

Физикохимия поверхности и защита материалов, 2020, T. 56, № 3, стр. 286-296
Химический состав и механические свойства покрытий на основе TiN, сформированных с использованием конденсации с ионной бомбардировкой
П. М. Корусенко 1, *, С. Н. Несов 1, С. Н. Поворознюк 1, 2, П. В. Орлов 3, Д. Н. Коротаев 3, К. Н. Полещенко 3
1 Омский государственный технический университет
644050 Омск, пр. Мира, 11, Россия
2 Омский научный центр СО РАН
644024 Омск, пр. К. Маркса, 15, Россия
3 Сибирский государственный автомобильно-дорожный университет
644080 Омск, пр. Мира, 5, Россия
* E-mail: korusenko_petr@mail.ru
Поступила в редакцию 01.01.2020
После доработки 02.01.2020
Принята к публикации 10.01.2020
Аннотация
С использованием методов SEM, EDX, XPS исследованы морфология, состав, химическое состояние элементов покрытий на основе нитрида титана (TiN), сформированных методом конденсации с ионной бомбардировкой, при различных режимах на подложках из стали Гадфильда (110Г13Л). Установлено, что в формируемых покрытиях наибольшее количество титана находится в составе нитрида и оксинитрида, а также присутствует некоторое количество оксидов титана и титана, химически связанного с углеродом. Показано, что титан в металлическом состоянии отсутствует для всех исследованных режимов формирования покрытий и, следовательно, при данных режимах не происходит образования капельной макрофракции. Послойный XPS анализ показал, что покрытия обладают геторогенностью состава по глубине. Методом Виккерса исследована микротвердость образцов. Наибольшие значения микротвердости имеют образцы с покрытиями сформированными при 35 и 50 min, предположительно, вследствие более высокого содержания соединений по типу TiNxCyOz, имеющих более высокую твердость, чем TiN.
1. ВВЕДЕНИЕ
Износостойкие покрытия на основе нитрида титана, получаемые вакуумными ионно-плазменными методами, находят широкое применение в различных областях. Распространенными физическими методами формирования покрытий из нитрида титана являются магнетронное распыление и метод конденсации с ионной бомбардировкой (КИБ) [1–3]. Преимуществами метода КИБ являются высокая скорость формирования покрытий, а также адгезия покрытия к подложке за счет достаточно высокой энергии ионов как на этапе предварительной очистки, так и в процессе нанесения покрытия. К недостаткам данного метода относят возможность образования при катодно-дуговом осаждении капельной фазы металлического титана (капельной макрофракции), снижающей механические характеристики покрытий. Однако, в настоящее время практически во всех применяемых источниках используются [2, 4] методы магнитного управления положением и динамикой катодного пятна, а также магнитная фокусировка потока эрозионной плазмы, снижающих вероятность осаждения капельной макрофракции. Так, с увеличением скорости перемещения катодного пятна, масса испаряемого материала значительно уменьшается, а фокусировка плазменного потока магнитным полем позволяет добиться дополнительного снижения количества и размеров макрочастиц в плазме [2, 5]. Помимо этого известно [6], что макрочастицы в плазме приобретают заряд в результате столкновений с электронами. Подача на подложку отрицательного напряжения смещения позволяет препятствовать осаждению значительной части макрофракций (особенно мелких), движущихся в сторону подложки [7]. В случае Ti катодов наличие азотосодержащей атмосферы дополнительно снижает выход макрофракций вследствие формирования поверхностного термостойкого TiN слоя [2, 7]. Все это позволяет в значительной степени устранить проблему осаждения капельной фазы, в особенности, применительно к TiN покрытиям.
Покрытия на основе нитрида титана, полученные методом КИБ, как правило имеют нано- или субмикрокристаллическую структуру [3, 8, 9] и, обычно, обладают улучшенными физико-механическими свойствами по сравнению с поликристаллическим материалами. Причинами улучшения свойств являются измельчение размера зерен и, следовательно, увеличение протяженности границ зерен [3, 10], а также торможение пластической деформации. Формирование нано- и субмикрокристаллических структурных состояний в тонких покрытиях, является следствием крайне неравновесных условий синтеза таких соединений как TiN. Нитридотитановые покрытия обладают высокой твердостью и износостойкостью, однако при температуре >500°С они достаточно быстро окисляются, что снижает их характеристики [11]. В последнее время стали широко использоваться покрытия TiN легированные Al. Такие покрытия обладают более высокой сопротивляемостью окислению и износостойкостью [11, 13–15]. Высокая эффективность легирования нитридотитановых покрытий подобными, нерастворимыми в равновесных условиях элементами, связана [13, 16] с термодинамически контролируемой сегрегацией по границам нанозерен TiN зернограничной фазы, часто в аморфном состоянии, ограничивающей рост зерна. Такая структура покрытий близка к концепции сверхтвердых нанокомпозитных покрытий, предложенной в работах [10, 16].
Нитрид титана часто входит в состав широкого спектра нанокомпозитных покрытий сложного состава, формируемых методом КИБ, таких как TiAlN, AlCrTiN, (TiZrNbAlYCr)N и др. [3, 10–15]. Универсализация комплекса свойств подобных покрытий достигается посредством увеличения числа элементов в данных нитридных системах [11]. Весьма высокие физико-химические свойства достигаются за счет деформационного эффекта упрочнения твердых растворов вследствие вариативности их состава, искажений кристаллической решетки и даже нанокомпозитной структуры. В ряде работ [10–12, 17, 18] подобные многоэлементные соединения рассматриваются, как новый класс материалов – так называемых высокоэнтропийных сплавов. Данные материалы характеризуются формированием неупорядоченных твердых растворов, стабилизация которых обеспечивается высокой энтропией смешения. Подобным материалам характерна высокотемпературная прочность, суперпарамагнетизм, сопротивление износу и коррозии, высокая твердость наряду не сопровождающаяся хрупкостью [11].
Не смотря на то, что покрытия на основе TiN достаточно давно применяются для повышения износостойкости режущих, штамповых инструментов, различных узлов трибосопряжений и в качестве диффузионных барьеров в микроэлектронике, разработчики технологий нанесения покрытий сталкиваются с проблемами надежного получения покрытий с заданными свойствами. Недостаточно изучены особенности и степень влияния физико-химических параметров (состав остаточной газовой среды в камере, продолжительность нанесения покрытия), сопровождающих процесс нанесения покрытия на его структурно-химическое состояние. Фазы внедрения переходных металлов IV группы, к числу которых относится и нитрид титана, обладают широкими областями гомогенности. Вариация параметров нанесения TiN покрытий приводит, как правило, к образованию фаз внедрения нестехиометрического состава, оказывающих сильное влияние на свойства материала покрытия. Помимо этого наличие остаточных газов в вакуумной камере в процессе нанесения покрытия способно влиять на фазовый состав и приводить к образованию таких фаз как оксиды, оксинитриды, карбиды и, таким образом, также влиять на их эксплуатационные свойства. Для исследования влияния данных факторов на распределение элементного и фазового состава по толщине покрытия необходимо использование аналитического оборудования, обладающего высокой пространственной локальностью.
В данной работе исследовались образцы из стали 110Г13Л с ионно-плазменным покрытиями на основе TiN, легированными Al. Покрытия были сформированы методом КИБ с варьированием времени нанесения (15, 35 и 50 min). Для анализа элементного состава и химического состояния элементов покрытий использовалась рентгеновская фотоэлектронная спектроскопия (X-ray photoelectron spectroscopy – XPS). При исследовании изменения состава по толщине покрытия, применялось травление ионами аргона. Для анализа морфологии, структуры и усредненного элементного состава покрытий использовалась методы сканирующей электронной микроскопии (Scanning Electron Microscopy – SEM) и энергодисперсионной рентгеновской спектроскопии (Energy-dispersive X-ray spectroscopy – EDX). Влияние элементного и фазового состава от времени формирования покрытий на механические характеристики образцов исследовалось посредством измерения микротвердости методом Виккерса с приложением различной нагрузки на индентор.
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Формирование покрытий на основе TiN осуществлялось методом КИБ на установке ННВ-6.6. В качестве подложки была использована легированная сталь марки 110Г13Л, известная как сталь Гадфильда. Она обладает высоким сопротивлением износу (истиранию) при больших давлениях или ударных нагрузках, при которых происходит значительное ее упрочнение вследствие деформированного упрочнения аустенита и образования мартенсита с гексагональной решеткой. Исходные образцы обладали шероховатостью не более 1.25 µm. При нанесении покрытий использовался титановый катод с включением порядка 5% алюминия, наличие которого, как указывалось ранее, способствует повышению износостойкости и сопротивляемости окислению. Присутствие алюминия также снижает вероятность образования капельной фазы металлического титана в покрытии за счет препятствования длительному горению и разрастанию катодных пятен, с тенденцией к слиянию, увеличивая скорость перемещения катодного пятна [2, 4, 6, 7]. До нанесения ионно-плазменных покрытий осуществлялась очистка подложек с образцами посредством ионного травления с подачей ускоряющего напряжения ~900–1000 V при давлении в камере ~5 × 10–5 Torr. В процессе ионной очистки происходит нагрев образцов до температуры ~450–550°С и активация их поверхности за счет формирования структурных дефектов. Формирование покрытий проводилось в атмосфере сухого азота при давлении в камере ~2–3 × 10–3 Torr. Ток дугового разряда составлял ~100–110 A. На подложку подавалось напряжение ~200–220 V. Использовалось три времени формирования покрытий – 15, 35 и 50 min. Оценка скорости осаждения TiN покрытия показала, что за одну минуту формируется слой толщиной ~30 nm. Таким образом, оценочная толщина покрытий, сформированных при 15, 35 и 50 min составляет ~0.5, 1.0 и 1.5 µm, соответственно.
Для анализа морфологии и структуры покрытий использовалась SEM, осуществленная на электронном микроскопе JEOL JSM 6610 LV в центре коллективного пользования ОНЦ СО РАН (ОмЦКП СО РАН, г. Омск). Регистрация изображений осуществлялась при ускоряющем напряжении 20 kV, с пространственным разрешением 20 µm. Анализ усредненного элементного состава покрытий методом EDX был выполнен на данном микроскопе с использованием приставки INCA-350 Oxford Instruments. Диаметр зондирующего пучка электронов при исследовании методом EDX составлял ~1.5 µm, а глубина анализа до ~2–3 µm.
Для исследования элементного, фазового состава и химического состояния покрытий с высокой локальностью по глубине использовался метод XPS, реализованный на аналитическом комплексе Surface Science Center (Riber). Для возбуждения рентгеновского излучения использовался источник с Al-анодом с энергией 1486.6 eV. Съемка спектров осуществлялась в условиях сверхвысокого вакуума (~10–9 Torr) с использованием анализатора типа двухкаскадного цилиндрического зеркала – MAC-2. Глубина XPS анализа составляла ~1–3 nm. Разрешение по энергии при регистрации спектров остовных линий находилось на уровне 0.7 eV, обзорных спектров – 1.2 eV и было постоянно во всем диапазоне измеряемых энергий. Диаметр рентгеновского пучка составлял ~5 mm, мощность источника 240 W. Для получения информации об изменении состава и химического состояния по толщине покрытий был использован послойный XPS анализ, который осуществлялся непосредственно в камере спектрометра. Слой покрытия стравливался пучком ионов аргона со средней энергией 3 keV при давлении в камере спектрометра ~3 × 10–5 Torr.
Исследование механические характеристик образцов осуществлялось посредством измерения микротвердости методом Виккерса на микротвердомере ПМТ-3 с приложением различной нагрузки на индентор и, соответственно, с различной толщиной анализируемого слоя. Нагрузка на индентор составляла 0.2, 0.5 и 1.0 N, что соответствовало глубине вдавливания от 0.9 до 3.0 µm. Осуществлялось по 5 измерений при каждой нагрузке для всех вариантов покрытий.
3. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
3.1. Морфология и элементный состав покрытий
На рис. 1 приведены SEM изображения покрытий, полученных при различном времени формирования.
Рис. 1.
SEM изображения покрытий, полученных при различном времени формирования и карты распределения Ti, N, Al: (а), (г), (ж), (к) – 15 min; (б), (д), (з), (л) – 35 min; (в), (е), (и), (м) – 50 min.

Как видно (рис. 1) покрытия обладают достаточно близкой текстурой поверхности: наблюдается наличие кратеров и возвышенностей, а также кластеров сферической формы. Сравнение изображений рис. 1а–1в показывает, что покрытие с максимальным временем формирования имеет более гладкую поверхность и наименьшее количество сферических кластеров. Элементный EDX анализ, приведенный в табл. 1, показал наличие в составе покрытий титана, кислорода, азота, углерода, алюминия и элементов, принадлежащих подложке (железа и марганца). Присутствие кислорода и углерода в покрытиях, по-видимому, связано с их наличием в остаточной атмосфере камеры установки при их формировании. В связи с тем, что энергии рентгеновских квантов, эмитируемых с К-уровней азота и кислорода, имеют достаточно близкие значения, является затруднительным раздельное определение их концентрации. Поэтому в табл. 1 приведены суммарные значения концентрации азота и кислорода. Из табл. 1 видно, что концентрация элементов подложки значительно снижается с увеличением времени формирования покрытия. Последнее связано с увеличением толщины покрытия. Из данных EDX анализа следует, что отношение средних значений концентрации азота с кислородом к титану имеет наибольшее значение в случае самого тонкого покрытия и наименьшее значение для самого толстого покрытия. Кроме этого, результаты количественного EDX анализа, полученные непосредственно с участков поверхности, содержащих сферические кластеры, показывают, что концентрация азота и кислорода практически не отличается от их концентрации на участках, не содержащих указанные кластеры. Следовательно, данные кластеры не являются капельной фазой металлического титана.
Таблица 1.
Состав покрытий, полученных при различном времени формирования, по данным EDX
| № точки | Концентрация, ат. % | |||||
|---|---|---|---|---|---|---|
| [C] | [N] + [O] | [Al] | [Ti] | [Mn] | [Fe] | |
| 15 min | ||||||
| 1 | 12.18 | 39.54 | 0.69 | 39.56 | 1.54 | 6.49 |
| 2 | 12.22 | 42.03 | 0.71 | 37.67 | 1.38 | 5.99 |
| 3 | 12.66 | 41.55 | 0.73 | 37.42 | 1.31 | 6.33 |
| Среднее значение | 12.35 | 41.05 | 0.71 | 38.21 | 1.41 | 6.27 |
| 35 min | ||||||
| 1 | 17.61 | 40.07 | 0.43 | 41.40 | – | 0.49 |
| 2 | 14.96 | 46.03 | 0.72 | 37.79 | – | 0.50 |
| 3 | 13.86 | 41.95 | 0.73 | 43.02 | – | 0.44 |
| Среднее значение | 15.48 | 42.69 | 0.62 | 40.74 | 0.47 | |
| 50 min | ||||||
| 1 | 16.72 | 36.93 | 0.40 | 45.60 | – | 0.35 |
| 2 | 16.05 | 42.52 | 1.26 | 39.77 | – | 0.40 |
| 3 | 11.33 | 28.58 | 0.81 | 58.77 | – | 0.51 |
| Среднее значение | 14.71 | 36.01 | 0.82 | 48.04 | – | 0.42 |
3.2 Состав и химическое состояние покрытий
На рис. 2 представлены обзорные XPS спектры для покрытий с различным временем формирования. В спектрах (рис. 2) присутствуют линии титана: Ti 2s (~555 eV), Ti 2p (~455 eV), Ti 3s (~57 eV) и Ti 3p (~57 eV); кислорода: O 1s (~530 eV); азота: N 1s (~400 eV); углерода: C 1s (~285 eV); железа: Fe 2p (~710 eV). Фотоэлектронные линии Al, входящего в состав катода и зафиксированного посредством EDX анализа, не наблюдаются ввиду низкого значение сечения фотоионизации для Al 2p уровня при использовании возбуждающего излучения с энергией 1486.6 eV, а также его низкой концентрацией в покрытиях. Наличие пика железа Fe 2p может быть связано как с частичным испарением стальных конструкционных элементов ННВ-6.6, так и ионным перемешиванием материалов подложки и покрытия.
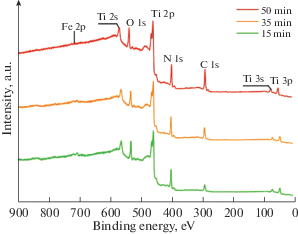
Элементный состав покрытий, определенный из обзорных XPS спектров, приведен в табл. 2. Как следует из табл. 2, содержание основных элементов и примесей (Ti, N, O, C) в покрытиях по данным XPS (табл. 2) существенно отличается от данных EDX анализа (табл. 1), что связано со значительным различием глубины зондирования этих методов (~2–3 µm для EDX и ~1–3 nm для XPS). Это свидетельствует о том, что состав покрытий в тонком поверхностном слое отличается от среднего по толщине покрытия. При этом наблюдается повышение концентрации углерода, и снижение концентрации азота в тонком слое вблизи поверхности с увеличением времени формирования покрытия (табл. 2). Высокая концентрация кислорода и углерода позволяет предположить наличие оксинитридов, карбонитридов, оксикарбонитридов и оксидов титана в составе покрытий.
Таблица 2.
Элементный состав TiN покрытий, полученных при различном времени формирования, по данным XPS
| Образец | Концентрация, ат. % | ||||
|---|---|---|---|---|---|
| [Ti] | [N] | [O] | [C] | [Fe] | |
| 15 min | 28.3 | 30.3 | 18.2 | 22.0 | 1.2 |
| 35 min | 22.6 | 26.0 | 17.0 | 33.5 | 0.9 |
| 50 min | 21.4 | 21.6 | 17.2 | 39.8 | 0.0 |
Анализ химического состояния элементов покрытий: азота, углерода и титана проводился по спектрам остовных линий N 1s, C 1s и Ti 2p.
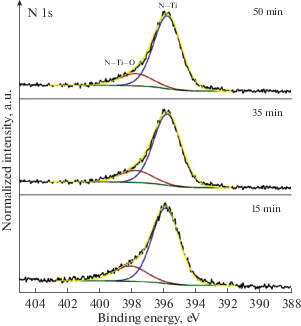
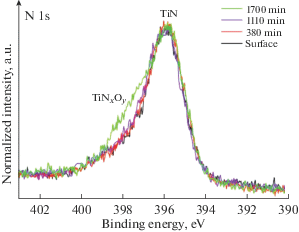
На рис. 3 приведены XPS спектры азота для покрытий с различным временем формирования. Положение основного максимума N 1s линии для всех вариантов покрытий находится на энергии ~396 eV. Это соответствует азоту химически связанному с титаном в нитриде титана [19]. Состояния в высокоэнергетической области спектра N 1s в диапазоне энергий связи ~397–399 eV дает возможность предположить наличие атомов азота в связи с титаном и кислородом, Это может свидетельствовать о присутствии оксинитридов титана в составе покрытий. Результаты разложения спектров N 1s показали (рис. 3), что максимум высокоэнергетического компонента находится на энергии ~397.8 eV, что соответствует азоту в составе оксинитридов титана (TiNxO1 –x) [19, 20]. По-видимому, имеет место замещение части атомов азота в ГЦК решетке нитрида титана кислородом. Возможно также залечивание вакансий азота атомами кислорода в решетке TiN, так как известно [21], что для твердых растворов, образующихся в интервале составов TiN0.6O0.4–TiN1.0 доля дефектов вакансионного типа составляет не многим более процента. Соотношение интегральных площадей компонентов, отвечающих азоту в составе TiN и TiNxO1 –x, для всех вариантов покрытий имеют близкие значения. Доля азота в TiN составляет ~82–80%, доля азота в TiNxO1 –x ~18–20%.
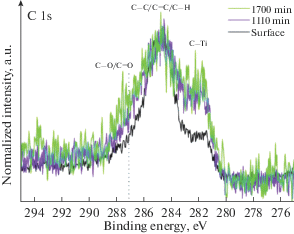
Форма XPS спектров остовной линий углерода C 1s, представленных на рис. 4, позволяет говорить, что углерод присутствует в покрытиях в трех химически неэквивалентных состояниях. Компонент спектров углерода на энергии связи ~284.6 eV имеет наибольшую интенсивность и отвечает углероду, входящему в состав соединений обладающих С–С/C=C/С–Н химическими связями [22]. По-видимому, данный углерод присутствует в виде аморфных включений. Компонент спектра с максимумом расположенным на энергии связи ~281.7 eV соответствует углероду, химически связанному с титаном [23]. Из рис. 4 видно, что при наибольшем времени формирования покрытия (50 min) данный компонент имеет несколько более высокое значение ~19%, в то время как для 15 и 35 min ~15%. Компонент с максимумом на энергии связи ~287.4 eV отвечает углероду химически связанному с кислородом [24]. Источником углерода в процессе формирования покрытий является остаточная атмосфера камеры установки. На основе полученных данных нельзя утверждать о формировании новой фазы карбида титана в зернах нитрида титана. Более вероятно образование твердых растворов углерода с постепенным замещением по диффузионному механизму атомов азота атомами углерода. Учитывая изоморфизм и возможность образования непрерывного ряда твердых растворов для систем TiN–TiO–ТiC [21] весьма вероятно формирование карбонитридов и оксикарбонитридов титана. Образование таких соединений, главным образом, определяется температурой и длительностью процесса. Следовательно, наблюдаемое в нашем случае, некоторое увеличение относительного содержания Ti–C связей при увеличении времени формирования покрытия является закономерным.

На рис. 5 приведены спектры Ti 2p покрытий с различным временем формирования.
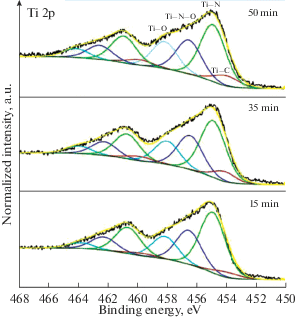
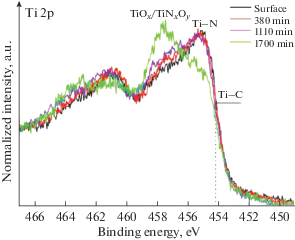
Основные максимумы линии Ti 2p в спектрах покрытий локализованы на энергиях связи ~455 eV (Ti 2p3/2) и ~461 eV (Ti 2p1/2), что соответствует нитриду титана (рис. 5) [25]. При этом наличие протяженного плеча в высокоэнергетической области спектра указывает на присутствие титана в различном химическом окружении. Состояния на энергии связи ~457 и ~458 eV относятся к титану в составе оксинитрида и оксида соответственно [26]. Наличие состояний в низкоэнергетической области спектра на энергии связи ~454 eV связано с присутствием химических связей титана с углеродом [27]. Данный пик становится более заметным при увеличении времени формирования покрытия, что свидетельствует об увеличении содержания углерода в соединениях по типу TiNxCy и TiNxCyOz. За исключением данной особенности форма и энергетические положения основных максимумов практически совпадают. Это позволяет говорить о достаточно близком химическом окружении титана в поверхностном слое покрытий, сформированных за различное время. Отличие, главным образом, заключается в относительной доле связи Ti–C, которая составляет 4.3% для времени формирования покрытия 15 min, 7.2% для 35 min и 8.0% для 50 min. Детальный анализ спектров Ti 2p покрытий показал отсутствие титана в металлическом состоянии. Таким образом, формирование капельной фазы титана в покрытиях при указанных параметрах получения можно полностью исключить.
С учетом зафиксированной неоднородности состава покрытий (см. табл. 1 и 2) по толщине представляется актуальным проанализировать его изменение по мере удаления от поверхности. Для исследования изменения химического состояния элементов по глубине был использован образец с покрытием, сформированным при минимальном времени (15 min).
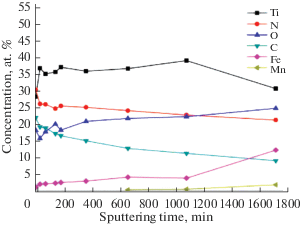
На рис. 6 представлены результаты послойного XPS анализа. С увеличением времени травления наблюдается постепенное увеличение концентрации Fe, который является основным элементом подложки. Начиная от времени травления ~700 min в спектрах присутствует Mn, который является основным легирующим элементом стали 110Г13Л. Как следует из рис. 6 концентрации химических элементов, полученные из данных XPS существенно отличается от результатов EDX анализа только в самом тонком поверхностном слое, не превышающем несколько десятков нанометров. Это, по-видимому, связано с адсорбцией углерода и кислорода на разогретую после завершения процесса формирования покрытия поверхность образца из остаточной атмосферы вакуумной камеры с образованием химических связей. С увеличением расстояния от поверхности величины концентраций, полученные данными методами, принимают достаточно близкие значения (см. табл. 1 и 2). В целом, за исключением тонкого поверхностного слоя, в процессе ионного травления наблюдается тенденция к синхронному снижению концентрации N и C, при этом концентрация O возрастает.
Рис. 6.
Профили распределения концентрации элементов в зависимости от времени ионного травления по данным XPS анализа для покрытия, сформированного за 15 min.
На рис. 7 представлены спектры Ti 2p до и после ионного травления различной продолжительности. Как видно, с увеличением времени травления наблюдается повышение интенсивности состоянии на энергии связи ~458 eV, отвечающих титану в составе TiO2 –y, TiNxO1 –x и, соответственно, уменьшению интенсивности состояний, отвечающих TiN. После 1700 min травления интенсивность данных компонентов начинает превалировать над интенсивностью компонента TiN.
Рис. 7.
XPS спектры Ti 2p для покрытия, сформированного за 15 min, до и после ионного травления различной продолжительности.
Анализ спектров N 1s (рис. 8) подтверждает постепенное повышение доли оксинитридов при увеличении времени травления покрытия. Об этом свидетельствует увеличение ширины линий N 1s и повышение интенсивности состояний, отвечающих TiNxO1 –x (~397.8 eV) при продолжительности травления от 380 до 1700 min. Данные результаты позволяют предположить, что образование оксинитридов и оксидов титана наиболее интенсивно протекает на начальной стадии формирования покрытия вследствие более низких температур образования этих соединений относительно температур образования нитридов титана [28]. При этом источником кислорода, очевидно, является остаточная атмосфера в реакционной камере.
Рис. 8.
XPS спектры N 1s для покрытия, сформированного за 15 min, до и после ионного травления различной продолжительности.
При анализе профилей распределения концентрации элементов (рис. 6) было показано, что с увеличением времени травления наблюдается значительное снижение концентрации углерода в покрытии. Разложение спектра углерода C 1s (рис. 9) после различного времени травления, позволило установить, что соотношение интенсивности состояний, соответствующих аморфному углероду (C–C/C=C/C–H) и состояниям углерода связанного с титаном изменяется в сторону увеличения последнего. При этом наблюдается тенденция снижения общей концентрации углерода и роста доли углерода в связях Ti–C, с увеличением расстояния от поверхности покрытия. Это, предположительно, связано с тем, что диффузия углерода происходит от поверхности в глубь покрытия. При этом слой с высоким содержанием доли Ti–C связи в соединениях будет являться эффективным барьером для дальнейшей диффузии углерода на глубину [29]. Вероятно, поэтому после достижения максимума доли углерода в составе Ti–C связей (после 1110 min травления) наблюдается заметное снижение общей концентрации углерода.
Рис. 9.
XPS спектры C 1s для покрытия, сформированного за 15 min, до и после ионного травления различной продолжительности.
Следует отметить, что при длительных временах травления ионами аргона с энергией в несколько keV может происходить перераспределение атомов углерода, кислорода и азота в изучаемых слоях поверхности, что связано с селективностью распыления атомов и внедрением атомов отдачи. Это, в частности, может приводить к внедрению поверхностного углерода в состав основного материала. Однако, в нашем случае, с увеличением времени травления наблюдается постепенный рост доли углерода в составе карбида титана, который, как известно, формируется при достаточно высоких температурах [30]. Поэтому, формирование карбида титана вследствие ионно-стимулированной диффузии маловероятно. В случае ионного травления оксинитрида титана можно предполагать некоторую селективность распыления атомов азота и кислорода. Однако, незначительное различие атомной массы и энергии связи данных элементов с титаном [31] позволяет говорить, что различие скорости распыления азота и кислорода при ионном травлении не оказывает значительного влияния на результаты анализа состава покрытия по глубине.
3.3. Исследование микротвердости образцов с покрытиями
В табл. 3 приведены значения микротвердости, измеренные методом Виккерса при нагрузках на индентор 0.2, 0.5 и 1.0 N, для исходной подложки стали 110Г13Л и образцов с различным временем нанесения покрытия. Как следует из таблицы глубина проникновения индентора hср в исходную подложку для средних значений микротвердости находилась в пределах от 1.29 до 3.06 µm. Разброс значений микротвердости у исходного образца стали 110Г13Л связан с разницей твердости зерен аустенита (твердость ниже) и межзеренной прослойки, включающей избыточные карбиды марганца и железа (Fe, Mn)3С и обладающей более высокой твердостью. Для образцов с покрытиями наименьшая средняя глубина проникновения hср соответствовала самой низкой нагрузке на индентор для покрытия с наибольшим временем формирования (50 min), а наибольшая hср наблюдалась у исходного образца при нагрузке на индентор 1 N. Это связано с тем, что в случае покрытия с максимальным временем формирования при нагрузке на индентор 0.2 N измеряется твердость в первую очередь покрытия, поскольку глубина проникновения индентора не превышает оценочное значение его толщины. В этом случае подложка из стали незначительно влияет на значение твердости. Однако, в случае покрытий полученных при 15 и 35 min глубина проникновения индентора сопоставима либо превышает их оценочную толщину. При этом значение микротвердости для данных покрытий увеличивается по сравнению с образцом исходной стали при всех используемых нагрузка на индентор. Известно [21], что карбонитриды, оксинитриды и оксикарбонитриды титана обладают более высокой твердостью, чем нитрид титана. Следовательно, высокие значения микротвердости для покрытий с временем формирования 35 и 50 min могут быть связаны с увеличением доли Ti–C связей в них, что было зафиксировано посредством XPS анализа. Кроме того, методом XPS было показано, что с увеличением глубины от поверхности повышается доля оксинитридов. Очевидно, что покрытие с большим временем формирования имеет более протяженный слой с высоким содержанием оксинитридов титана и, следовательно, обладает более высоким значением микротвердости. Сравнение значений микротвердости (табл. 3) показывает, что покрытие, сформированное при 35 min, практически не уступает покрытию с большей толщиной (сформированному при 50 min).
Таблица 3.
Значения микротвердости образцов из стали 110Г13Л с различным временем нанесения покрытия
| Время нанесения покрытий, min |
Нагрузка на индентор 0.2 N | Среднее значение микротвердости, N/mm2 |
hср, µm | ||||
|---|---|---|---|---|---|---|---|
| Микротвердость, N/mm2 | |||||||
| 0 (Исходный) | 4820 | 4030 | 4580 | 4820 | 4360 | 4522 | 1.29 |
| 15 | 5010 | 7580 | 7130 | 6080 | 5790 | 6318 | 1.09 |
| 35 | 8470 | 7130 | 8030 | 7580 | 9050 | 8052 | 0.97 |
| 50 | 8510 | 9050 | 8510 | 9650 | 7580 | 8660 | 0.94 |
| Время нанесения покрытий, min |
Нагрузка на индентор 0.5 N | Среднее значение микротвердости, N/mm2 |
hср, µm | ||||
| Микротвердость, N/mm2 | |||||||
| 0 (Исходный) | 4730 | 4230 | 4480 | 4730 | 4230 | 4480 | 2.05 |
| 15 | 5480 | 7410 | 6430 | 5480 | 7660 | 6492 | 1.71 |
| 35 | 6430 | 9270 | 6260 | 8580 | 7410 | 7590 | 1.58 |
| 50 | 7660 | 7410 | 6010 | 7410 | 7130 | 7124 | 1.63 |
| Время нанесения покрытий, min |
Нагрузка на индентор 1.0 N | Среднее значение микротвердости, N/mm2 |
hср, µm | ||||
| Микротвердость, N/mm2 | |||||||
| 0 (Исходный) | 4200 | 3830 | 4130 | 3900 | 4130 | 4038 | 3.06 |
| 15 | 5720 | 5850 | 5140 | 6260 | 6570 | 590,8 | 2.53 |
| 35 | 8470 | 7820 | 7240 | 8030 | 6890 | 7690 | 2.22 |
| 50 | 8020 | 7240 | 8020 | 7240 | 8700 | 7844 | 2.2 |
ЗАКЛЮЧЕНИЕ
Методами SEM, EDX, XPS осуществлено исследование морфологии, состава, химического состояния элементов покрытий на основе нитрида титана, сформированных методом конденсации с ионной бомбардировкой, при различном времени на подложках из стали 110Г13Л. Установлено, что покрытия с различным временем формирования обладают достаточно близким химическим составом. Наибольшее количество титана находится в составе нитрида и оксинитрида. При этом в составе покрытий присутствует некоторое количество оксидов титана, а также титана, химически связанного с углеродом. Наличие Ti–C связей позволяет говорить о присутствии более сложных соединений (оксикарбидов и оксикарбонитридов титана). Установлено, что с увеличением времени формирования, а, следовательно, и толщины покрытий, наблюдается повышение относительного содержания Ti–C связей. Показано, что для всех вариантов формирования покрытий отсутствует титан в металлическом состоянии. Следовательно, при использованных параметрах получения покрытий не происходит образования капельной макрофракции металлического титана. Проведенный послойный XPS анализ показал, что покрытия обладают гетeрогенностью состава по глубине. Установлено, что с увеличение расстояния от поверхности в покрытиях повышается содержание оксинитридов и оксидов титана.
Исследовано методом Виккерса значения микротвердости исходной подложки стали 110Г13Л и образцов с различным временем формирования покрытий, измеренные при нагрузках на индентор 0.2, 0.5 и 1.0 N. Показано, что микротвердость существенно повышается при нанесении покрытий. Одной из причин увеличения значений микротвердости на образцах с покрытиями является формирование соединений TiNxCyOz, обладающих более высокой твердостью, чем TiN. Покрытия, сформированные при 35 и 50 min, содержат большее количество указанных соединений и обладают максимальными значениями микроствердости. При этом значение микротвердости для покрытий при 35 и 50 min формирования отличается несущественно. Следовательно, достаточным временем формирования покрытий для повышения твердости изделий из стали 110Г13Л является 35 min.
Список литературы
Jafari A., Ghoranneviss Z., Elahi A.S. et al. // Advances in Mechanical Engineering. 2014. Article ID 373847. P. 1.
Андреев А.А., Саблев Л.П., Шулаев В.М. Вакуумно-дуговые покрытия. Харьков: ННЦ. ХФТИ, 2010. С. 318.
Fortuna S.V., Sharkeev Y.P., Perry A.J. et al. // Thin Solid Films. 2000. V. 377–378. P. 512.
Карпов Д.А., Бондарчук Э.Н., Кузнецов В.C., Литуновский В.Н. Снижение содержания макрофракций в вакуумно-дуговом осаждении покрытий. СПб: ФГУП НИИЭФА им. Д.В. Ефремова, 2009. С. 55.
Lomino N.S., Ovcharenko V.D., Andreev A.A. // IEEE Trans. on Plasma Science. 2005. V. 33. P. 1626.
Олеванов М.А., Манкелевич Ю.А., Рахимова Т.В. // ЖТФ. 2003. Т. 73. С. 51.
Хороших В.М. // Физическая инженерия поверхности. 2005. Т. 2. С. 200.
Pan W.L., Yu G.P., Huang J.H. // Surf. Coat. Technol. 1998. V. 110. P. 111.
Johnson C.A., Ruud J.A., Bruce R., Wortman D. // Surf. Coat. Technol. 1998. V. 108. P. 80.
Погребняк А.Д., Шпак А.П., Азаренков Н.А., Береснев В.М. // УФН. 2009. Т. 179. С. 35.
Погребняк А.Д., Багдасарян А.А., Якущенко И.В., Береснев В.М. // Успехи химии. 2014. Т. 83. С. 1027.
Погребняк А.Д., Береснев В.М., Бондар О.В. и др. // ПЖТФ. 2018. Т. 44. С. 25.
Коротаев А.Д., Мошков В.Ю., Овчинников С.В. и др. // Физическая мезомеханика. 2005. Т. 8. С. 103.
Hörlinga A., Hultman L., Odén M. et al. // Surf. and Coat. Technol. 2005. V. 191. P. 384.
Jang C.S., Jeon J.-H., Song P.K. et al. // Surf. and Coat. Technol. 2005. V. 200. P. 1501.
Veprek S., Veprek-Heijman M., Karvankova P., Prochazka J. // Thin Solid Films. 2005. V. 476. P. 1.
Yeh J.-W. // Annales de Chimie. Science des Materiaux. 2006. V. 31. P. 633.
Zhang Y., Zuo T.T., Tang Z. et al. // Progress in Materials Science. 2014. V. 61. P. 1.
Chan M., Lu F. // Thin Solid Films. 2009. V. 517. P. 5006.
Morales M., Cucatti S., Acuna J.J.S., Zagonel L.F. et al. // J. Phys. D: Appl. Phys. 2013. V. 46. P. 1.
Алямовский С.И., Зайнулин Ю.Г., Швейкин Г.П. Оксикарбиды и оксинитриды металлов IVA и VA подгрупп. М.: Наука, 1981. С. 144.
Dao V., Hoa N.T.Q., Larina L.L., Leed J. et al. // Nanoscale. 2013. V. 5. P. 12237.
Shah S.A., Habib T., Gao H., Gao P. et al. // Chem. Commun. 2017. V. 53. P. 400.
Несов С.Н., Корусенко П.М., Болотов В.В., Поворознюк С.Н. и др. // ФТТ. 2017. Т. 59. С. 2006.
Jaeger D., Patscheider J. // Surface Science Spectra. 2013. V. 20. P. 1.
Lin M.C., Chen M.-J., Chang L.-S. // Applied Surface Science. 2010. V. 256. P. 7242.
Nakatsuka O., Hisada K., Oida S., Sakai A. et al. // Japanese J. Applied Physics. 2016. V. 55. Article ID 06JE02. P. 1.
Lütjering G., Williams J.C. Titanium. Springer Berlin: Heidelberg, 2003. P. 442.
Замулаева Е.И., Левашов Е.А., Свиридова Т.А., Швындина Н.В. и др. // Известия вузов. Порошковая металлургия и функциональные покрытия. 2013. Т. 3. С. 73.
Zhu G., Wang W., Wang R., Zhao C.W. et al. // Materials. 2017. V. 10. Article ID 1007. P. 1.
Farhadizadeh A.R., Amadeh A.A., Ghomi H. // Communications in Theoretical Physics. 2017. V. 68. P. 678.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов