

Цитология, 2021, T. 63, № 4, стр. 322-334
Снижение эффективности репарации и антиоксидантной защиты способствует накоплению повреждений ДНК при старении клеток
П. И. Дерябин 1, А. В. Бородкина 1, *
1 Институт цитологии РАН
194064 Санкт-Петербург, Россия
* E-mail: borodkina618@gmail.com
Поступила в редакцию 22.03.2021
После доработки 30.03.2021
Принята к публикации 01.04.2021
Аннотация
Накопление стареющих клеток в организме положительно коррелирует с возрастом и рассматривается как один из факторов риска возраст-зависимого увеличения частоты опухолевых заболеваний. Во многом опухоль-промотирующую роль стареющих клеток связывают с паракринным влиянием на клетки микроокружения ассоциированного со старением секреторного фенотипа. Однако согласно недавним исследованиям, неопластическая трансформация также может быть обусловлена геномной нестабильностью стареющих клеток, возникающей вследствие накапливающихся повреждений ДНК. Настоящая работа посвящена исследованию молекулярных механизмов, которые могут обуславливать возникновение и накопление повреждений ДНК при старении клеток. В качестве моделей клеточного старения мы использовали репликативное и стресс-индуцированное старение эндометриальных стромальных клеток человека (эСК). Мы показали, что для обоих типов старения эСК характерно образование персистирующих фокусов повреждения ДНК. Используя генетически-кодируемый биосенсор HyPer, мы выявили снижение эффективности антиоксидантной защиты в стареющих эСК. Одновременно с этим в таких клетках существенно увеличивался уровень эндогенных активных форм кислорода (АФК), что в совокупности может приводить к возникновению фокусов повреждения ДНК в стареющих клетках. В свою очередь, накопление фокусов повреждения ДНК может быть опосредовано снижением эффективности работы систем репарации при старении эСК, о чем свидетельствуют как данные транскриптомного анализа, так и результаты оценки динамики репарации повреждений, вызванных окислительным стрессом или ионизирующим излучением. Таким образом, накопление повреждений ДНК в стареющих эСК связано, с одной стороны, с неэффективной антиоксидантной защитой и повышенным уровнем АФК, а с другой стороны, с низкой эффективностью репарации повреждений.
Старение принято рассматривать как однонаправленный и необратимый процесс ухудшения работы организма на молекулярном, клеточном, тканевом и органном уровнях (Hayflick, 2002). Помимо общего снижения функциональной активности всех систем организма, с возрастом существенно повышается риск развития рака, остеопороза, сердечно-сосудистых заболеваний, деменций и множества других патологий самой разной этиологии (Kirkwood, 2005). Сегодня для оценки реального биологического возраста человека и поиска корреляций с риском развития тех или иных патологий и (или) смертности проводят комплексный анализ, комбинируя самые разнообразные показатели, начиная от самых общих, таких как индекс массы тела, качество слуха и зрения, двигательная активность, признаки старческой астении, уровень кровяного давления, гемоглобина, глюкозы, холeстерина, аполипопротеина В, до более специфических – таких как, длина теломер и статус метилирования ДНК, измеренный при помощи эпигенетических часов Horvath/Hannum/GrimAge/PhenoAge (Levine, 2013; Li et al., 2020).
Отдельное место в ряду маркеров биологического возраста занимает клеточное старение. Во-первых, установлено, что практически у всех позвоночных с возрастом происходит накопление стареющих клеток в организме (Jeyapalan et al., 2007; Liu et al., 2009). Во-вторых, показано, что накопление стареющих клеток в тканях сопровождает прогрессию различных возраст-ассоциированных заболеваний, включая атеросклероз, остеопороз, рак, нейродегенеративные заболевания и множество других (Roos et al., 2016; Chinta et al., 2018; Zhang et al., 2019; Alimirah et al., 2020; Chandra et al., 2020; Kirkland, Tchkonia, 2020). Более того, экспериментальные работы с использованием трансгенных мышиных моделей демонстрируют, что клеточное старение скорее является причиной организменного старения, нежели его следствием (Baker et al., 2016). Последнее подчеркивает необходимость детального изучения молекулярных основ клеточного старения, что в перспективе может помочь не только устанавливать корреляции между возрастом и какими-либо функциональными изменениями, а понимать причины этих возрастных изменений.
Существует несколько механизмов появления стареющих клеток в тканях. Так, укорочение теломер опосредует запуск репликативного старения, мутации, приводящие к активации онкогенов или инактивации тумор-супрессоров, обуславливают индукцию онкоген-индуцированного старения и, наконец, воздействие различных стрессов (окислительный, УФ-облучение, γ-радиация, тепловой и др.) способствует развитию стресс-индуцированного старения (Hernandez-Segura et al., 2018). Независимо от разнообразных индукторов, выделяют ряд характерных признаков стареющих клеток, к которым относятся необратимая потеря пролиферативной активности, гипертрофия, активация сигнальных путей р53/р21 и р16/Rb, появление активности бета-галактозидазы, ассоциированной со старением (SA-β-Gal), дисфункция митохондрий, повышение уровня активных форм кислорода (АФК), аккумуляция липофусцина, изменение состава секретируемых факторов и накопление повреждений ДНК (Hernandez-Segura et al., 2018). Стоит подчеркнуть, что это далеко не полный перечень модификаций, которые сопровождают старение клеток (Hernandez-Segura et al., 2018).
Несмотря на драматические внутриклеточные изменения и необратимую остановку клеточного цикла, в течение длительного времени стареющие клетки остаются жизнеспособными и сохраняют метаболическую активность. При благоприятном сценарии, стареющие клетки, несущие повреждения, распознаются и удаляются из организма клетками иммунной системы (Prata et al., 2018; Kale et al., 2020). В противном случае, эти клетки накапливаются и за счет своей паракринной активности негативно влияют на микроокружение, постепенно приводя к дисфункции тканей, в которых они локализуются (Ovadya, Krizhanovsky, 2014; Zhu et al., 2014; The Tabula Muris Consortium, 2020).
Вплоть до недавнего времени эти два сценария очень упрощенно описывали вклад стареющих клеток в прогрессию организменного старения. Важно, что оба сценария предполагают, что клеточное старение по аналогии со старением организма – это запрограммированный и однонаправленный процесс постепенной дисрегуляции всех внутриклеточных систем, который в конечном итоге заканчивается клеточной гибелью. Однако работы последних лет свидетельствуют о том, что постепенное накопление персистирующих повреждений ДНК в стареющих клетках и “адаптация” к этим повреждениям в ряде случаев могут приводить к возвращению стареющих клеток в цикл и, таким образом, способствовать онкотрансформации (Gosselin et al., 2009; Graziano, Gonzalo, 2017; Coutelier et al., 2018; Zampetidis et al., 2021).
В связи с этим, вопрос поддержания геномной стабильности по мере развития старения клеток является принципиально важным как в контексте общего старения организма, так и в контексте развития рака. В настоящей работе мы сфокусировались на исследовании молекулярных причин, которые могут обуславливать геномную нестабильность стареющих клеток – эндометриальных стромальных клеток (эСК). А именно, мы постарались разобраться с тем, что может приводить к возникновению и накоплению персистирующих повреждений ДНК в стареющих эСК.
МАТЕРИАЛ И МЕТОДИКА
Культивирование эндометриальных стромальных клеток человека (эСК). В настоящей работе использовали эСК человека (линия 2804), ранее полученные и охарактеризованные сотрудниками ИНЦ РАН (Санкт-Петербург) (Земелько и др., 2011). ЭСК культивировали в среде DMEM/F12 (Gibco, США), содержащей 10% эмбриональной сыворотки (HyClone, США), 1% гентамицина и 1% глутамакса, в атмосфере 5% СО2 при 37°С.
Моделирование репликативного и стресс-индуцированного старения эСК. Клетки (эСК) считали репликативно старыми после достижения 28-ого пассажа, когда клетки практически полностью утрачивали пролиферативную активность. Для индукции стресс-индуцированного старения эСК на ранних пассажах (6–9) подвергали сублетальному окислительному воздействию, добавляя в бессывороточную среду Н2О2 (конечная концентрация 200 мкМ), раствор которой готовили из 30%-ной Н2О2 непосредственно перед использованием. Обработку клеток проводили в течение 1 ч при 37°С в атмосфере 5% СО2, после чего клетки дважды промывали фосфатно-солевым буферным раствором (PBS) и далее культивировали в свежей ростовой среде (клетки не пересевали, так как сразу после обработки Н2О2 клетки останавливались в цикле и полностью утрачивали способность пролиферировать). Наличие маркеров старения анализировали не ранее, чем на 28-м пассаже в случае репликативного старения эСК и не ранее, чем через 14 сут после окислительного воздействия в случае стресс-индуцированного старения.
Анализ пролиферативной активности, размера и автофлуоресценции клеток. Прикрепленные клетки снимали с чашек смесью растворов трипсина и Версена (0.05%), осаждали центрифугированием и промывали раствором PBS. Для построения кривых роста образец окрашивали йодистым пропидием (PI; 50 мкг/мл) и анализировали количество PI-негативных (живых) клеток в указанные временные точки на цитофлуориметре CytoFlex или CytoFLEX S (Backman Coulter, США). Изменение размера клеток оценивали по изменению прямого светорассеяния (FS) живых клеток. Накопление липофусцина (агрегатов поврежденных макромолекул, накапливающихся в лизосомах стареющих клеток) оценивали по среднему значению его автофлуоресценции. Данные анализировали с помощью программы CytExpert (версии 1.2 и 2.0).
Оценка уровня внутриклеточных АФК. Использовали флуоресцентный краситель 2,7-дихлорфлуоресцеиндиацетат (H2DCF-DA) (Molecular Probes, США). Прикрепленные к поверхности чашки клетки инкубировали в течение 20 мин при 37°С в темноте в среде без сыворотки, содержащей 10 мкМ красителя. После окончания времени инкубации с красителем клетки промывали PBS, переводили в суспензионное состояние при помощи смеси растворов трипсина и Версена (0.05%) и анализировали на проточном цитометре. В каждом образце анализировали не менее 10 тыс. клеток.
Количественный и качественный анализ активности SA-β-Gal. Для количественной оценки активности SA-β-Gal использовали флуоресцентный краситель C12FDG (5-dodecanoylaminofluorescein Di-β-D-galactopyranoside) (Invitrogen, США). Проникая в клетки, этот субстрат расщепляется β-галактозидазой и превращается во флуоресцирующий продукт, который удерживается в клетке. Процедуру пробоподготовки проводили в соответствии с инструкцией производителя, последующий анализ интенсивности флуоресценции осуществляли на проточном цитометре. Качественно активность SA-β-Gal выявляли с помощью фирменного набора Senescence-galactosidase staining kit (Cell Signaling, США). Все процедуры осуществляли в соответствии с инструкцией фирмы-производителя. Клетки на чашках промывали PBS, фиксировали в течение 10 мин при комнатной температуре 1-кратным фиксирующим раствором, после чего дважды промывали PBS и окрашивали в β-галактозидазном растворе при 37°С в течение ночи. Об активности SA-β-Gal судили по появлению синих гранул в цитоплазме клеток.
Функциональная характеристика антиоксидантных систем клетки. Оценивали скорость восстановления генетически кодируемого сенсора перекиси водорода HyPer (Belousov et al., 2006) после однократного добавления Н2О2 в клеточную среду. В экспериментах использовали линию эСК, экспрессирующую HyPer в клеточной цитоплазме (Lyublinskaya et al., 2018). Оценивали динамику изменения флуоресцентного сигнала HyPer, отражающего уровень окисления сенсора. Клетки ресуспензировали в свежей ростовой среде (50 тыс. кл./мл), 20 мин выдерживали при 5% СО2 и 37°С, добавляли в суспензию 200 мкМ Н2О2, аликвотировали в микропробирки по 200 мкл, после чего каждые 5 мин последовательно анализировали пробы на проточном цитометре CytoFLEX S (Backman Coulter, США) на протяжении 45 мин. Для калибровки флуоресцентного сигнала биосенсора использовали образцы клеток, инкубированные в течение 10 мин с избыточными дозами Н2О2 (1 мМ) и дитиотреитола (30 мМ), соответственно принимая уровень сигнала в этих пробах за 1 (полностью окисленный сенсор) и 0 (полностью восстановленный сенсор) (Lyublinskaya et al., 2018). В ходе предварительных экспериментов проверяли скорость самопроизвольного восстановления сенсора: к клеткам после 5-минутного действия Н2О2, которая приводила к полному окислению HyPer, добавляли каталазу (1000 U), которая мгновенно элиминирует Н2О2 в клеточной среде. В этих условиях флуоресцентный сигнал HyPer возвращался к своему базальному уровню в течение 3–5 мин, что позволило нам использовать этот сенсор для анализа кинетики существенно более медленных процессов восстановления сенсора при элиминации экзогенного Н2О2 клетками.
Электрофорез и иммуноблотинг. Пробоподготовку, электрофорез и иммуноблотинг осуществляли в соответствии с процедурой подробно описанной в нашей предыдущей работе (Дерябин и др., 2015). Для специфического выявления белков использовали антитела против ингибитора циклин-зависимых киназ р21, глицеральдегид-3-фосфатдегидрогеназы (GAPDH, clone 14C10), фосфорилированной формы белка Rb (p-Rb; Ser807/811), фосфорилированной формы гистона H2AX (p-(H2AX; Ser139), белков ламинов А и С (LMNA/C) и ядерного негистнового белка (high-mobility group protein B1, HMGB1 (D3E5)). В качестве вторичных антител применяли козьи антитела, выработанные против иммуноглобулинов кролика (GAR-HRP) или мыши (GAM-HRP). Все антитела приобретены в фирме Cell Signaling (США).
Иммунофлуоресцентный анализ. Клетки выращивали на покровных стеклах 8 × 8 мм (Menzel-Glaser, Германия), помещенных в пластиковые чашки диаметром 35 мм (Corning, США). Стекла с клетками дважды промывали теплым РBS, затем клeтки фиксировали 4%-ным раствором формалина (Sigma, США) в течение 15 мин при комнатной температуре. После фиксации клетки промывали PBS и пермеабилизовали 10 мин при комнатной температуре раствором PBS, содержащим 0.1% Тритона. Далее стекла инкубировали в 1%-ном растворе BSA в РBS для блокирования неспецифического связывания. Аналогичный по составу раствор использовали для разведения первичных антител. Для специфического выявления белков использовали следующие первичные антитела: моноклональные мышиные антитела против киназы АТМ, фосфорилированной по Ser1981 (Invitrogen, США); поликлональные кроличьи антитела против γH2AX (Ser139) (Cell Signaling, США). С первичными антителами клетки инкубировали в течение ночи при 4°С. После окончания инкубации клетки промывали раствором PBS, содержащим 0.01% Tween-20. Далее клетки инкубировали с вторичными антителами в течение 30 мин при 37°С в темноте. В качестве вторичных антител применяли козьи антитела, выработанные против иммуноглобулинов кролика, конъюгированные с Alexa Fluor 568 (Life Technologies, США) или козьи антитела, выработанные против иммуноглобулинов мыши, конъюгированные с Alexa Fluor 488 (Life Technologies, США). Ядра клеток окрашивали с помощью DAPI (Sigma, США) в концентрации 1 мкг/мл. Далее покровные стекла заключали в глицериновый буфер с 2% пропилгаллата. Распределение флуоресцентно-меченных белков в клетках изучали с помощью флуоресцентного микроскопа Zeiss LSM 5 Pascal (Carl Zeiss, Германия), оборудованного цифровой камерой DCF 420C (Leica, Германия). Использовали объективы с увеличением 40× и 100×.
Обратная транскрипция и ПЦР в реальном времени. Выделение РНК, обратную транскрипцию и ПЦР в реальном времени проводили в соответствии с процедурой, подробно описанной в нашей предыдущей работе (Griukova et al., 2019). Для проведения ПЦР в реальном времени использовали следующие специфические праймеры к генам каталазы (CAT): прямой 5'-TTAATCCATTCGATCTCACC-3' и обратный 5'-GGCGGTGAGTGTCAGGATAG-3', температура отжига 59°C (210 н.п.); супероксиддисмутазы 1 (SOD1): прямой 5'-GGTCCTCACTTTAATCCTCTAT-3' и обратный 5'-CATCTTTGTCAGCAGTCACATT-3', температура отжига 59°C (97 п.н.); глутатионпероксидазы 1 (GPX1): прямой 5'-CGCCACCGCGCTTATGACCG-3' и обратный 5'-GCAGCACT-GCAACTGCCAAGCAG-3', температура отжига 59°C (238 п.н.). В качестве количественного и качественного контроля кДНК использовали олигонуклеотидные праймеры для GAPDH прямой 5'-GAGGTCAATGAAGGGGTCAT-3' и обратный 5'-AGTCAACGGATTTGGTCGTA-3', температура отжига 59°C (100 п.н.). Все праймеры получены из фирмы Евроген (Россия).
Биоинформатический анализ. В работе использовали набор данных РНК-секвенирования, полученный нами ранее и доступный в публичном репозитории Gene Expression Omnibus по идентификатору GSE160702. Обработка сырых данных, квантификация единиц экспрессии транскриптов, суммирование транскриптов до уровня генов и статистическое тестирование дифференциальной экспрессии проводили с использованием инструментов FastqPuri (версия 1.0.7), BBtools (версия 38.75), salmon (версия 1.1.0), tximeta (верися 1.4.5) и пакета DESeq2 (версия 1.26.0) для программы R. Подробное описание параметров анализа приведено в нашей опубликованной работе (Deryabin et al., 2021). Анализ обогащения по функциональной принадлежности (GSEA) выполняли с использованием пакетов R‑Studio clusterProfiler (версия 3.14.3) и fgsea (версия 1.12.0) на основании баз Gene Ontology (GO) и Kyoto Encyclopedia of Genes and Genomes Pathway (KEGG Pathway).
Статистическая обработка данных. Статистический анализ выполнен с использованием программы R. Данные представлены в виде средних значений и их стандартных отклонений (n = 3). При установлении достоверности различий данных между двумя группами использовали t-тест Стьюдента, для множественных сравнений между группами применяли однофакторный дисперсионный анализ (ANOVA) с поправкой Тьюки.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
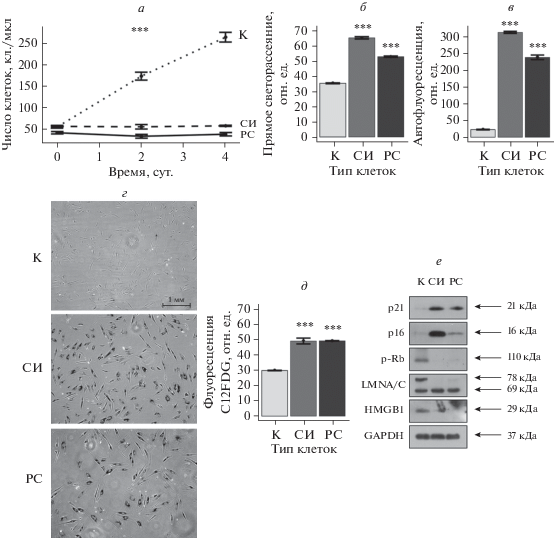
Характеристика репликативно- и стресс-индуцированно стареющих эСК. Для исследования молекулярных причин, способствующих возникновению и накоплению повреждений ДНК в стареющих клетках, мы использовали две наиболее распространенные модели клеточного старения – репликативное и стресс-индуцированное. Для индукции репликативного старения эСК пассировали до достижения 28 пассажа, когда клетки практически полностью утрачивали способность пролиферировать. Для запуска стресс-индуцированного старения эСК на ранних пассажах подвергали однократному сублетальному окислительному воздействию (200 мкМ H2O2, 1 ч) с последующим культивированием в свежей ростовой среде в течение минимум 14 сут. Для верификации состояния старения мы проанализировали панель классических маркеров клеточного старения. Как показано на рис. 1а–е, вне зависимости от типа, стареющие эСК характеризовались потерей пролиферативной активности, увеличением размера, накоплением липофусцина, появлением активности SA-β-Gal, активацией классических сигнальных путей p21/Rb и p16/Rb, а также снижением экспрессии ламина А/С (LMNA/C) и ядерного негистонового HMGB1 белка (рис. 1а–е). Таким образом, выбранные нами модели адекватно отражают репликативную и стресс-индуцированную формы старения эСК, и, соответственно, могут быть использованы для анализа механизмов, опосредующих накопление повреждений ДНК при старении клеток.
Рис. 1.
Основные характеристики стареющих эндометриальных стромальных клеток человека (эСК). а – Кривые роста контрольных (К), репликативно состаренных (РС) и стресс-индуцированно (СИ) состаренных эСК. б – Средний размер клеток, определенный по прямому светорассеянию. в – Уровень автофлуоресценции клеток, отражающий накопление липофусциновых гранул. г, д – Активность SA-β-Gal в контрольных, РС и СИ-состаренных эСК, выявленная цитохимически или по интенсивности флуоресценции красителя C12FDG соответственно. е – Уровни экспрессии белков p16, p21, LMNA/C, HMGB1 и фосфорилирования Rb, выявленные с помощью специфических антител; справа указаны мол. массы; в качестве контроля нагрузки использовали GAPDH. а, б, в, д – Данные представлены в виде средних значений и стандартных отклонений (n = 3); различия достоверны (***) при р < 0.001: при наличии внутригрупповых отличий для каждого типа клеток (а, тест ANOVA) и по сравнению с контролем (б, в, г, тест ANOVA с поправкой по Тьюки).
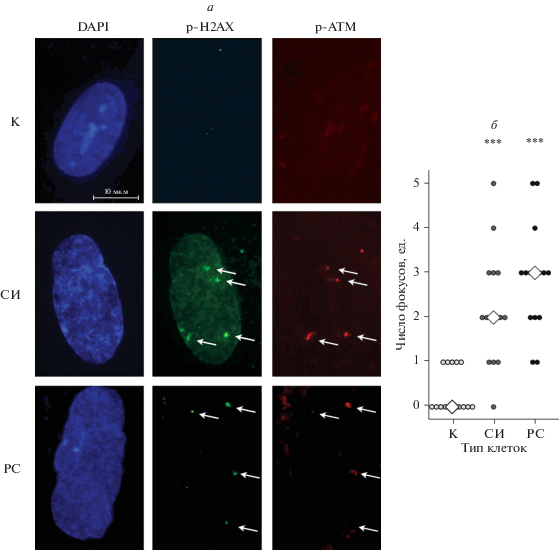
Репликативный и стресс-индуцированный типы старения эСК сопровождаются накоплением повреждений ДНК. На следующем этапе мы оценили наличие маркеров повреждения ДНК в стареющих эСК. Известно, что персистирующие фокусы повреждения ДНК характерны для всех типов клеточного старения, однако первоначальные источники их появления при репликативном и стресс-индуцированном старении различаются. Так, в случае репликативного старения незащищенные концы теломер, образующиеся в результате их укорочения до критического уровня, распознаются клеткой как двойные разрывы ДНК (Takai et al., 2003). В случае стресс-индуцированного старения сами ДНК-повреждающие стимулы непосредственно приводят к формированию двойных разрывов, часть из этих повреждений затем может успешно репарироваться, а часть может сохраняться (Borodkina et al., 2014; Shmulevich, Krizhanovsky, 2021). Вне зависимости от источника, в местах повреждения ДНК собираются уникальные белковые комплексы, которые, с одной стороны, распознают повреждения, а с другой стороны, запускают каскад сигнальных событий, называемый ответом на повреждение ДНК (Shmulevich, Krizhanovsky, 2021).
Важнейшим участником этих комплексов является киназа ATM, которая связывается с сайтами повреждения и способствует фосфорилированию гистона H2AX, фланкирующего эти сайты (Hernandez-Segura et al., 2018). И действительно, при помощи иммунофлуоресцентного анализа мы обнаружили фокусы повреждения ДНК, содержащие фосфорилированные ATM и H2AX в обоих типах старых эСК (рис. 2). Считается, что именно повреждения ДНК, которые накапливаются в старых клетках, приводят к постоянной активации ответа на повреждения ДНК и поддержанию необратимого блока клеточного цикла (Hernandez-Segura et al., 2018). Тем не менее вопрос, что является причиной формирования фокусов повреждения ДНК в стареющих эСК и почему эти повреждения не репарируются, остается открытым.
Рис. 2.
Накопление повреждений ДНК при старении эСК. а – Иммунофлуоресцентный анализ внутриклеточной локализации фосфорилированных форм киназы АТМ (р-АТМ: красный цвет, стрелки указывают внутриядерную локализацию белка) и гистона H2AX (p-H2AX: зеленый цвет, стрелки указывают внутриядерную локализацию белка) в контрольных (К), репликативно (РС) и стресс-индуцированно (СИ) состаренных эСК; синий цвет – DAPI, об.: 100×. б – Квантификация фокусов повреждений ДНК в клетках (число фокусов γH2AX на ядро). Данные представлены в виде дискретных значений и их моды (n = 15); различия достоверны (***) при р < 0.001 по сравнению с контролем (тест ANOVA с поправкой по Тьюки).
Из наших предыдущих результатов по изучению преждевременного старения эСК, индуцированного сублетальными дозами перекиси водорода, мы знаем, что значительная часть исходных повреждений ДНК, которые возникают непосредственно в результате повреждающего действия пероксида, практически полностью репарируется, однако в ходе дальнейшего развития старения количество фокусов в стареющих эСК заметно увеличивается (Borodkina et al., 2014). Следовательно, должны существовать дополнительные молекулярные механизмы, обуславливающие возникновение и накопление повреждений ДНК при старении клеток.
Повышение эндогенного уровня АФК и изменение способности нейтрализовать окислители могут способствовать возникновению повреждений ДНК в старых эСК. Наиболее вероятной молекулярной причиной формирования повреждений ДНК в стареющих клетках в настоящее время считают повышенный уровень эндогенных АФК (Borodkina et al., 2014; Davalli et al., 2016; Liguori et al., 2018). Так, показано, что высокий уровень АФК характерен для репликативного, стресс-индуцированного и онкоген-индуцированного типов старения (Lu, Finkel, 2008; Jeong, Cho, 2015; Shmulevich, Krizhanovsky, 2021). В связи с этим, мы в первую очередь сравнили уровень внутриклеточных АФК в молодых клетках и эСК, состаренных репликативно или в результате действия сублетального окислительного стресса. Используя флуоресцентный краситель H2DCF-DA, мы показали, что уровень внутриклеточных АФК в обоих типах старых эСК значительно выше, чем в молодых клетках (рис. 3а). Стоит подчеркнуть, что интенсивность флуоресценции окисленного красителя, отражающая уровень АФК, была сопоставима в обоих типах старых эСК (рис. 3а). Сходный высокий уровень АФК в репликативно- и сресс-индуцированно-состаренных эСК может свидетельствовать в пользу некой универсальности внутриклеточных механизмов генерации АФК при различных типах старения, не зависящих от индуктора.
Рис. 3.
Модуляция уровня внутриклеточных АФК (а), экспрессии генов CAT, SOD1, GPX1(б) и эффективности антиоксидантной защиты (в) в контрольных (К), репликативно (РС) и стресс-индуцированно (СИ) стареющих эСК. а – Интенсивность флуоресценции в клетках DCF после введения АФК-зависимого зонда Н2DCF-DA. б – Экспрессия мРНК генов SOD1, CAT и GPX1; значения экспрессии нормированы на уровень референсного гена GAPDH. в – Динамика временного восстановления генетически кодируемого сенсора перекиси водорода HyPer в контрольных и состаренных эСК после индукции окислительного стресса добавлением 200 мкМ Н2О2 в клеточную среду (серая зона вдоль кривых – доверительные интервалы). г – Константа скорости восстановления сенсора в эСК, определенная при аппроксимации экспоненциальной зависимостью графиков, представленных на рисунке, в. Данные (а–г) представлены в виде средних значений и стандартных отклонений (n = 3); различия достоверны при р < 0.05 (*), р < 0.01 (**) или р < 0.001 (***) по сравнению с контролем: а, г, – тест ANOVA с поправкой по Тьюки, б – t-критерий Стьюдента.
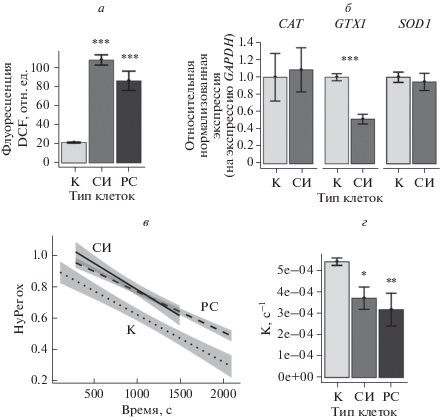
Основным источником повышенного уровня АФК в стареющих клетках считаются митохондрии (Kaplon et al., 2013; Korolchuk et al., 2017). Согласно нашим предыдущим результатам и данным из литературы, старение клеток, и в частности эСК, сопровождается дисфункцией митохондрий и нарушением окислительного фосфорилирования, что и приводит к росту уровня внутриклеточных АФК (Borodkina et al., 2014, Kwon et al., 2019).
Повышенная генерация в результате нарушенной работы митохондрий может оказаться не единственным условием для столь значительного накопления АФК в стареющих эСК. Хорошо известно, что в клетке функционируют различные системы антиоксидантоной защиты, которые в норме должны нейтрализовать избыток АФК. В связи с этим, мы оценили уровень экспрессии ряда генов ферментов антиоксидантной защиты, включая ген CAT, SOD1 и GPX1, в молодых и старых эСК. Оказалось, что уровни экспрессии CAT и SOD1 практически не изменялись, а уровень GPX1 был заметно ниже в старых эСК по сравнению с молодыми клетками (рис. 3б).
Стоит отметить, что данные из литературы, касающиеся эффективности работы антиоксидантных систем при старении, весьма противоречивы. Так, например, было показано значительное снижение экспрессии CAT и глутатионтрансферазы в сателитных клетках, полученных от пожилых доноров, по сравнению с таковой в клетках молодых доноров (Fulle et al., 2005). С другой стороны, в клетках, полученных от пациентов с синдромом Вернера (генетического заболевания, характеризующегося преждевременным старением), не было выявлено никаких дефектов в системе антиоксидантной защиты (Poot, 1991). Результаты сравнительно недавней работы, в которой оценивали уровень экспрессии и активность работы ферментов антиокисдантной защиты в молодых и репликативно старых миофибробластах, также свидетельствуют об отсутствии значимых различий уровней экспрессии SOD1, CAT и GPX1 в молодых и старых клетках (Khor et al., 2017). Одновременно с этим показано, что в эндотелиальных прогениторных клетках, полученных от пожилых доноров, экспрессия SOD1 и CAT не изменяется, тогда как экспрессия и активность GPX1 существенно снижена по сравнению с клетками молодых доноров (He et al., 2009). Согласно данным этих авторов, такое изменение экспрессии и активности GPX1 приводило тому, что клетки пожилых доноров хуже справлялись с окислительным воздействием, чем клетки молодых доноров (He et al., 2009).
Мы также предположили, что сниженная экспрессия GPX1 в стареющих эСК может приводить к снижению эффективности систем антиоксидантной защиты клеток. Для проверки этого предположения мы применили подход, основанный на использовании генетически кодируемого сенсора перекиси водорода HyPer (Belousov et al., 2006). HyPer является химерным белком, который получен путем внедрения пермутированной формы желтого флуоресцентного белка (cpYFP) в регуляторный домен OxyR бактериального транскрипционного фактора, чувствительного к H2O2. Под действием H2O2 домен OxyR претерпевает конформационные изменения, которые влияют на структуру cpYFP и приводят к изменению его флуоресцентных свойств. Отслеживая флуоресцентный сигнал HyPer в клетках, подвергнутых Н2О2-индуцированному окислительному воздействию, можно оценивать, как быстро восстанавливается сенсор и, следовательно, как быстро клетки справляются с окислительным стрессом и элиминируют экзогенный пероксид (Malinouski et al., 2011).
В настоящей работе мы использовали проточную цитометрию для анализа реакции на экзогенный пероксид молодых и состаренных эСК, экспрессирующих HyPer в клеточной цитоплазме (Lyublinskaya et al., 2018). Оказалось, что в состаренных эСК скорость восстановления HyPer была существенно замедлена по сравнению с молодыми клетками (рис. 3в, г). Полученные результаты могут свидетельствовать как о замедлении процесса элиминации внутриклеточных окислителей, так и о замедлении процесса восстановления окисленных белков в стареющих клетках, что вкупе или по отдельности приводит к снижению эффективности их систем антиоксидантной защиты.
Интересно отметить, что обе использованные нами модели клеточного старения характеризовались одинаково низкими скоростями восстановления сенсора HyPer, что подтверждает наше предположение о схожем функционально-метаболическом редокс-статусе эСК (их одинаково низкой способности противостоять окислительным воздействиям) при развитии репликативного и стресс-индуцированного старения.
Суммируя результаты, полученные в этой части работы, можно предполагать следующий сценарий возникновения повреждений ДНК в ходе репликативного и стресс-индуцированного старения эСК. Дисфункция митохондрий приводит к повышенной генерации АФК, однако несмотря на увеличение уровня внутриклеточных АФК, экспрессия генов ферментов антиоксидантной защиты не возрастает, а остается неизменной или даже падает, что коррелирует со снижением эффективности работы защитных систем. Это способствует накоплению окислителей, которые, в свою очередь, и могут приводить к повреждению ДНК в ходе развития старения эСК. Соответственно, накопление внутриклеточных АФК может негативно сказываться на геномной стабильности стареющих клеток. И действительно, в одной из работ было показано, что именно АФК, накапливающиеся в стареющих клетках, могут выступать в качестве тумор-промотирующих факторов, которые способствуют переходу стареющих клеток в трансформированные тумурогенные клетки (Gosselin et al., 2009).
Снижение эффективности репарации может способствовать накоплению повреждений ДНК в стареющих эСК. Выше мы постарались разобраться с тем, что может обуславливать возникновение повреждений ДНК в стареющих эСК, в этой части мы постараемся выяснить, что же может способствовать накоплению этих повреждений в процессе клеточного старения. В отличие от большинства макромолекул в клетке, которые в случае повреждения достаточно быстро удаляются при помощи различных механизмов деградации и заменяются вновь синтезированными, ДНК при повреждении не может быть деградирована и синтезирована вновь, поскольку именно она и является матрицей практически для всех синтетических процессов (Yousefzadeh et al., 2021). В связи с этим в клетке постоянно функционируют механизмы эффективной репарации различных повреждений ДНК, включая эксцизионную репарацию оснований (base excision repair, BER), эксцизионную репарацию нуклеотидов (nucleotide excision repair, NER), репарацию ошибочно спаренных нуклеотидов (mismatch repair, MMR), гомологичную рекомбинацию (homologous recombination, HR) и негомологичное соединение концов (non-homologous end joining, NHEJ). Такое многообразие систем репарации ДНК обусловлено необходимостью коррекции самых различных повреждений, например поперечных сшивок, одноцепочечных и двуцепочечных разрывов ДНК и так далее, которые могут возникать в клетках как спонтанно, так и в ответ на различные стрессы (Yousefzadeh et al., 2021).
Мы предположили, что одной из причин накопления повреждений ДНК в стареющих эСК как раз может оказаться снижение эффективности работы систем, обеспечивающих репарацию ДНК. Для проверки этого предположения мы использовали данные РНК-секвенирования образцов молодых и старых эСК (идентификационный номер в базе данных Gene Expression Omnibus – GSE160702). Функциональный анализ транскриптомных данных в категориях биологических процессов и путей баз данных GO и KEGG Pathway однозначно свидетельствует о том, что активность процессов репарации ДНК, репарации двуцепочечных разрывов ДНК (DSBs), и в частности путей BER, NER, MMR, HR, NHEJ и Fanconi anemia pathway подавлена в стареющих эСК (рис. 4а, б).
Рис. 4.
Эффективность репарации ДНК снижена в стареющих эСК. Анализ GSEA в терминах баз данных GO:BP (а) и KEGG Pathway (б).
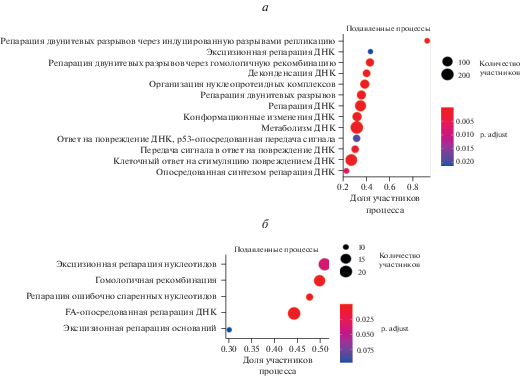
Существенное снижение эффективности работы всех систем репарации в стареющих эСК хорошо согласуется с результатами, недавно описанными для других моделей клеточного старения, включая онкоген-индуцированное старение эпителиальных клеток молочной железы, репликативное старение эндотелиальных клеток, этопозид-индуцированное старение фибробластов WI38 и Ras-индуцированное старение фибробластов IMR90 (Collin et al., 2018). При помощи биоинформатического анализа данных РНК-секвенирования авторы обнаружили подавление экспрессии генов, вовлеченных в NER, BER, MMR, HR во всех типах стареющих клеток (Collin et al., 2018). Таким образом, можно сделать вывод о том, что снижение эффективности репарации ДНК, является общей чертой для различных типов стареющих клеток, в том числе и для стареющих эСК.
Для валидации данных биоинформатического анализа мы сравнили экспериментально функционирование систем репарации в молодых и старых эСК на примере репарации DSBs. В качестве индукторов мы выбрали пероксид водорода (200 мкМ) и ионизирующее γ-излучение (5 Гр), которые, согласно данным из литературы, приводят к возникновению DSBs, и оценивали динамику активации гистона H2AX (Driessens et al., 2009; Vignard et al., 2013). Известно, что динамика образования и элиминации фокусов γH2AX хорошо отражает кинетику репарации DSBs (Sedelnikova et al., 2008). Мы обнаружили, что действие Н2О2, равно как и γ-излучение приводят к формированию фокусов повреждения ДНК и в молодых, и в старых эСК (рис. 5а). Важно, что через 24 ч после стрессовых воздействий в контрольных клетках уровень фосфорилированного гистона H2AX уменьшается практически до базального уровня, тогда как в старых эСК он оставался повышенным, что свидетельствует об уменьшении эффективности репарации ДНК при старении эСК (рис. 5а, б). Эти результаты хорошо подтверждают данные биоинформатического анализа, описанные выше.
Рис. 5.
Анализ динамики образования и элиминации фокусов γH2AX в контрольных (К) и стресс-индуцированно (СИ) стареющих эСК после окислительного стресса или ионизирующего облучения. К и СИ-состаренные эСК подвергали окислительному воздействию (200 мкМ, 1 ч) или ионизирующему облучению (5 Гр) и в указанные временные точки анализировали фосфорилирование γH2AX методами иммуноблотинга (а) или иммунофлуоресценции (б; красный и синий цвет – соответственно p-H2AX и DAPI, об.: 40×). В качестве контроля нагрузки использовали GAPDH.
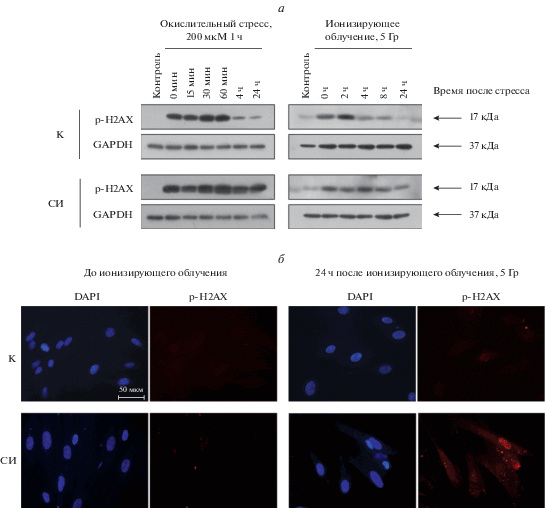
В литературе также есть свидетельства, что в молодых пролиферирующих клетках повреждения ДНК эффективно репарируются за счет BER, NER и HR, а в клетках, полученных от пожилых доноров, эти системы репарации работают значительно менее эффективно (Dizdaroglu, 2012; Pole et al., 2016). Более того, экспериментально установлено, что подавление экспрессии какого-либо из генов, участвующих в репарации, может само по себе приводить к клеточному старению, а оверэкспрессия этих генов, наоборот, в некоторых случаях может ингибировать старение (Mao et al., 2012; Sedic et al., 2015; Collin et al., 2018). Таким образом, снижение эффективности репарации может обуславливать накопление повреждений ДНК в стареющих эСК. Важно, что такие нерепарируемые повреждения ДНК могут крайне негативно сказываться на геномной стабильности стареющих клеток. Яркое подтверждение этого предположения было описано для стареющих эпителиальных клеток (Nassour et al., 2016). Так, было показано, что накопление нерепарируемых повреждений ДНК может обуславливать возвращение пролиферативной активности в эпителиальных клетках, достигших состояния репликативного старения, и, соответственно, может являться первым шагом к злокачественным новообразованиям (Nassour et al., 2016).
По результатам проведенного нами исследования можно сделать следующее заключение. Процесс старения эСК сопровождается повышением уровня эндогенных АФК, в частности за счет неэффективной антиоксидантной защиты; АФК могут приводить к возникновению новых повреждений ДНК, а низкая эффективность репарации способствует накоплению образующихся повреждений, что, в конечном итоге, может привести к геномной нестабильности стареющих клеток.
Список литературы
Дерябин П.И., Бородкина А.В., Никольский Н.Н., Бурова Е.Б. 2015. Взаимное влияние р53/р21/Rb и МАР-киназных сигнальных путей в эндометриальных стволовых клетках человека в условиях окислительного стресса. Цитология. Т. 57. № 11. С. 788. (Deryabin P.I., Borodkina A.V., Nikolsky N.N., Burova E.B. 2015. Relationship between p53/p21/rb and mapk signaling pathways in human endometrium-derived stem cells under oxidative stress. Tsitologiia. V. 57. P. 788.)
Земелько В.И., Гринчук Т.М., Домнина А.П., Арцыбашева И.В., Зенин В.В., Кирсанов А.А., Бичевая Н.К., Корсак В.С., Никольский Н.Н. 2011. Мультипотентные мезенхимные стволовые клетки десквамированного эндометрия. Выделение, характеристика и использование в качестве фидерного слоя для культивирования эмбриональных стволовых линий человека. Цитология. Т. 53. № 12. С. 919. (Zemelko V.I., Grinchuk T.M., Domnina A.P., Artzibasheva I.V., Zenin V.V., Kirsanov A.A., Bichevaia N.K., Korsak V.S., Nikolsky N.N. 2011. Multipotent mesenchymal stem cells of desquamated endometrium: Isolation, characterization, and application as a feeder layer for maintenance of human embryonic stem cells. Tsitologiia. V. 53. P. 919.)
Alimirah F., Pulido T., Valdovinos A., Alptekin S., Chang E., Jones E., Diaz D.A., Flores J., Velarde M.C., Demaria M., Davalos A.R., Wiley C.D., Limbad C., Desprez P.Y., Campisi J. 2020. Cellular senescence promotes skin carcinogenesis through p38MAPK and p44/42MAPK signaling. Cancer Res. V. 80. P. 3606.
Baker D.J., Childs B.G., Durik M., Wijers M.E., Sieben C.J., Zhong J., Saltness R.A., Jeganathan K.B., Verzosa G.C., Pezeshki A., Khazaie K., Miller J.D., van Deursen J.M. 2016. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. V. 530. P. 184.
Belousov V.V., Fradkov A.F., Lukyanov K.A., Staroverov D.B., Shakhbazov K.S., Terskikh A.V., Lukyanov S. 2006. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Met. V. 3. P. 281.
Borodkina A., Shatrova A., Abushik P., Nikolsky N., Burova E. 2014. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging. V. 6. P. 481.
Chandra A., Lagnado A.B., Farr J.N., Monroe D.G., Park S., Hachfeld C., Tchkonia T., Kirkland J.L., Khosla S., Passos J.F., Pignolo R.J. 2020. Targeted reduction of senescent cell burden alleviates focal Radiotherapy-Related bone loss. J. Bone Miner. Res. V. 35. P. 1119.
Chinta S.J., Woods G., Demaria M., Rane A., Zou Y., McQuade A., Rajagopalan S., Limbad C., Madden D.T., Campisi J., Andersen J.K. 2018. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. V. 22. P. 930.
Collin G., Huna A., Warnier M., Flaman J.M., Bernard D. 2018. Transcriptional repression of DNA repair genes is a hallmark and a cause of cellular senescence. Cell Death Dis. V. 9. P. 259.
Coutelier H., Xu Z., Morisse M.C., Lhuillier-Akakpo M., Pelet S., Charvin G., Dubrana K., Teixeira M.T. 2018. Adaptation to DNA damage checkpoint in senescent telomerase-negative cells promotes genome instability. Genes Dev. V. 32. P. 1499.
Davalli P., Mitic T., Caporali A., Lauriola A., D’Arca D. 2016. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid. Med. Cell. Longev. 2016. P. 3565127. https://doi.org/10.1155/2016/3565127
Deryabin P., Domnina A., Gorelova I., Rulev M., Petrosyan M., Nikolsky N., Borodkina A. 2021. “All-In-One” genetic tool assessing endometrial receptivity for personalized screening of female sex steroid hormones. Front. Cell Dev. Biol. 9. P.624053. https://doi.org/10.3389/fcell.2021.624053
Dizdaroglu M. 2012. Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Lett. V. 327. P. 26.
Driessens N., Versteyhe S., Ghaddhab C., Burniat A., De Deken X., Van Sande J., Dumont J.E., Miot F., Corvilain B. 2009. Hydrogen peroxide induces DNA single- and double-strand breaks in thyroid cells and is therefore a potential mutagen for this organ. Endocr. Relat. Cancer. V. 16. P. 845.
Fulle S., Di Donna S., Puglielli C., Pietrangelo T., Beccafico S., Bellomo R., Protasi F., Fanò G. 2005. Age-dependent imbalance of the antioxidative system in human satellite cells. Exp. Gerontol. V. 40. P. 189.
Gosselin K., Martien S., Pourtier A., Vercamer C., Ostoich P., Morat L., Sabatier L., Duprez L., T’kint de Roodenbeke C., Gilson E., Malaquin N., Wernert N., Slijepcevic P., Ashtari M., Chelli F., et al. 2009. Senescence-associated oxidative DNA damage promotes the generation of neoplastic cells. Cancer Res. V. 69. P. 7917.
Graziano S., Gonzalo S. 2017. Mechanisms of oncogene-induced genomic instability. Biophys. Chem. V. 225. P. 49.
Griukova A., Deryabin P., Shatrova A., Burova E., Severino V., Farina A., Nikolsky N., Borodkina A. 2019. Molecular basis of senescence transmitting in the population of human endometrial stromal cells. Aging. V. 11. P. 9912.
Hayflick L. 2002. Handbook of the biology of aging. Amsterdam: Elsevier. 416 pp.
He T., Joyner M.J, Katusic Z.S. 2009. Aging decreases expression and activity of glutathione peroxidase-1 in human endothelial progenitor cells. Microvasc. Res. V. 78. P. 447.
Hernandez-Segura A., Nehme J., Demaria M. 2018. Hallmarks of cellular senescence. Trends Cell Biol. V. 28. P. 436.
Jeong S.G., Cho G.W. 2015. Endogenous ROS levels are increased in replicative senescence in human bone marrow mesenchymal stromal cells. Biochem. Biophys. Res. Commun. V. 460. P.971.
Jeyapalan J.C., Ferreira M., Sedivy J.M., Herbig U. 2007. Accumulation of senescent cells in Mitotic tissue of aging primates. Mech. Ageing Dev. V. 128. P. 36
Kale A., Sharma A., Stolzing A. Desprez P-Y., Campisi J. 2020. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing. 17. https://doi.org/10.1186/s12979-020-00187-9
Kaplon J., Zheng L., Meissl K., Chaneton B., Selivanov V.A., Mackay G., van der Burg S.H., Verdegaal E.M., Cascante M., Shlomi T., Gottlieb E., Peeper D.S. 2013. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. V. 498. P. 109.
Khor S.C., Ngah W.Z.W., Yusof Y.A.M., Karim N.A., Makpol S. 2017. Tocotrienol-rich fraction ameliorates antioxidant defense mechanisms and improves replicative senescence-associated oxidative stress in human myoblasts. Oxid. Med. Cell. Longev. 2017. P. 3868305. https://doi.org/10.1155/2017/3868305
Kirkland J.L., Tchkonia T. 2020. Senolytic drugs: From discovery to translation. J. Intern. Med. V. 288. P. 518.
Kirkwood T.B. 2005. Understanding the odd science of aging. Cell. V. 120. P. 437.
Korolchuk V.I., Miwa S., Carroll B., von Zglinicki T. 2017. Mitochondria in cell senescence: Is mitophagy the weakest link? EbioMedicine. V. 21. P. 7.
Kwon S.M., Hong S.M., Lee Y.K., Min S., Yoon G. 2019. Metabolic features and regulation in cell senescence. BMB Rep. V. 52. P. 5.
Levine M.E. 2013. Modeling the rate of senescence: can estimated biological age predict mortality more accurately than chronological age? J. Gerontol. A Biol. Sci. Med. Sci. V. 68. P. 667.
Li X., Ploner A., Wang Y., Magnusson P.K., Reynolds C., Finkel D., Pedersen N.L., Jylhävä J., Hägg S. 2020. Longitudinal trajectories, correlations and mortality associations of nine biological ages across 20-years follow-up. Elife. 9: e51507. https://doi.org/10.7554/eLife.51507
Liguori I., Russo G., Curcio F., Bulli G., Aran L., Morte D.D., Gargiulo G., Testa G., Cacciatore F., Bonaduce D., Abete P. 2018. Oxidative stress, aging, and diseases. Clin. Interv. Aging. V. 13. P. 757.
Liu Y., Sanoff H.K., Cho H., Burd C.E., Torrice C., Ibrahim J.G., Thomas N.E., Sharpless N.E. 2009. Expression of p16 (INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell. V. 8. P. 439.
Lu T., Finkel T. 2008. Free radicals and senescence. Exp. Cell Res. V. 314. P. 1918.
Lyublinskaya O.G., Antonov S.A., Gorokhovtsev S.G., Pugovkina N.A., Kornienko J.S., Ivanova J.S., Shatrova A.N., Aksenov N.D., Zenin V.V., Nikolsky N.N. 2018. Flow cytometric HyPer-based assay for hydrogen peroxide. Free Rad. Biol. Med. V. 128. P. 40.
Mao Z., Tian X., Van Meter M., Ke Z., Gorbunova V., Seluanov A. 2012. Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. PNAS. V. 109. P. 11800.
Malinouski M., Zhou Y., Belousov V.V., Hatfield D.L., Gladyshev V.N. 2011. Hydrogen peroxide probes directed to different cellular compartments. PLoS One. 6: e14564. https://doi.org/10.1371/journal.pone.0014564
Nassour J., Martien S., Martin N., Deruy E., Tomellini E., Malaquin N., Bouali F., Sabatier L., Wernert N., Pinte S., Gilson E., Pourtier A., Pluquet O., Abbadie C. 2016. Defective DNA single-strand break repair is responsible for senescence and neoplastic escape of epithelial cells. Nat. Commun. V. 7. P. 10399.
Roos C.M., Zhang B., Palmer A.K., Ogrodnik M.B., Pirtskhalava T., Thalji N.M., Hagler M., Jurk D., Smith L.A., Casaclang Verzosa G., Zhu Y., Schafer M.J., Tchkonia T., Kirkland J.L., Miller J.D. 2016. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. V. 15. P. 973.
Ovadya Y., Krizhanovsky V. 2014. Senescent cells: SASPected drivers of age-related pathologies. Biogerontol. V. 15. P. 627.
Pole A., Dimri M., Dimri G.P. 2016. Oxidative stress, cellular senescence and ageing. AIMS Mol. Sci. V. P. 300.
Poot M. 1991. Oxidants and antioxidants in proliferative senescence. Mutation Res./DNAging. V. 256. P.177.
Prata L.G.P.L., Ovsyannikova I.G., Tchkonia T., Kirkland J.L. 2018. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 40:101275. https://doi.org/10.1016/j.smim.2019.04.003
Sedelnikova O.A., Horikawa I., Redon C., Nakamura A., Zimonjic D.B., Popescu N.C.,Bonner W.M. 2008. Delayed kinetics of DNA double–strand break processing in normal and pathological aging. Aging Cell. V. 7. P. 89.
Sedic M., Skibinski A., Brown N., Gallardo M., Mulligan P., Martinez P., Keller P.J., Glover E., Richardson A.L., Cowan J., Toland A.E., Ravichandran K., Riethman H., Naber S.P., Näär A.M., et al. 2015. Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nat. Commun. V. 6. P. 7505.
Shmulevich R., Krizhanovsky V. 2021. Cell Senescence, DNA damage, and metabolism. Antioxid. Redox Signal. V. 34. P. 324.
Takai H., Smogorzewska A., de Lange T. 2003. DNA damage foci at dysfunctional telomeres. Curr. Biol. V. 13. P. 1549.
The Tabula Muris Consortium. 2020. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature. V. 583. P. 590.
Vignard J., Mirey G., Salles B. 2013. Ionizing-radiation induced DNA double-strand breaks: a direct and indirect lighting up. Radiother. Oncol. V. 108. P. 362.
Yousefzadeh M., Henpita C., Vyas R., Soto-Palma C., Robbins P., Niedernhofer L. 2021. DNA damage-how and why we age?. Elife. 10: e62852. https://doi.org/10.7554/eLife.62852
Zampetidis C., Galanos P., Angelopoulou A., Zhu Y., Karamitros T., Polyzou A., Mourkioti I., Lagopati N., Mirzazadeh R., Polyzos A., Garnerone S., Gusmao E.G., Sofiadis K., Pefani D.E., Demaria M., et al. 2021. Genomic instability is an early event driving chromatin reorganization and escape from oncogene-induced senescence. BioRxiv preprint. https://doi.org/10.1101/2020.12.20.423639
Zhang P., Kishimoto Y., Grammatikakis I., Gottimukkala K., Cutler R.G., Zhang S., Abdelmohsen K., Bohr V.A., Misra Sen J., Gorospe M., Mattson M.P. 2019. Senolytic therapy alleviates Ab-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. V. 22. P. 719.
Zhu Y., Armstrong J.L., Tchkonia T., Kirkland J.L. 2014. Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr. Opin. Clin. Nutr. Metab. Care. V. 17. P. 324.
Дополнительные материалы отсутствуют.