

Физикохимия поверхности и защита материалов, 2020, T. 56, № 3, стр. 238-242
Методические особенности определения цис-жирных кислот эндогенного происхождения в биологических объектах
З. Б. Хесина 1, *, С. Д. Ярцев 1, А. К. Буряк 1
1 Институт физической химии и электрохимии им. А.Н. Фрумкина Российской академии наук
119071 Москва, Ленинский проспект, 31, корп. 4, Россия
* E-mail: maldi-ms@yandex.ru
Поступила в редакцию 30.01.2019
После доработки 05.01.2020
Принята к публикации 10.01.2020
Аннотация
Разработан подход, позволяющий проводить количественное определение эндогенных цис-жирных кислот в присутствии цис- и транс-жирных кислот, не включающий трудоемкую и длительную стадию их хроматографического разделения. Подход основан на использовании ингибитора, блокирующего действие фермента Elovl6, участвующего в синтезе жирных кислот в организме. Методом жидкостной хроматографии, масс-спектрометрии последовательно исследуются образцы, полученные без использования ингибитора и с его использованием. Разработанный подход применен для анализа эндогенных цис-жирных кислот, синтезирующихся в раковых клетках.
ВВЕДЕНИЕ
На протяжении нескольких десятков лет большое внимание уделяется обнаружению и количественному определению жирных кислот (ЖК) в биологических жидкостях, клетках и тканях человека. Ведутся исследования о влиянии ЖК на ожирение, диабет II типа, сердечно-сосудистые и онкологические заболевания [1, 2].
Одним из типов рака, ассоциированных с содержанием ЖК, является рак молочной железы (РМЖ). Он занимает первое место среди злокачественных заболеваний у женщин по частоте и смертности. Ежегодно регистрируется более 1.5 миллиона новых случаев РМЖ и около трети из них заканчиваются летальным исходом [3].
Так как ЖК входят в состав липидов, образующих клеточные мембраны, они могут оказывать значительное влияние на метастатический потенциал раковых клеток. Различия в составе липидов часто коррелируют с агрессивностью и молекулярным подтипом РМЖ.
Показано, что большую роль в распространении рака играет гидрогенизирование полиненасыщенных жирных кислот с образованием “транс-жиров” [4, 5]. Этот процесс применяется при изготовлении маргарина, а также кондитерских и хлебопекарных изделий низкого качества. При их производстве жидкие цис-ненасыщенные жиры, такие как растительные масла, частично гидрируются с образованием насыщенных жиров, которые имеют более желательные физические свойства. В ходе частичного гидрирования ненасыщенных жиров происходит побочный процесс изомеризации под действием катализатора, применяемого для гидрирования: цис-двойные связи частично переходят в транс-двойные [6].
В организмах млекопитающих все синтезируемые ЖК образуются исключительно в изогнутой цис-конфигурации, что придает этим молекулам дополнительную жесткость. Попадание в организм живых существ транс-ЖК, образовавшихся в результате частичного гидрирования ненасыщенных цис-ЖК, может иметь негативные последствия. Молекулы транс-ЖК имеют не изогнутую, а прямую конфигурацию, вследствие чего состоящие из таких молекул липиды не способны должным образом выполнять свои функции в составе клеточных мембран [7].
В настоящее время механизм биосинтеза ЖК в организме млекопитающих, а также катализирующие этот процесс ферменты изучены достаточно полно. Установлено, что в цитоплазме клеток печени синтезируется пальмитиновая кислота (С16 : 0), а в митохондриях из уже синтезированной в цитоплазме пальмитиновой кислоты или из жирных кислот экзогенного происхождения образуются ЖК из 18, 20 и 22 углеродных атомов.
При изучении метаболизма жирных кислот важно отделить цис-изомеры, образующиеся в организме эндогенно, от цис- и транс-изомеров, поступающих из пищи.
Для того, чтобы различить цис- и транс-изомеры ЖК, традиционно используют ИК-спектроскопию [8], однако этот метод не позволяет проводить количественное определение, и в целом характеризуется невысокой точностью. Альтернативным подходом является применение газовой хроматографии с использованием капиллярных колонок большой длины с сильнополярной неподвижной фазой. Указанный подход также не лишен недостатков, среди которых необходимость дериватизации (эта дополнительная стадия усложняет пробоподготовку образцов, приводит к появлению на хроматограммах пиков компонентов, не имеющих отношения к разделяемым кислотам) [9], плохое разделение большого числа изомеров [10]. Указанные недостатки могут быть частично устранены применением масс-спектрометрического детектирования [11], а в отношении плохого разделения некоторых изомеров может быть использована двумерная хроматография [12].
Также применяются методы, основанные на использовании жидкостной хроматографии. Так как прямое разделение кислот методом ВЭЖХ затруднено (вследствие близости их времен удерживания), применяют различные косвенные методы, такие как разделение фенациловых [13] или метиловых [14] эфиров кислот на ион-обменной неподвижной фазе, содержащей ионы серебра, с выделением фракций и их последующим анализом методом ГХ-МС в режиме off-line.
Использование капиллярного зонного электрофореза позволяет провести разделение изомеров небольшого числа жирных кислот [15].
Описано использование спектроскопии ионной подвижности для разделения цис- и транс-изомеров кислот [16, 17], этот метод используется для разделения пространственных изомеров, и, будучи соединенным с хроматографическим разделением и масс-спектрометрическим детектированием, позволяет разделить большое число изомеров жирных кислот. Однако вследствие сложности и высокой стоимости подобного оборудования, метод в настоящее время не может быть применен для массовых анализов.
В целом, следует отметить, что, несмотря на постоянное совершенствование инструментальных методов аналитической химии, проблема разделения пространственных изомеров жирных кислот не решена, и разработанные к настоящему времени подходы либо не обеспечивают желаемого разделения, либо сопряжены с трудоемкой пробоподготовкой и использованием сложного оборудования.
Поэтому стоит задача разработки подхода, позволяющего определять цис-изомеры жирных кислот эндогенного происхождения в присутствии цис- и транс-изомеров, попавших в организм из пищи, при этом не включающего стадию хроматографического разделения изомеров.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Подготовка образцов раковых клеток для ВЭЖХ-МС анализа
Клеточные линии рака РМЖ MDA-MB-231 (эстроген-рецептор отрицательная) и MCF7 (эстроген-рецептор положительная) культивировали в 24-луночном планшете (по 2 × 105/лунку) в присутствии 100 мкМ ингибитора фермента элонгазы Elovl6 (ингибитор был предоставлен НТЦ “Биоклиникум”) и в его отсутствии согласно методике, описанной в статье [18]. Лунки промывали фосфатным буфером (Dulbecco’s Phosphate-Buffered Saline- DPBS), после чего проводили кислотный гидролиз суспензии раковых клеток. Для этого вносили 500 мкл смеси ацетонитрил-соляная кислота (4 : 1 по объему), инкубировали 2 ч при 90°С в термошейкере и оставляли на ночь при комнатной температуре [19].
Затем в виалы вносили по 1 мл гексана и инкубировали на шейкере в течение 1 ч, отбирали верхний слой (экстракт ЖК) и переносили его в пластиковые флаконы на 2 мл, после чего упаривали досуха на роторном вакуумном испарителе. В микропробирки вносили по 150 мкл смеси 20 мМ ацетата аммония с метанолом (15 : 85 по объему) и инкубировали 1 ч в термошейкере Thermomixer (Eppendorf) при 30°С и 900 об./мин.
ВЭЖХ-МС анализ ЖК в раковых клетках
Полученные растворы ЖК переносили в стеклянные виалы и проводили анализ методом жидкостной хроматографии с масс-спектрометрическим детектированием (ВЭЖХ-МС). Разделение выполняли с использованием колонки Eclipse Plus C18 (Agilent) (4.6 × 250 мм, 5 мкм) на хроматографе Agilent 1260 Infinity I, оснащенном градиентным четырехканальным насосом, дегазатором, автосэмплером и термостатом колонок. Подвижная фаза А – 20 мМ водный раствор ацетата аммония, фаза В – метанол. Хроматографирование проводили в градиентом режиме: 0–11 мин – 87% В; далее 11–13 мин – линейно повышали до 99% В; 13–15 мин – линейно понижали до 87% В; 15–20 мин – 87% В. Температура термостата колонок: 40°С; скорость расхода подвижной фазы: 0.6 мл/мин.
Детектирование проводили с использованием масс-спектрометра Bruker Maxis Impact в режиме ионизации отрицательных ионов и детектирования выбранных ионов, соответствующих анионам исследуемых кислот (С16 : 0 – 255.23 Да; C18 : 0 – 283.27 Да; C16 : 1 – 253.22 Да; C18 : 1 – 281.25 Да). Частота детектирования – 1 Гц; температура источника – 180°С; давление газа в распылителе – 0.6 бар; поток осушающего газа – 4 л/мин; напряжение на капилляре – 4500 В).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Методические особенности хроматографического разделения ЖК, входящих в состав клеточных мембран
Фосфолипиды и триглицериды, в состав которых входят цис-ЖК, являющиеся объектом исследования, составляют мембрану клетки. Первоначальная предполагаемая схема анализа ЖК заключалась в хроматографическом разделении триглицеридов и фосфолипидов, их фрагментации и определении ЖК по MRM-переходам (мониторинг заданных реакций), где масса иона-предшественника – это масса липида клеточной мембраны, а масса иона-продукта – это масса аниона ЖК (рис. 1а). Однако хроматографическое разделение фосфолипидов и триглицеридов оказалось крайне затруднительным: близкие по строению и свойствам липиды, в состав которых входят исследуемые ЖК, образовывали на хроматограммах большую область неразрешенных пиков (рис. 2а). На масс-спектрах фрагментных ионов, зарегистрированных во время выхода описанной области неразрешенных пиков, наблюдали сигналы, соответствующие анионам исследуемых кислот, однако из-за плохого хроматографического разделения достоверно определить содержания кислот не представлялось возможным.
Рис. 1.
Схема пробоподготовки образцов раковых клеток для определения содержания жирных кислот без стадии кислотного гидролиза (а) и со стадией кислотного гидролиза (б).

Рис. 2.
Хроматограммы, полученные при анализе образцов раковых клеток. Пробоподготовка без стадии кислотного гидролиза (а) и со стадией кислотного гидролиза (б). Детектирование по полному ионному току отрицательных ионов.

Для того чтобы успешно выполнить количественное определение ЖК, внесли изменение в процедуру пробоподготовки с целью выделить ЖК из липидов и перевести их в свободную форму. В качестве дополнительной стадии пробоподготовки предложен кислотный гидролиз исходных образцов с последующей экстракцией ЖК гексаном (рис. 1б). Предложенный вариант пробоподготовки позволил выделить ЖК из липидов в свободном виде, после чего хроматографически разделить ЖК (рис. 2б). Количественное определение ЖК проводили методом масс-спектрометрии в режиме детектирования выбранных ионов (SIM). Это дало возможность не проводить длительное хроматографическое разделение липидов с последующим детектированием их фрагментов (ЖК), а получить непосредственно сами ЖК с гораздо меньшей массой и большими различиями в строении, чем у липидов.
Подход к определению цис-ЖК (С16 : 1 и С18 : 1), образующихся эндогенно под действием ферментов, в присутствии экзогенных цис- и транс-ЖК
В отличие от растительных, ткани животных обладают весьма ограниченной способностью превращать насыщенные ЖК в ненасыщенные.
Установлено, что две наиболее распространенные мононенасыщенные ЖК – пальмитоолеиновая (С16 : 1) и олеиновая (C18 : 1) – синтезируются из пальмитиновой (C16 : 0) и стеариновой (С18 : 0) кислот под действием ферментов десатураз и элонгаз. Процессы десатурации и элонгации (удлинения) происходят попеременно и многократно повторяются. На рис. 3 представлены пути превращения пальмитиновой кислоты в реакциях десатурации и элонгации.
Рис. 3.
Схема образования цис-ненасыщенных кислот в организме из пальмитиновой кислоты под действием ферментов десатуразы (SCD1) и элонгазы (Elovl6).
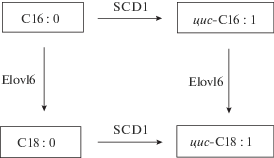
Стеароил-КоА-десатураза (SCD1) – интегральный фермент, который катализирует образование двойной связи между 9 и 10 атомами углерода в ЖК. Десатурация с SCD1 всегда ведет к образованию цис- двойной связи [20].
Элонгаза жирных кислот Elovl6 была идентифицирована, клонирована и охарактеризована в 2001–2002 годах [21, 22]. Elovl6 удлиняет насыщенные и мононенасыщенные ЖК. Этот фермент, так же как и десатураза SCD1, играет определяющую роль в изменении состава липидов клеточных мембран и процессах метаболизма ЖК в клетках, так как его действие влияет на соотношение С16 и С18 ЖК [23, 24]. Ингибирование действия фермента элонгации (Elovl6) приводит к преимущественной десатурации насыщенных ЖК с образованием мононенасыщенных цис-ЖК.
При проведении ВЭЖХ-МС анализа кислоты С18 : 1 в раковых клетках, культивированных в отсутствии ингибитора, можно определить суммарное содержание эндогенной цис-ЖК С18 : 1 и экзогенных цис- и транс-С18 : 1. При этом не представляется возможным отделить эндогенно образовавшуюся кислоту от экзогенных. Однако это возможно в случае, если дополнительно провести анализ тех же клеток, но культивированных в присутствии ингибитора фермента элонгазы. При этом образование кислоты С18 : 1 по эндогенному механизму практически подавлено и, следовательно, площадь хроматографического пика, соответствующего этой кислоте, уменьшается. В этом случае указанный пик соответствует уже не суммарному содержанию эндогенной и экзогенной кислот, а только цис- и транс-С18 : 1, поступившим из пищи. Таким образом, по разности площадей хроматографических пиков кислоты С18 : 1, выделенной из клеток, культивированных в отсутствии ингибитора и в присутствии его, можно определить суммарное содержание эндогенной цис-ЖК С18 : 1 в клетках. То есть, таким способом возможно отделить цис-ЖК, образующиеся в организме, от смеси цис- и транс-ЖК, поступающих из пищи.
Аналогично эксперименту, описанному выше, можно также провести ингибирование фермента десатуразы SCD1. При этом эндогеннное образование цис-C16 : 1 подавлено и весь хроматографический пик, соответствующий этой кислоте, будет отвечать только экзогенным цис- и транс-С16 : 1. Необходимо вычислить площади пиков, соответствующих этой кислоте, в случае анализа клеток, культивированных без ингибитора и с ним. Разность найденных площадей будет соответствовать эндогенно образующейся цис-С16 : 1.
Определение эндогенной цис-ЖК С18 : 1 в присутствии экзогенных цис- и транс-ЖК С18 : 1 в раковых клетках
На хроматограмме, полученной при исследовании образцов клеток, культивированных в отсутствии ингибитора (рис. 4), пик, соответствующий ЖК С16 : 1, отвечает смеси эндогенно образовавшихся цис-ЖК и экзогенно поступивших в организм цис- и транс-ЖК. Аналогично для кислоты С18 : 1: соответствующий ей пик отвечает цис-ЖК С18 : 1, образовавшейся из С16 : 1 под действием Elovl6 и из С18 : 0 под действием SCD1. Отделить только по этой хроматограмме эндогенные цис-ЖК от экзогенных цис- и транс-ЖК не представляется возможным.
Рис. 4.
Наложение хроматограмм образцов раковых клеток, полученных в присутствии и в отсутствии ингибитора элонгазы Elovl6 на стадии культивирования. Детектирование в режиме выбранных ионов, соответствующих анионам кислот.

На хроматограмме образцов, культивированных в присутствии ингибитора Elovl6, пик, соответствующий кислоте С16 : 1, значительно выше, чем в первом случае, за счет того, что удлинение ЖК было практически подавлено, а, значит, почти все кислоты подвергались десатурации с образованием мононенасыщенной ЖК. Площадь пика, соответствующего кислоте С18 : 1, напротив, падает в эксперименте, проведенном в присутствии ингибитора, за счет того, что ЖК теперь преимущественно десатурирует, а не удлиняется, доля эндогенных цис-С18 : 1 оказывается незначительной. А значит, можно утверждать, что разница площадей пиков С18 : 1 в первом и втором экспериментах (культивирование в отсутствии и в присутствии ингибитора) отвечает эндогенно образующейся в организме кислоте цис-С18 : 1. Такой подход дает возможность проводить определение эндогенной цис-кислоты в присутствии экзогенных цис- и транс-кислот С18 : 1.
Список литературы
Christie W.W. // Lipids. 1998. Is. 4. V. 33. P. 343.
Zelles L. // Biology and Fertility of Soils. 1999. I. 2. V. 29. P. 111.
Jemal A., Bray F., Ward E. et al. // A Cancer J. Clinicians. Global Cancer Statistics. 2011. I. 2. V. 61. P. 69.
Slattery M.L., Benson J., Ma K. et al. // Nutrition and Cancer. 2001. I. 2. V. 39. P. 170.
Gebauer S.K., Chardigny J.M., Jakobsen M.U. et al. // Advances in Nutrition. 2011. I. 4. V. 2. P. 332.
Liu W.H., Inbaraj B.S., Chen B.H. // Food Chemistry. 2007. I. 4. V. 104. P. 1740.
Katan M.B., Zock P.L., Mensink R.P. // Annual Review of Nutrition. 1995. V. 15. P. 473.
Costa T.G., Castilho H.S., Sanches S.A. // Trends in Food Science and Technology. 2011. V. 22. P. 543.
Roach J.A., Mossobaa M.M., Yurawecza M.P. et al. // Analytica Chimica Acta. 2002. V. 465. P. 207.
Berdeaux O., Dutta P.C., Dobarganes M.C. et al. // Food Analytical Methods. 2009. V. 2. P. 30.
Zhang M., Yang X., Zhao H.T. et al. // Food Control. 2015. V. 57. P. 293.
Kulsing C., Nolvachai Y., Zeng A.X. et al. // ChemPlusChem 2014. I. 79. V. 6. P. 790.
Christie W.W., Grace H.M. // J. Chromatography A. 1989. V. 469. 1989. P. 261.
Kraft J., Kramer J.K., Hernandez G. et al. // Lipid Technology. 2014. I. 26. V. 2. P. 39.
Stolyhwo A., Rutkowska J. // Food Anal. Methods. 2013. V. 6. P. 457.
Kyle J.E., Zhang X., Weitz K.K. et al. // Analyst. 2016. I. 141. V. 5. P. 1649.
Damen C.W., Isaac G., Langridge J. et al. // J. Lipid Research. 2014. V. 22. P. 382.
Захарова Г.С., Полозников А.А., Астахова Л.А. и др. // Известия АН. Серия химическая. № 12. С. 2307.
Mutay A., Özcan F., Aslan I. et al. // Lipids in Health and Disease. 2013. I. 3. V. 169. P. 189.
Gutiérrez-Juárez R., Pocai A., Mulas C. // J. Clin. Invest. 2006. V. 169. P. 1686.
Moon Y.A., Shah N.A., Mohapatra S. et al. // J. Biol. Chem. 2001. V. 276. P. 45358.
Matsuzaka T., Shimano H., Yahagi N. et al. // J. Lipid Res. V. 43. P. 911.
Green C.D., Ozguden-Akkoc C.G., Wang Y. et al. // J. Lipid Res. 2010. V. 51. P. 1871.
Shi H.B., Wu M., Zhu J.J., Zhang C.H. // J. Dairy Sci. 2017. V. 100. P. 4987.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов