первые очаги коррозии появлялись уже на 4-й день экспозиции в камере солевого тумана. Следовательно, на основании проведенных предварительных испытаний можно сделать вывод о том, что защитная способность разработанных покрытий должна быть удовлетворительной.

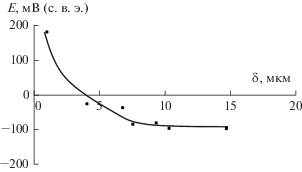
Однако, учитывая катодный характер разработанного электролитического покрытия по отношению к стали, представляется целесообразным его нанесение не непосредственно на стальную основу, а на предварительно сформированный из пирофосфатного электролита меднения беспористый подслой меди (5 мкм). Поскольку стационарный потенциал меди во всех исследованных коррозивных средах более положительный, чем у электролитических сплавов Cr–P–W, даже при возникновении сквозных пор в покрытиях не будет происходить окисления основы. Однако и в этом случае наличие сквозных трещин следует признать нежелательным: их появление будет приводить к растворению покрытий. Толщина покрытия, при которой влияние медного подслоя становится пренебрежимо малым, была оценена с помощью измерения бестокового потенциала образцов с покрытиями Cr–P–W различной толщины. Из полученных данных (рис. 7) видно, что значение стационарного потенциала становится постоянным при δ > 10 мкм, приближаясь к бестоковому потенциалу электролитического сплава Cr–P–W, отделенного от основы. Этот результат находится в соответствии с ранее полученными данными о пористости разработанных электролитических покрытий.
Рис. 7.
Зависимость бестокового потенциала Cr–P–W/Cu-образцов от толщины нанесенного покрытия в 0.1 М HCl.
Микротвердость Cr–P–W-покрытий находится в пределах 3.8–6.0 ГПа, что несколько меньше по сравнению с Co–Cr–W-покрытиями, разработанными нами ранее [32] (4.8–5.9 ГПа). Следует отметить, что микротвердость разработанных покрытий может быть существенно повышена до 7.5–9.8 ГПа после термообработки, проведенной при 200°С в атмосфере воздуха в течение 2 ч.
ВЫВОДЫ
1. Покрытия Cr–P–W, содержащие до 8 мас. % фосфора и 1.5 мас. % вольфрама могут быть получены электроосаждением из водно-диметилформамидных растворов в гальваностатических условиях при 0.22–0.25 А см–2.
2. Электролитические сплавы Cr–P–W характеризуются удовлетворительной коррозионной стойкостью в кислых средах: lg iкор в 0.5 М H2SO4 составляет –5.6 [A см–2], а в 1.0 М HCl – –4.9.
3. При анодной поляризации Cr–P–W покрытий продолжительная область потенциалов пассивного состояния наблюдается как в коррозионной среде не содержащей хлорид ионов (0.5 М H2SO4), так и в присутствии хлорид-ионов (1.0 M HCl, 0.5 М NaCl).
4. Сквозные поры и трещины в Cr–P–W-покрытиях на стали перестают фиксироваться при их толщине выше 15 мкм. Для обеспечения высокой защитной способности необходимо применять осаждать их достаточно толстые слои; целесообразно также применение медного подслоя.
Список литературы
Zhang H., Liu L., Bai J., Liu Z. // Thin Solid Films. 2015. V. 395. P. 36–40.
Tharamani C.N., Hoor F.S., Begum N.S., Mayanna S.N. // J. Electrochem. Soc. 2006. V. 153. P. C164–C169.
Surviliene S., Cesuniene A., Selskis A., Juskenas R. // Trans IMF. 2010. V. 88. P. 100–106.
Sheu H.-H., Lu Ch.-E., Lee H.-B., Pu N.-W., Wu P.-F., Hsieh Sh.-H., Ger M.-D. // J. Taiwan Inst. Chem. Eng. 2016. V. 59. P. 496–505.
Кузнецов В.В., Матвеев Д.В. // Электрохимия. 2008. Т. 44. С. 796–801.
Kuznetsov V.V., Pavlov L.N., Vinokurov E.G., Filatova E.A., Kudryavtsev V.N. // J. Solid State Electrochemistry. 2015. V. 19. № 9. P. 2545–2553.
Кузнецов В.В., Павлов Л.Н., Винокуров Е.Г., Филатова Е.А., Кудрявцев В.Н. // Электрохимия. 2015. Т. 51. С. 205–210.
Винокуров Е.Г., Антошкин М.К., Кудрявцев В.Н. // Заводская лаборатория. 1991. Т. 57. С. 18–19.
Винокуров Е.Г., Кудрявцев В.Н., Бондарь В.В. // Защита металлов. 1992. Т. 28. С. 659–664.
Кузнецов В.В., Чепелева С.А., Кодинцев А.И. // Успехи химии и хим. технологии. ФБГОУ РХТУ им. Д.И. Менделеева. 2000. Т. 14. С. 41.
Wienberg J.H.O.J., Steegh M., Aarnts M.P., Lammers K.R., Mol J.M.C. // Electrochim. Acta. 2015. V. 173. P. 819–826.
Lu Ch.-E., Lee J.-L., Sheu H.-H., Hou K.-H., Tseng Ch.-Ch., Ger M.-D. // Int. J. Electrochem. Sci. 2015. V. 10. P. 5405–5419.
Шлугер М.А., Кабина А.Н. // Гальванотехника и обработка поверхности. 1994. Т. 3. № 4. С. 11–15.
Целуйкин В.Н. // Физикохимия поверхности и защита материалов. 2016. Т. 52. С. 171–184.
Баканов В.И., Нестерова Н.В., Якупов А.А. // Физикохимия поверхности и защита материалов. 2017. Т. 53. С. 270–277.
Винокуров Е.Г., Кузнецов В.В., Бондарь В.В. // Координационная химия. 2004. Т. 30. С. 530–539.
Кузнецов В.В., Винокуров Е.Г., Кудрявцев В.Н. // Электрохимия. 2000. Т. 36. С. 853–858.
Винокуров Е.Г. // Физикохимия поверхности и защита материалов. 2010. Т. 46. С. 543–547.
Кузнецов В.В., Винокуров Е.Г., Азарко О.Е., Кудрявцев В.Н. // Электрохимия. 1999. Т. 35. С. 779–780.
Zeng Zh., Liang A., Zhang J. // Electrochim. Acta. 2008. V. 53. P. 7344–7349.
Li B., Lin A., Gan F. // Surf Coat. Technol. 2006. V. 201. P. 2578–2586.
Zhang J., Gu Ch., Tong Y., Gou J., Wang X., Tu J. // RSC Adv. 2015. V. 5. P. 71268–71277.
Li B., Lin A., Wu X., Zhang Y., Gan F. // J. Alloys and Compounds. 2008. V. 453. P. 93–101.
Lin L., Li Y., Wang F. // J. Mater. Sci. Technol. 2010. V. 26. P. 1–14.
Кузнецов В.В., Тележкина А.В., Демаков А.Г., Баталов Р.С. // Гальванотехника и обработка поверхности. 2017. Т. 25. № 1. С. 16–22.
Лосев В.В., Пчельников А.П. // Итоги науки и техники. Электрохимия. М.: ВИНИТИ, 1979. Т. 15. С. 62.
El-Sharif M., Watson A., Chisholm C.U. // Trans. IMF. 1988. V. 66. P. 34–41.
ГОСТ 9.302-88. Единая система защиты от коррозии и старения. Покрытия металлические и неметаллические неорганические. Методы контроля. Т. 1. М.: Изд. Стандартов, 1990.
Сафонов В.А., Выходцева Л.Н., Едигарян А.А., Алиев А.Д., Молодкина Е.Б., Данилов А.И., Лубнин Е.Н., Полукаров Ю.М. // Электрохимия. 2001. Т. 37. С. 148–156.
Тетерин Ю.А., Соболев А.В., Пресняков И.А., Маслаков К.А., Тетерин А.Ю., Морозов И.В., Чернявский И.О., Иванов К.Е., Шевельков А.В. // ЖЭТФ. 2017. Т. 151. С. 293–304.
McCafferty E. // Corr Sci. 2005. V. 47. P. 3202−3215.
Kuznetsov V.V., Filatova E.A., Telezhkina A.V., Kruglikov S.S. // J. Solid State Electrochem. 2018. V. 22. P. 2267–2276.
Burstein G.T., Sazon D. // Reference Module in Materials Science and Materials Engineering. 2016. https://doi.org/10.1016/B978-0-12-803581-8.01589-7
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов