

Физикохимия поверхности и защита материалов, 2020, T. 56, № 3, стр. 252-263
Влияние морфологии углеродной подложки на электрохимическое поведение электроактивных композитных покрытий на основе поли-(3,6-ди(3-аминофенилен)амино-2,5-дихлор-1,4-бензохинона
В. В. Абаляева 1, *, Г. В. Николаева 1, Е. Н. Кабачков 1, О. Н. Ефимов 1
1 Учреждение Российской академии наук Институт проблем химической физики Российской академии наук
142432 Черноголовка, Московская обл., просп. Академика Н.Н. Семенова, 1, Россия
* E-mail: avva@icp.ac.ru
Поступила в редакцию 30.01.2019
После доработки 06.01.2020
Принята к публикации 10.01.2020
Аннотация
На основе мономера 3,6-ди(3-аминофенилен)амино-2,5-дихлор-1,4-бензохинона (ДАФАДХБ) и соответствующего полимера ПДАФАДХБ получены электроактивные композитные покрытия (КП). Были использованы 2 типа углеродных подложек: 1) с гладкой поверхностью – стеклоуглерод (СУ), графитовая фольга “Графлекс” (ГФ) и 2) активированная графитовая фольга (АГФ) с шероховатой поверхностью. В первом варианте проводили электрохимический синтез (ЭС) покрытия окислением мономера на поверхности подложки. Во втором полимерное композитное покрытие наносили путем полива суспензии ПДАФАДХБ в муравьиной кислоте (МК), в которой предварительно полимеризовали мономер ДАФАДХБ (в дальнейшем химический синтез – ХС). Исследовано влияние введенных в состав КП одностенных углеродных нанотрубок (ОНТ) и оксида графена (ОГ). Получены КП с высокими (до 2800 Ф/г) значениями удельной электрохимической емкости и расширенными интервалами циклирования до 2 В.
1. ВВЕДЕНИЕ
В последние годы растет интерес к гибридным нанокомпозитам на основе полианилина (ПАни) и его производных с углеродными наполнителями, которые рассматриваются как перспективные электродные материалы для суперконденсаторов. Действительно, оба компонента (полимер и углеродная составляющая) вносят свой аддитивный вклад в электрохимические характеристики полученных материалов. КП из таких компонентов имеют двойнослойную емкость и Фарадеевскую псевдоемкость. Электродное покрытие, содержащее только ПАни, при длительных зарядо-разрядных циклах теряет емкость из-за уплотнения электродного материала (ухудшение пористости) и роста диффузионных затруднений для транспорта противоионов, которые даже в исходном состоянии довольно велики. Ранее нами была показана возможность повышения стабильности электродов полианилинового ряда путем введения электроактивных заместителей у атома азота, что препятствует уплотнению полимерных цепей [1]. Мы также нашли, что проведение окислительной полимеризации анилина и его N-замещенных производных удобно проводить с перекисью бензоила в МК с образованием суспензии полимера. В этой же среде можно проводить гетерофазную полимеризацию, вводя ОНТ или ОГ. Полученную суспензию наносят путем полива на токопроводящую подложку, что позволяет получать полимерное нанокомпозитное покрытие [2]. Другой важной особенностью данной работы было использование в качестве подложки АГФ, которая представляет собой углеродный материал на основе терморасширенного графита (ТРГ). Образцы ГФ были предоставлены ООО Унихимтек, МГУ. Метод приготовления ГФ подробно описан в [3]. ГФ имеет хорошую электропроводность, низкую плотность 1 г/см3 и однородную пористую структуру, образованную щелевидными мезопорами с характеристическим радиусом около 20 Å [4, 5]. Фольга содержит незначительное количество примесей тяжелых металлов в окклюдированном состоянии, что не сказывается на электрохимической активности. Для улучшения смачивания поверхности ГФ ее активируют анодной обработкой.
В данной работе проведено сопоставительное исследование электрохимических свойств композитного полимерного покрытия, полученного на углеродной подложке электросинтезом из мономера ДАФАДХБ, либо поливом из дисперсии в МК, полученной в ХС, в том числе c углеродным наполнителем (10 вес. % ОНТ или 15 вес. % ОГ) на подложки из ГФ и АГФ. Представлялось интересным выяснить влияние морфологии поверхности углеродного материала, на котором формовали полимерное покрытие, на электрохимические свойства КП в зависимости от состава КП и способа его изготовления.
2.ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
2.1. ХС мономера и полимера
Мономер ДАФАДХБ(III) синтезировали по реакции алкилирования м-фенилендиамином (I) хлоранила (ХА) (II) в растворе диоксана при 15°С (Схема 1 ).

Схема 1.
ХС полимера (IV) ПДАФАДХБ проводили полимеризацией мономера с помощью перекиси бензоила в МК. 1.2 × 10–4 моля мономера с 10 вес. % ОНТ или с 15 вес. % ОГ или без дополнительных включений были введены при интенсивном перемешивании в 20 мл 99.8% МК при комнатной температуре, при этом мономер хорошо растворялся в МК. К раствору при перемешивании добавляли 2.4 × 10–4 моля перекиси бензоила в 1 мл ацетона (мольное соотношение мономера к окислителю 1 : 2). Содержимое реактора перемешивали магнитной мешалкой в течение 2-х часов и оставляли на ночь. Образовавшийся вязкий раствор использовали для приготовления покрытий на исследуемой подложке. Для исследования подложку с покрытием сушили в вакуумном шкафу при 60°С до постоянного веса. При ХС полимера ОНТ или ОГ в его состав вводили главным образом для улучшения адгезии получаемого продукта (что было замечено экспериментально) и для увеличения проводимости.
2.2. ЭС и электрохимические исследования
ЭС и электрохимические исследования проводили в потенциодинамическом или в гальваностатическом режиме в интервале потенциалов ‒0.7 В…+1.3 В (относительно Ag/AgCl) в трехкамерной стеклянной электрохимической ячейке (используемый объем электролита 15 мл) при комнатной температуре в 1 М H2SO4 на потенциостате PS-7 (фирма Элинс, Россия) с программным обеспечением. Все потенциалы приведены относительно указанного электрода сравнения. Пространства рабочего и вспомогательного электродов были разделены пористой стеклянной перегородкой. Рабочим электродом служила пластина ГФ, АГФ или СУ размером (1 × 5 см) с нанесенным электрохимическим или химическим покрытием 1 × 1 см (как описано в п. 2.1), вспомогательным электродом служила чистая СУ пластина той же площади. Для ЭС ПДАФАДХБ на соответствующую подложку наносили навеску насыщенного раствора мономера ДАФАДХБ в МК площадью 1 × 1 см. Электрод сушили при 60°С, взвешивали и ЭС проводили в 1 М H2SO4.
2.3. Подготовка графитовой фольги
Анодирование ГФ толщиной 0.6 мм проводили в 0.1% (NH4)2SO4 при 3 В в течение 1–5 мин. Рабочим анодом является исходная пластина ГФ, катодом служит пластина из нержавеющей стали. После анодирования АГФ осторожно вынимали из раствора, промывали дистиллированной водой, высушивали при 60°С до постоянного веса, взвешивали и наносили на поверхность исследуемую смесь (мономера или полимера), сушили до постоянного веса для определения точной навески.
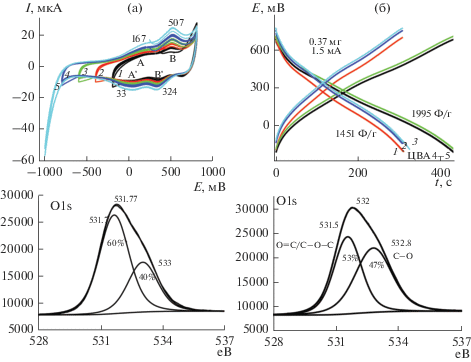
Необходимость активации ГФ возникла при нанесении раствора полимера на поверхность ГФ (рис. 1а), что явилось технически сложной задачей, т.к. большая часть наносимого раствора практически “скатывалась” с гладкой поверхности ГФ. И только разрыхление поверхности при анодировании позволило получить полноценное покрытие для электрохимического исследования. Как можно видеть из рис. 1б, АГФ представляет собой пористую неоднородную поверхность, образовавшуюся при разрыве графеновых слоев во время анодирования. Разрыхление поверхности ГФ начинается с 1 мин анодирования, а после 5 минут наблюдается разрушение (отслоение) поверхностных слоев. Все промежуточные времена определяют степень и величину разрыхления. Для данной работы АГФ получена со временем разрыхления 3 мин (рис. 1б). Как видно из рис. 1б, полученная АГФ состоит из плотно упакованных чешуек. При нанесении дисперсии композита АГФ чешуйки полностью покрываются наносимым раствором, и разделяются, образуя многофазную систему, которая состоит из собственно графитовых частиц, полимерного покрытия на этих частицах и пустот между ними (рис. 1в). В дальнейшем мы будем использовать термин межфазные пустоты. В отличие от гладких электродных подложек на основе СУ и неактивированной исходной ГФ полученная АГФ может поглощать значительное количество наносимого раствора и стабильно удерживать его. При изготовлении электрода на АГФ старались избежать образования свободной композитной пленки на поверхности АГФ после сушки (чтобы не препятствовать проникновению электролита в межфазные пустоты). Исследование поверхности ГФ и АГФ методом РФЭС показало значительное увеличение содержания кислородсодержащих групп. Из обзорного спектра (рис. 2а) видно, что анодированная поверхность АГФ содержит 6.12% кислорода, тогда как на исходной поверхности до анодирования присутствует лишь 1.5% кислорода. В нашем случае разрыхление поверхности ГФ и образование на ней дополнительных кислородных групп во-первых открывает доступ для впитывания наносимой суспензии полимера внутрь подложки с заполнением межфазных пустот и, во-вторых, способствует дополнительному закреплению групп-доноров N-содержащего полимера на поверхности. Этот факт подтверждается наличием пика 533.9 эВ в спектре РФЭС, относящимся к новым связям С=О и О=С–ОН, появившемся на поверхности АГФ (отсутствующим на поверхности ГФ) после окисления поверхности во время анодирования (рис. 2в) [6]. Энергия связи 535.8 эВ на спектре ГФ (рис. 2б) относится к присутствию в образце связанной воды а также возможных примесей [7].

Таким образом, для исследования были получены следующие 6 вариантов КП.
КП1, КП2, КП3 получены из мономера во время ЭС на трех подложках:
1. КП1 получен осаждением мономера на СУ из раствора в МК с последующим ЭС КП1.
2. КП2 получен аналогично п. 1 на ГФ.
3. КП3 получен аналогично п. 1 на АГФ.
КП4, КП5, КП6, получены из дисперсии полимера в МК после его ХС (согласно п. 2.1).
4. КП4 получен поливом дисперсии полученного в ХС полимера на АГФ.
5. КП5 получен поливом дисперсии полученного в ХС полимера с 10 вес. % ОНТ на АГФ.
6. КП6 получен поливом дисперсии полученного в ХС полимера с 15 вес. % ОГ на АГФ.
2.4. Исследования КП
Удельную электрохимическую емкость (Ф/г) рассчитывали по формуле Cm или СS = it/ΔV, где i – удельная плотность тока (А/г или А/см2), рассчитанная из веса или площади композитной пленки на электроде; t – время разряда (с); ΔV – область потенциалов (В), в которой происходит разряд. Важное внимание для последующих расчетов уделялось определению навески и точной площади покрытия подложки.
Электронномикроскопические исследования проводили на растровом электронном автоэмиссионном микроскопе Supra 25 производства Zeiss с рентгеноспектральной энергодисперсионной приставкой INCAEnergy производства Oxford Instruments для определения элементного состава образцов. Разрешение на получаемых изображениях составляет величину 1–2 нм.
ИК Фурье спектры снимали на ИК Фурье спектрометре Perkin 100, оснащенном приставкой многократного нарушенного полного внутреннего отражения с германиевой призмой в диапазоне 4000–675 см–1. Сухие образцы помещали на призму приставки и прижимали с помощью механического прижимного устройства.
РФЭ спектры были сняты как обзорные спектры низкого разрешения в диапазоне 0–1000 эВ с рассчитанным поверхностным содержанием элементов и спектры высокого разрешения С1s, O1s и N1s. Прибор – электронный спектрометр для химического анализа Specs PHOIBOS 150 MCD, рентгеновская трубка с магниевым анодом (MgKα-излучение 1253.6 эВ). При съемке спектров вакуум в камере спектрометра не превышал 3 × 10–9 мбар. Мощность источника составляла 225 Вт. Спектры регистрировались в режиме постоянной энергии пропускания (10 эВ для отдельных линий). Cпектры отдельных линий записаны с шагом 0.05 эВ. Обзорный спектр записывался с шагом 1 эВ.
3. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
3.1. Электрохимические характеристики используемых подложек в 1 М H2SO4
Все электрохимические процессы в КП проходят на поверхности проводящей подложки – СУ, ГФ или АГФ. Изучение электрохимического поведения самой подложки, ее влияние на поведение КП и определение ее количественного вклада в значение электрохимической емкости КП в целом явилось важной задачей в данном исследовании. На рис. 3а и 3б приводятся примеры электрохимического поведения в 1 М H2SO4 чистых СУ, ГФ и АГФ, не содержащих КП.

На ЦВА СУ и ГФ электродов присутствуют размытые, плохо выраженные редокс-пики, возможно, связанные с поверхностными кислородсодержащими группами. По форме ЦВА можно судить, что основной вклад в емкость вносит двойнослойное заряжение. Достаточно стабильно протекает циклирование в течение ряда циклов на подложке из СУ (рис. 3а), причем в разных интервалах циклирований, при этом ΔЕ = Ea – Eк составляет около 95 мВ. При расширении интервала циклирования до катодного потенциала –300 мВ наблюдается резкий изгиб ЦВА, связанный с выделением водорода.
Образец ГФ (рис. 3б, ЦВА 1) показывает неустойчивое поведение, с увеличением от цикла к циклу площади ЦВА и ΔЕ от 247 до 296 мВ за 5 циклирований. Такое поведение, вероятно, можно отнести к “разработке” электрода. В нашем случае в отличие от СУ поверхность ГФ гладкая, но материал состоит из графеновых пластин, уплотненных при формовании ГФ прокаткой терморасширенного графита. В ходе электродной реакции верхний слой разрушается и открывается доступ к пористой структуре ТРГ. Интересно отметить, что в этом случае перегиба на ЦВА, связанного с выделением водорода, не наблюдается также как и на АГФ электроде, что позволяет исследовать электрохимическое поведение КП, не осложненное побочными реакциями связанными с газовыделением.
АГФ, которая имеет шероховатую поверхность после анодной активации, напротив, показывает стабильное циклирование (рис. 3б, ЦВА АГФ) с первого цикла с постоянным ΔЕ = 172 мВ и также постоянным от цикла к циклу значением тока и положениями анодного и катодного пиков. Также из зарядо-разрядных кривых были рассчитаны СS для этих подложек, которые приведены в табл. 1 и на рис. 3а и 3б.
Таблица 1.
Результаты расчетов Суд по экспериментальным данным для КП1–КП6 и подложек
| Номер КП | Состав, навеска (мг) | Рабочий интервал, В |
i, А/г | Сm, Ф/г макс. | СS, Ф/см2 макс. |
|---|---|---|---|---|---|
| КП1 | ДАФАДХБ/СУ ЭС | –0.4…+1 | 3 | 11 | 0.07 |
| КП2 | ДАФАДХБ/ГФ ЭС 0.31 | –0.4…+1.2 | 6 | 1437 | 0.45 |
| КП3 | ДАФАДХБ/АГФ ЭС 0.3 | –0.7…+1.2 | 6.5 | 2587 | 0.74 |
| КП4 | ПДАФАДХБ/АГФ 0.31 | –0.4…+1.2 | 6 | 919 | 0.37 |
| –0.4…+1.4 | 6 | 1180 | 0.4 | ||
| КП5 | ПДАФАДХБ/ОНТ/АГФ 0.34 | –0.7…+1.3 | 6 | 1320 | 0.41 |
| КП6 | ПДАФАДХБ/ОГ/АГФ 0.37 | –0.75…+0.8 | 4.5 | 2027 | 0.67 |
| СУ | –0.2…+0.8 | 3 | 0.07 | ||
| ГФ | –0.2…+0.8 | 3 | 0.15 | ||
| –0.7…+1.3 | 3 | 0.135…–0.16 | |||
| АГФ | –0.2…+0.8 | 3 | 0.23 | ||
| –0.7…+1.3 | 3 | 0.21…–0.24 |
Интересно отметить, что в отличие от СУ ЦВА электродов ГФ и АГФ сохраняют форму характерную для двойнослойного заряжения суперконденсаторов, то есть имеют большее окно стабильности по потенциалам.
3.2. Электродные реакции ДАФАДХБ на подложках СУ, ГФ и АГФ
3.2.1. ЭС КП1 (электродные реакции ДАФАДХБ на СУ). Электросинтез КП1 протекает сходно с ростом ПАни покрытия. Он включает окисление мономера при достижении потенциала 750 мВ (рис. 4), которое сопровождается увеличением тока.
Рис. 3.
(а) ЦВА СУ электрода в 1 М H2SO4, 20 мВ/с; (б) ЦВА ГФ и АГФ электродов в 1 М H2SO4, 20 мВ/с.

При увеличении анодного потенциала циклирования до 0.9 В появляется пара редокс-пиков Ea = = 456 мВ и Eк = 384 мВ, ΔЕ = 72 мВ, которая связана с ростом полимерного покрытия. Этот процесс сопровождается сначала ростом а впоследствии снижением анодного тока в районе 900 мВ от цикла к циклу. Рост редокс-пиков происходит очень медленно и в конце концов прекращается из-за высокого сопротивления КП1. Общая картина ЭС после 50 циклов (с изменением интервала циклирования в катодную и анодную стороны) показана на рис. 4. Сm, полученная из разрядных данных составляла 11 Ф/г.
3.2.2. ЭС КП2 (электродные реакции ДАФАДХБ на ГФ). КП2 был получен покрытием 1 см2 поверхности ГФ из раствора мономера ДАФАДХБ в МК, высушен, взвешен и замерена площадь покрытия. Общая картина ЭС при циклировании в интервале потенциалов –0.2…+0.7 В (рис. 5а, ЦВА 1) сходна с ЭС на СУ. Также наблюдается рост площади ЦВА от цикла к циклу и появление редокс-пары Ea = 563 мВ и Eк = 321 мВ. ΔЕ в этом интервале составляет 242 мВ и при увеличении интервала циклирования в анодную сторону постоянно увеличивается, что связано с ростом сопротивления КП.
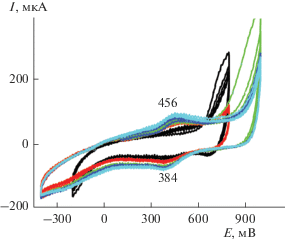
При достижении анодного потенциала 1 В рассчитанная из разрядных кривых электрохимическая емкость продолжает увеличиваться от 726 до 1437 Ф/г (0.45 Ф/см2). Также продолжают раздвигаться анодный и катодный пики (ΔЕ = 503 мВ). Все дальнейшие изменения интервалов циклирования приводят только к снижению величины Суд. При увеличении катодного потенциала на 200 мВ вид ЦВА стабилизируется, и площадь ЦВА от цикла к циклу перестает увеличиваться (ЦВА 4), ΔЕ достигает постоянной величины 503 мВ. При дальнейшем изменении интервала потенциалов сохраняется среднее значение Сm в районе 1300–1450 Ф/г. Такое поведение КП можно объяснить с точки зрения синтеза на графитовых стенках в межфазных полостях полимера или, что более вероятно, олигомеров. Этот процесс может сопровождаться расширением межфазных полостей вследствие расклинивающего действия растущей органической фазы. Но в конечном результате процесс прекращается из-за высокого сопротивления полимерной фазы.
3.2.3. ЭС КП3 (электродные реакции ДАФАДХБ на АГФ). КП3 был получен покрытием 1 см2 поверхности АГФ из раствора мономера ДАФАДХБ в МК, высушен, взвешен и замерена покрытая площадь. Циклирование мономера ДАФАДХБ началось с интервала –0.2…+0.8 В с дальнейшим расширением в катодную и анодную стороны. В отличие от гладкой поверхности ГФ после анодирования поверхность АГФ шероховатая и суспензия может проникать в межфазные полости разрыхленного графита. Это приводит к появлению основной редокс-пары B–B' (Eа = 450 мВ, Eк = 360 мВ с ΔЕ = 90 мВ, рис. 5а). Она сдвинута в область менее положительных потенциалов по сравнению с ЦВА на ГФ (рис. 5а), а низкая величина ΔЕ указывает на более высокую обратимость электродной реакции. Эти особенности ЦВА могут быть связаны с редокс-переходом в молекулах мономера, связанных с графитовой матрицей за счет ковалентного и нековалентного взаимодействия в межфазных полостях в отличие от ГФ, где реакция электрополимеризации, по-видимому, протекает на поверхности. При расширении интервала циклирования и в анодную и в катодную стороны вид ЦВА практически сохраняется, но появляется вторая пара редокс-пиков А–А' (Eа = = 135 мВ, Eк = 85 мВ, ΔЕ = 50 мВ) на плече редокс-пары B–B', которая, возможно, связана с образованием олигомеров.
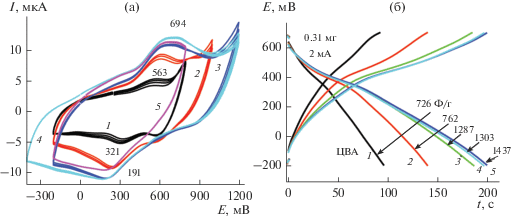
Конечно, необходимы дополнительные исследования для выяснения природы этих электродных реакций. Важно отметить, что положение редокс-пар пиков и ΔЕ при всех интервалах циклирования остаются практически постоянными. При этом времена разрядов (рис. 6б), снятые после каждого интервала циклирования меняются незначительно. Также необходимо отметить, что только в первом цикле в интервале –0.2…+0.8 В Суд несколько отличается от Суд остальных циклирований. Рассчитанные значения Суд для данного КП3 после ЦВА 1–3 и 4–6 составляют соответственно 2470 и 2590 Ф/г или 0.74 Ф/см2.
3.3. Химическая полимеризация ДАФАДХБ, получение и исследование КП4-КП6
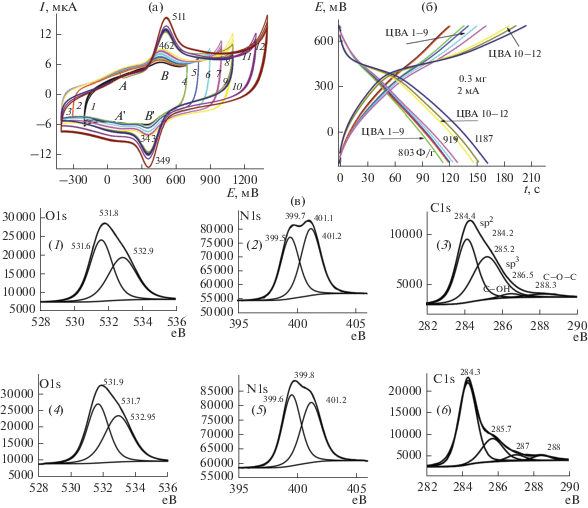
3.3.1. Исследование КП4 (ПДАФАДХБ на АГФ). Нанесение ПДАФАДХБ на АГФ было осуществлено по методике, описанной в п. 2.1. Циклирование КП4 проводилось в разных интервалах потенциалов с расчетом после каждого циклирования значений удельной электрохимической емкости Сs и Сm. Даже при большом изменении интервала потенциалов в катодном и анодном направлениях (рис. 7а) потенциалы основных редокс-пиков В–В' (Eа = 511 мВ, Eк = 349 мВ с ΔЕ = 62 мВ) практически не смещаются. На их фоне пики А–А' сильно размыты и выглядят как плечо у пары В–В'.
Рис. 6.
(а) ЭС ПДАФАДХБ на АГФ, 1 М H2SO4, 20 мВ/с; (б) зарядно-разрядные кривые после ЭС ПДАФАДХБ на АГФ.

Следуя логике размещения полимера в межфазных полостях в графите, можно связать природу основной электродной реакции для пары В–В' с редокс-переходом в молекулах полимера, связанных ковалентными и нековалентными связями с графитом. Как можно видеть из зарядо-разрядных кривых (рис. 7б) в интервале циклирования –0.4…+1 В (ЦВА 1–9) значения Сm меняются плавно. Но при достижении анодного потенциала +1.3 В площади ЦВА 10–12 заметно возрастают и Сm скачкообразно увеличивается до 1187 Ф/г (СS = 0.61 Ф/см2). На рис. 6б на кривых заряда-разряда после описанных ЦВА 10–12 наблюдается появление “полки”, которая далее снова становится наклонной. Природа этого явления требует дополнительного исследования. По данн ым РФЭС (рис. 7в) образцов КП4 до (верхний ряд) и после (нижний ряд) циклирований и снятия зарядо-разрядных кривых в спектрах О1s (спектры 1, 4) существенных различий в состояниях не наблюдается. Немного снизилось количество кислородных связей С(О) с энергией связи 531.6 еВ. Такой эффект может быть следствием взаимодействия полимера с кислородсодержащими группами в разрыхленном графите, например из-за образования межмолекулярных водородных связей. В то же время спектры N1s до и после циклирования отличаются в значительной степени. В образцах 2 и 5 нет пиков, отвечающих за иминные связи N атома (398 еВ) [8]. Энергии связей 399.4–400 еВ соответстуют аминному характеру связи, а связи с более высокими энергиями 401–402 еВ соответствуют положительно заряженному имину в биполяронном состоянии и протонированному амину в поляронном состоянии [9]. Такие спектры характерны для протонированной солевой формы ПАни. В литературе нет сведений об особенностях связей N атома в полимере типа ПДАФАДХБ, но можно думать, что в ПДАФАДХБ (схема 1 ) образование иминных структур должно быть затруднено. После всех циклирований и зарядо-разрядов (нижний ряд, спектр 5) пики N1s 399–400 еВ увеличили свою интенсивность, т.е. количество атомов N в протонированном состоянии в процессе циклирования возросло. Сравнение С1s до (спектр 3) и после (спектр 6) циклирований показывыает, что увеличилось количество связей С–С 284.3 еВ, характеризующих sp2 [10] cостояние поверхности, количество связей sp3 285.3 еВ уменьшилось. Эти связи, возможно, образовались на торцах графитовой пластины или при ее разрыве во время анодирования. На этом фоне увеличилось процентное содержание на поверхности связей С–ОН и С(О)ОН, которые образуют внутренние водородные связи, поддерживающие устойчивость изучаемых структур при циклировании и обеспечивающих высокие значения Суд изучаемых композитных покрытий.
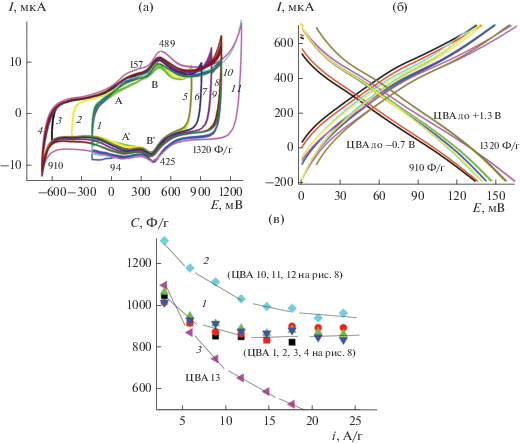
3.3.2. Исследование КП5 (ПДАФАДХБ/ОНТ на АГФ). КП5, полученный поливом раствора ПДАФАДХБ-ОНТ в МК на поверхность АГФ, показывает устойчивое циклирование во всех исследуемых интервалах потенциалов с их расширением как в анодную, так и в катодную стороны. (рис. 8а). После каждого интервала циклирования проводилось зарядо-разрядное исследование (рис. 8б) и расчет С для каждого интервала. Как следует из рис. 8а, КП5, полученный поливом раствора ПДАФАДХБ-ОНТ в МК на поверхность АГФ, показывает устойчивое циклирование во всех исследуемых интервалах потенциалов с их расширением как в анодную, так и в катодную стороны (рис. 8а). После каждого интервала циклирования проводилось зарядо-разрядное исследование (рис. 8б) и расчет Суд для каждого интервала.
Рис. 7.
(а) ЦВА КП4 в 1 М H2SO4 20 мВ/c; (б) зарядно-разрядные кривые для КП4 после указанных ЦВА; (в) РФЭС КП4 (1), (2), (3) – до всех испытаний, (4), (5), (6) – после ЦВА и З–Р.
В процессе всех испытаний при изменении интервалов циклирования в любом направлении остается практически постоянным значение ∆ЕА–К для обеих окислительно-восстановительных пар пиков, т.е. 60 мВ для пары А–А' и 64 мВ для основной пары пиков В–В'. Результаты такого зарядо-разрядного процесса приведены на рис. 8в, на котором обобщены рассчитанные значения С. Из рисунка следует следующий вывод: все значения С, рассчитанные при продвижении интервала циклирования в катодную сторону от –200 до –700 мВ укладываются на одну общую кривую 1 (ЦВА 1–4 на рис. 8а) и находятся в интервале 900–1300 Ф/г. Все значения С, рассчитанные при продвижения интервала циклирования в анодную сторону от 800 до 1300 мВ укладываются на одну общую кривую 2 (ЦВА 6–11 на рис. 8а и находятся в интервале 1050–1350 Ф/г). Если такой электрод остается до следующего испытания в электролите, результаты циклирований могут быть неизменными достаточно длительное время. Если электрод был вынут из электролита и оставался на воздухе продолжительное время, чтобы успеть высохнуть, то последующие результаты при электрохимическом исследовании значительно отличаются. Согласно кривой 3, при таком циклировании (после выдержки электрода на воздухе) в интервале –700…+1300 мВ, которое повторяет предыдущее циклирование, электрод теряет свою электрохимическую емкость значительно более быстрыми темпами. Возможно, это связано с “схлопыванием” графеновых слоев при высушивании и при последующем погружении в электролит неполным их расправлением для поступления электролита в прежних объемах. Из этого эксперимента следует один важный вывод: работа электрода, описанного на рис. 8а, важна своей высокой стабильностью в очень широком интервале потенциалов при соблюдении описанных экспериментальных условий.
3.3.3. Исследование КП6 (ПДАДХБ/ОГ на АГФ). КП6, полученное поливом р-ра ПДАФАДХБ-ОГ в МК на поверхность АГФ, показывает устойчивое циклирование во всех катодных интервалах потенциалов без их расширения в анодную сторону после 0.8 В (рис. 9а).
Рис. 8.
(а) ЦВА КП5, 1 М H2SO4, 20 мВ/с; (б) зарядно-разрядные кривые КП5; (в) результаты расчета С для разных интервалов циклирования КП5.
При этом расширение катодного потенциала постоянно увеличивает площадь ЦВА (рис. 9а) и, соответственно, время разряда (рис. 9б), поэтому при продвижении в катодную область возрастает значение Сm и СS. В районе –0.8…–0.9 В наблюдается скачкообразное увеличение времени разряда и, соответственно, величины Сm до 1995 Ф/г. Это связано с возможным восстановлением в этой области ОГ и следующим за этим значительным увеличением проводимости всей системы в целом. Кроме ЦВА (рис. 9а) о возможности восстановления ОГ при используемых катодных потенциалах также свидетельствует уменьшение кислородной составляющей в РФЭС. при 533 еВ в образце (б) после ЦВА5 (рис. 9а) по сравнению с исходным образцом (а) до начала его катодного восстановления (ЦВА 1, рис. 9а).
4. ИК-СПЕКТРЫ ЭЛЕМЕНТОВ, СОСТАВЛЯЮЩИХ КОМПОЗИТНОЕ ПОКРЫТИЕ
Полосы связи С–Cl в ХА 707 и 730 см–1 [11] присутствуют в спектрах (рис. 10) твердых образцов ХА и мономера (сп. 1 и 5) но отсутствуют в спектрах КП, посаженных на какой-либо образец графитовой фольги (сп. 2–5). Также как и мономер, также и полимеры, которые образуются на поверхности графитовой фольги, вступают в π–π* взаимодействие своими хинодииминными звеньями с поверхностью графитовой фольги [12]. При этом происходит значительное снижение интенсивности характеристических пиков а их полосы сдвигаются в длинноволновую область. Следует отметить, что образцы на спектрах 2–5 были сняты в виде соскоба с фольги, и потому концентрация вещества в этих образцах достаточно низкая, но отдельные характеристические пики все же можно идентифицировать. Связь С–Cl, таким образом не видна на снятых спектрах, т.к. ее полоса выходит за рамки снимаемого интервала см–1, и это касается только образцов, которые вступают во взаимодействие с поверхностью фольги (спектры 2–5).
Рис. 9.
(а) ЦВА КП6 в катодной области, 1 М H2SO4, 20 мВ/с; (б) зарядно-разрядные кривые для разных интервалов циклирования КП6; (в) РФЭС для КП6 (ПДАФАДХБ/ОГ на АГФ): исходного (а) и после ЦВА5 (б).
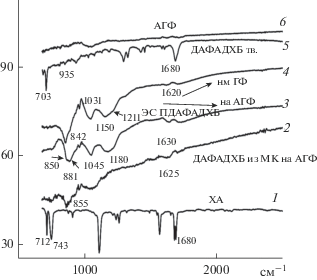
Следующая характеристическая полоса относится к С=О связи ХА и активно просматривается в ХА (спектр 1), мономере (спектр 5) при 1680 см–1, также эта полоса видна, но сдвинута в длинноволновую область на 50–60 см–1 в полимерах, полученных в ЭС на ГФ (спектр 4) и АГФ (спектр 3), прошедшем все ЦВА и зарядо-разрядные испытания. Исходя из структурной формулы на схеме 1 , это обстоятельство может быть связано не только с малой концентрацией образца на поверхности фольги, но и с образованием внутренних водородных связей [13, 14]. Очень активные полосы 840–850 см–1 свидетельствует о сильной деформации кольца в области С–N–С [15]. Также полосы 730 и 870 см–1 указывают соответственно на деформацию кольца и частично относятся к C–H колебаниям выходящим из плоскости кольца [16–18]. Очень широкие полосы в области 1030–1211 см–1 содержат в себе несколько полос, которые разделить из-за сильного взаимодействия полимера с графеновой поверхностью практически невозможно. Но все эти полосы относятся к образованию полимера. Например, полоса 1211 см–1 могла бы быть отнесена к колебаниям С–N связи третичного амина [18], которая присутствует только в полимере. Образец 3 в районе 850–880 см–1 показал сложный спектр, в котором на основании данных [17, 18] можно предположить деформацию как бензоидного, так и хиноидного колец. Образование в данном полимере иминных структур свидетельствует об очень высокой активности такого образца. Как свидетельствует из предыдущих расчетов, этот образец КП3 показал максимальное в данной работе значение Суд.
5. ВЫВОДЫ
Проведенные исследования показывают, что мономер ДАДХБ на гладкой СУ подложке можно полимеризовать с образованием покрытия с низкой емкостью. Намного большую эффективность в качестве подложки для получения композитного покрытия с высокими электрохимическими характеристиками проявляют графитовые материалы – ГФ и АГФ. Использование АГФ с высокой шероховатостью показало самые высокие электрохимические характеристики КП благодаря хорошему смачиванию поверхности раствором суспензии полимера и заполнению межфазных пустот. Сравнительные данные приведены в таблице. Наиболее высокая емкость 2587 Ф/г (0.74 Ф/см2) при токовой нагрузке 6.5 А/г и достаточно широком окне потенциалов достигается при электрополимеризации ДАФАДХБ на АГФ, что обусловлено, скорей всего, хорошим контактом с высокопроводящим углеродным каркасом при заполнении межфазных пустот. Химически синтезированный полимер на АГФ подложке достаточно активен в катодной и анодной области циклирования, показывая стабильную ЦВА во всех исследованных интервалах. Введение в состав полимерного материала ОНТ и ОГ повысило значение Суд. Введение ОНТ в состав КП5 позволило еще и расширить интервал циклирования до 2 В. Пропитка АГФ суспензией полимера, содержащей оксид графена, позволяет получить КП с хорошей емкостью 2027 Ф/г (0.67 Ф/см2), но в узком окне потенциалов, в котором может произойти восстановление ОГ.
Авторы благодарят д. х. н. С.Г. Ионова (ООО Унихимтек, МГУ) за предоставление графитовой фольги “Графлекс” и сотрудников ИНХС РАН д. х. н. Г.П. Карпачеву, к. х. н. С.Г. Киселеву и к. х. н. А.В. Орлова за синтез 3,6-ди(3-аминофенилен)амино-2,5-дихлор-1,4-бензохинона.
Работа выполнена в соответствии с Госзаданием 0089-2019-0010.
Список литературы
Абаляева В.В., Ткаченко Л.И., Николаева Г.В., Орлов А.В., Киселева С.Г., Ефимов О.Н., Карпачева Г.П. // Высокомол. соед. Сер. Б. 2017. Т. 59. № 4. С. 299. [Abalyaeva V.V., Tkachenko L.I., Nikolaeva G.V., Orlov A.V., Kiseleva S.G., Efimov O.N., Karpacheva G.P. // Polymer Science. В. 2017. V. 59. № 4. P. 459–471].
Ткаченко Л.И., Николаева Г.В., Рябенко А.Г., Ефимов О.Н. // Физикохимия поверхности и защита материалов, 2018. Т. 54. № 4. С. 348–354. [Tkachenko L.I., Nikolaeva G.V., Ryabenko A.G., Efimov O.N. // Protection of Metals and Physical Chemistry of Surfaces, 2018. V. 54. № 4. Р. 617–623].
Savchenko D.V., Serdan A.A., Morozov V.A., Van Tendeloo G., Ionov S.G. // New Carbon Materials. 2012. V. 27. № 1. P. 12.
Simon P., Gogotsi Y. // Nature Mater. 2008. V. 7. (11). P. 845.
Malinauskas A., Malinauskiene J., Ramanavičius A. // Nanotechnology. 2005.V. 16 (10) P. R51.
Wilamowska M., Kujawa M., Michalska M., Lipi L., Lisowska-Oleksiaka A. // Synth. Met. 2016. V. 220. P. 334.
Al-Mashat L., Shin K., Kalantar-zadeh K., Plessis J.D., Han S.H., Kojima R.W. // J. Phys. Chem. C. 2010. 114(39). P. 16168.
Minkyu Kim, Choonghyeon Lee, Jyongsik Jang // Adv. Funct. Mater. 2014. V. 24. P. 2489.
Belmokhtar A., Benyoucef A., Zehhaf A., Yahiaoui A., Morallon E. // Synth. Met. 2012. V. 162. P. 1864.
Wilamowska M., Kujawa M., Michalska M., Lipinska L., Lisowska-Oleksiaka A. // Synth. Met. 2016. V. 220. P. 334.
Moamen S., Omar B., Hamdy Al-D., Khlood M., Lamia El-Z. // J. Saudi Chem. Soc. 2012. V. 16. P. 227.
Cong H.P., Ren X.C., Wang P., Yu S.H. // Energy Environ. Sci. 2013. V. 6. P. 1185.
Salvatierra R.V., Zitzer G., Savu S.-A., Alves A.P., Rocco M.L.M. // Synth. Met. 2015. V. 203. P. 16.
Moamen S., Lamia A., Okan Z. // Polyhedron. 2008. V. 27. Is. 2. P. 475.
Lindfors T., Kvarnström C., Ivaska A. // J. Electroanalyt. Chem. 2002. V. 518. Is. 2. P. 131.
Quillard S., Berrada K., Louarn G., Lefrant S., Lapkowski M., Pron A. // New J. Chem. 1995. V. 19. P. 365.
Berrada K., Quillard S., Louam G., Lefrant S. // Synth. Met. 1995. V. 69. Is. 1–3. P. 201.
Boyer M., Quillard S., Rebourt E., Louarn G., Buisson J.P., Monkman A., Lefrant S. // J. Phys. Chem. B. 1998. V. 102: P. 7382.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов