

Цитология, 2021, T. 63, № 6, стр. 548-556
Изменение числа CD38 и Cx43-иммунопозитивных клеток в нейроваскулярной единице головного мозга при экспериментальной болезни Альцгеймера
Е. Д. Хилажева 1, *, А. И. Мосягина 1, А. В. Моргун 1, Н. А. Малиновская 1, Я. В. Горина 1, Е. В. Харитонова 1, О. Л. Лопатина 1, А. Б. Салмина 1, 2
1 Научно-исследовательский институт молекулярной медицины и патобиохимии Красноярского государственного медицинского университета им. проф. В.Ф. Войно-Ясенецкого
660022 Красноярск, Россия
2 Отдел исследований мозга Научного центра неврологии
125367 Москва, Россия
* E-mail: elena.hilazheva@mail.ru
Поступила в редакцию 06.08.2021
После доработки 19.08.2021
Принята к публикации 20.08.2021
Аннотация
Известно, что митохондриальная дисфункция может являться триггером или сопутствующим механизмом при развитии болезни Альцгеймера. Также известно, что восстановление внутриклеточной митохондриальной активности нейронов и церебральных эндотелиоцитов возможно за счет переноса неповрежденных митохондрий из других клеток головного мозга, в частности, астроцитов, причем этот перенос митохондрий опосредован белком CD38 и функционально связанным с ним коннексином 43 (Cx43). Отсюда следует, что указанные молекулы перспективны для изучения как в отношении механизмов развития нейродегенерации, так и в отношении возможной модуляции их активности для коррекции неврологического дефицита. Цель настоящей работы заключалась в исследовании изменения числа CD38- и Cx43-иммунопозитивных клеток в составе нейроваскулярной единицы и гематоэнцефалического барьера (ГЭБ) головного мозга при экспериментальной болезни Альцгеймера. Нами показано, что токсическое действие бета-амилоида приводит к значительному увеличению числа CD38- и Cx43-позитивных клеток как в гиппокампе животных in vivo, так и в составе модели ГЭБ in vitro. Также нами показано, что культивирование изолированных астроцитов в присутствии бета-амилоида приводит к увеличению содержания Cx43 в клетках, причем проницаемость этих полуканалов достоверно увеличивается.
Болезнь Альцгеймера (БА) является одной из наиболее распространенных нейродегенеративных болезней и находится в фокусе многочисленных исследований уже на протяжении многих лет. Однако многие клеточно-молекулярные звенья патогенеза БА до сих пор остаются малоизученными (Compagnoni et al., 2020), что крайне затрудняет разработку эффективных и безопасных методов профилактики и лечения.
В последнее время в исследованиях процессов нейродегенерации особое внимание привлекают нарушения митохондриальной динамики в клетках нейрональной, астроглиальной и эндотелиальной природы. Известно, что в развитии нейродегенеративных процессов задействованы различные механизмы, связанные с митохондриями, в частности дефекты энергопродукции, мутации митохондриальной ДНК, изменения динамики митохондрий и аберрантный гомеостаз ионов Ca2+ (Franco-Iborra et al., 2018).
Показано, что митохондриальная дисфункция может также являться триггером или сопутствующим механизмом при развитии БА (Swerdlow, 2018). К сожалению, к настоящему времени не разработаны способы фармакологической модуляции для восстанавления аберрантной активности митохондрий и митохондриальной динамики в клетках головного мозга. Однако некоторые исследования показывают, что восстановление митохондриальной активности нейронов и церебральных эндотелиоцитов возможно с использованием репаративного потенциала других клеток, входящих в состав нейроваскулярной единицы (НВЕ) и гематоэнцефалического барьера (ГЭБ), например, астроцитов. В частности, было показано, что астроциты могут выступать в качестве доноров неповрежденных митохондрий при экспериментальной ишемии головного мозга, причем такой транспорт митохондрий связан с активностью CD38 в астроцитах, а также туннелирующих межклеточных нанотрубочек и экзосом (микровезикул) (Hayakawa et al., 2016).
CD38 – это трансмембранный гликопротеин, компонент клеточных сигнальных систем, сопряженных в клетках нервной системы с рецепторами многих нейротрансмиттеров. CD38 способен проявлять 2 типа ферментативной активности: АДФ-рибозилциклазную и цАДФ-рибозилгидролазную. Субстратами его являются НАД+, его фосфат НАДФ, а также цАДФ-рибоза. Кроме того, благодаря олигомерной структуре, CD38 может проявлять свою активность в качестве каталитически активного транспортера, ответственного за генерацию и вход цАДФ-рибозы в клетку. цАДФ-рибоза, генерируемая с помощью CD38, играет важную роль во многих биологических процессах в головном мозге и других тканях и органах. Примечательно, что такую же транспортную функциональную нагрузку дополнительно может нести и экспрессируемый в цитоплазматической мембране коннексин 43 (Cx43) (Салмина и др., 2012).
Известно, что Cx43 функционально связан с CD38, выполняя функцию канала для поступления НАД+ из цитозоля во внеклеточное пространство к активному сайту CD38, либо для поступления цАДФ-рибозы, синтезированной внеклеточно, в цитозоль для связывания с рианодиновыми рецепторами и индукции высвобождения кальция из внутриклеточных депо (Bruzzone et al., 2001; Song et al., 2011). Помимо этого, молекулы Cx43, находящиеся на поверхности астроцитов, обеспечивают формирование астроглиального синцития и могут служить в качестве каналов для секреции глиотрансмиттеров и ионов (Osipova et al., 2018). Показано, что в некоторых типах гликолитически активных клеток Cx43-опосредованный механизм секреции и (или) транспорта лактата достаточно эффективен (Dovmark et al., 2017), что позволяет предположить его наличие в астроцитах головного мозга.
Интересно, что в условиях подавления гликолиза и окислительного фосфорилирования астроциты снижают коммуникацию за счет Cx43-опосредованных межклеточных контактов (щелевых), но увеличивают активность Cx43-полуканалов, высвобождающих во внеклеточное пространство ионы и метаболиты (Contreras et al., 2002). Кроме того, несмотря на то, что экспрессия гена Cx43 долгое время считалась прерогативой астроцитов, существуют экспериментальные данные, демонстрирующие наличие этой молекулы в клетках нейрональной и эндотелиальной природы (Liu et al., 2012; Johnson et al., 2018), что может обеспечивать механизмы эффективной межклеточной коммуникации в пределах НВЕ головного мозга.
Вместе с тем, существуют экспериментальные данные, демонстрирующие повышение уровня содержания и активности белков CD38 и Cx43 при ряде нейродегенеративных заболеваний, что может говорить об их вкладе в развитие нейровоспаления и нейродегенерации (Camacho-Pereira, 2016; Yi et al., 2016).
В соответствии с вышеперечисленными обстоятельствами, изучение CD38 и сопряженного с ним Cx43 является перспективным как в отношении исследования механизмов развития нейродегенерации, в том числе альцгеймеровского типа, так и в отношении возможной модуляции процессов трансфера митохондрий в клетках НВЕ/ГЭБ для коррекции неврологического дефицита.
Таким образом, целью настоящей работы стало исследование изменения числа CD38 и Cx43-иммунопозитивных клеток в составе НВЕ/ГЭБ головного мозга при экспериментальной БА.
МАТЕРИАЛ И МЕТОДИКА
Объекты исследования. Животные. Для экспериментов in vivo использовали мышей-самцов линии C57BL/6 в возрасте 4 мес. Животным с экспериментальной БА вводили бета-амилоид (Aβ) в CA1 зону гиппокампа. Контрольную группу составляли ложно-оперированные животные после введения в гиппокамп растворителя для Aβ – фосфатно-солевого буферного раствора (PBS). Всего использовали 20 животных (n = 20). В экспериментах in vitro для получения первичных культур клеток использовали крыс линии Wistar. Общее число животных n = 14.
Животных содержали в клетках со свободным доступом к воде и корму при постоянной температуре 21 ± 1°С и регулярном световом цикле 12 ч день и 12 ч ночь. Исследования на животных проводили в соответствии с соблюдением принципов гуманности, изложенных в Директиве Европейского сообщества (2010/63/ЕС).
Клетки. Использовали первично-выделенные культуры клеток церебрального эндотелия, астроцитов и нейросфер (НСФ). Из НСФ путем направленной дифференцировки получали сокультуру астроцитов и нейронов. Контрольную группу составляли интактные клетки, экспериментальную – клетки, культивируемые в течение 1 сут в присутствии Аβ в конечной концентрации 100 нМ.
Моделирование нейродегенерации in vivo. Моделирование БА осуществляли интрагиппокампальным введением Aβ билатерально по 1 мкл в зону СА1 по стереотаксическим координатам ML = ±1.3 мм, AP = 2.0 мм, DV = –1.9 мм (Epelbaum et al., 2015). Aβ (Sigma-Aldrich, США) перед введением животным растворяли в PBS до концентрации 50 мкM с последующей агрегацией в термостате при 37°С в течение 7 сут. Оценку признаков БА проводили, начиная с 10-х сут (Sipos et al., 2007) после оперативного вмешательства. Модель БА верифицировали с помощью окрашивания тиофлавином S. После введения амилоида в ткани головного мозга наблюдали флуоресцирующие амилоидные бляшки зеленого цвета.
Приготовление срезов и иммуногистохимия. Для фиксации тканей осуществляли транскардиальную перфузию 4%-ным параформальдегидом с последующим забором головного мозга. Далее изолированный мозг фиксировали в 10%-ном нейтральном забуференном формалине, после чего погружали в 20%-ный раствор сахарозы.
С помощью микротома Microm HM 650 (Thermo Scientific, США) изготавливали срезы толщиной 50 мкм. Наличие в образцах исследуемых маркеров определяли методом непрямой иммуногистохимии для свободно плавающих срезов (Encinas, Enikolopov, 2008).
После промывки в PBS срезы блокировали 3%-ным козьим сывороточным альбумином в PBS и 1%-ном Тритоне X-100 в течение 1 ч при комнатной температуре с последующим инкубированием в течение ночи с первичными антителами в разведении 1 : 1000 в 3%-ном BSA в PBS и 0.2%-ном Тритоне X-100 при 4°С. Использовали первичные моноклональные крысиные антитела к CD31 (MBS532307; MyBioSource.com, США), куриные поликлональные антитела к нейронспецифической енолазе (NSE) (GTX85462; GeneTex, США), мышиные моноклональные антитела к глиальному фибриллярному кислому белку (GFAP) (MBS8241552; MyBioSource.com, США), кроличьи поликлональные антитела к Cx43 (MBS9600634; MyBioSource.com, США) и кроличьи поликлональные антитела к CD38 (GTX37752; GeneTex, США). После инкубации с первичными антителами срезы промывали и инкубировали со вторичными антителами в разведении 1 : 1000 в течение 2 ч при комнатной температуре. Использовали вторичные поликлональные антитела (все от Abcam, Великобритания): ослиные, специфичные к кролику, Alexa Fluor 555 (ab150074); козьи, специфичные к курице, Alexa Fluor 647 (ab150171); козьи, специфичные к мыши, Alexa Fluor 647 (ab150115); козьи, специфичные к крысе, Alexa Fluor 647 (ab150159. Изображения получали с помощью конфокального микроскопа Olympus FV 10i (Япония). В срезах головного мозга в НВЕ считали абсолютное число клеток в поле зрения нейрональной (NSE-позитивные), астроглиальной (GFAP-позитивные) и эндотелиальной (CD31-позитивные) природы, имеющих целевые молекулы (Cx43, CD38) на различных уровнях в гиппокампе. Каждый эксперимент проводили в 10 повторностях. Оценивали не менее пяти полей зрения в каждом срезе гиппокампа.
Выделение и культивирование церебральных эндотелиоцитов. Использовали модифицированный протокол (Liu et al., 2013). Выделение мозга с удалением мозговых оболочек и крупных церебральных сосудов осуществляли c помощью роллинга на фильтровальной бумаге. Выделяли кору головного мозга и удаляли крупные сосуды в холодном растворе Хенкса (ПанЭко, Россия). Мелконарезанную кору головного мозга центрифугировали в пробирке объемом 15 мл в течение 3 мин при 150 g и комнатной температуре. Последующие этапы включали:
1) добавление к осадку в двукратном объеме 25%-ной FBS (HyClone, США), тритурирование 25 раз пипеткой 5 мл с последующим центрифугированием гомогената в течение 10 мин при 600 g при комнатной температуре;
2) забор самого нижнего слоя и перенос в новую коническую пробирку 15 мл;
3) повторение этапов тритурирования и центрифугирования три раза, после чего проводили ферментативную обработку пеллета в растворе 0.1%-ной коллагеназы II (ПанЭко, Россия) в течение 35 мин при 37°С с периодическим перемешиванием и дальнейшим центрифугированием в течение 5 мин при 200 g;
4) умеренное ресуспензирование осадка в питательной среде и посев в культуральные флаконы.
Культивирование эндотелиальных клеток осуществляли при 37°С, 5% CO2 в культуральных флаконах, предварительно покрытых 0.1%-ным раствором желатина (Biological Industries, США) в культуральной среде DMEM (ПанЭко, Россия), содержащей 20% FBS (HyClone, США), а также 0.58 мг/мл глутамина, 100 Ед/мл пенициллина и 100 мг/мл стрептомицина (ПанЭко, Россия). Смену среды проводили каждые 3 сут.
Выделение и культивирование нейросфер. Животных декапитировали после охлаждения на льду и производили забор головного мозга. Мозг помещали в ледяной раствор 2%-ной глюкозы в PBS (ПанЭко, Россия). Выделяли интересующие регионы (гиппокамп, стенки боковых желудочков) и иссекали до размеров 1 мм3. После окончания диссекции выделенную ткань переносили в свежий раствор 2%-ной глюкозы в PBS (в конической пробирке 14 мл) на 1 мин. После осаждения ткани удаляли супернатант. Оставшуюся ткань ресуспензировали в 1 мл среды NeuroCult NS-A Proliferation (StemCell, США). Проводили тритурацию ткани (25–30 раз) стерильным пластиковым наконечником до получения однородной суспензии клеток. К полученной суспензии добаляли 1 мл свежей среды NeuroCult NS-A Proliferation. Через 2 мин после осаждения неразделенных кусочков ткани под силой тяжести собирали супернатант и переносили его в новую стерильную пробирку объемом 14 мл. Собранный супернатант центрифугировали при 150 g в течение 5 мин, после чего удаляли супернатант и добавляли 1 мл свежей среды NeuroCult NS-A Proliferation с последующей тритурацией. Полученную клеточную суспензию переносили в культуральные флаконы T-75 см2 с 25 мл среды NeuroCult NS-A Proliferation. Культивирование осуществляли в условиях инкубатора при 5% CO2 и 37°C. Через 24–48 ч после выделения наблюдали образование нейросфер.
Получение культуры астроцитов. Использовали направленную дифференцировку нейросфер в среде Astrocyte Medium (ScienCell, США) следующего состава: базальная средa (Basal Medium, ScienCell, США), 10% FBS (HyClone, США), AGS (ScienCell, США), раствор пенициллина-стрептомицина (ScienCell, США) в конечной концентрации 50 ед./мл (Хилажева и др., 2015). Через 7–9 сут наблюдали образование монослоя астроцитов.
Получение сокультуры астроцитов и нейронов. Нейросферы направленно дифференцировали в астроциты и нейроны. Нейросферы ресуспендировали в культуральной среде NeuroCult NS-A Differentiation Kit (StemCell, США) и засевали в культуральные флаконы T-75 см2. Через 5–7 сут наблюдали образование сокультуры астроцитов и нейронов.
Получение модели ГЭБ in vitro. Для получения модели ГЭБ сокультуру астроцитов и нейронов засевали на дно лунок культурального планшета, затем в лунки устанавливали культуральные вставки (Corning-Costar, США), на которые помещали эндотелиоциты. Соотношение количества эндотелиоцитов, астроцитов и нейронов составляло 1 : 2 : 1 соответственно. Смесь клеток культивировали в среде DMEM (ПанЭко, Россия), содержащей 20% FBS (HyClone, США), а также 0.58 мг/мл глутамина, 100 ЕД/мл пенициллина и 100 мг/мл стрептомицина (все от ПанЭко, Россия) в условиях инкубатора при 37°C и 5% CO2.
Оценка Cx43-опосредованного транспорта в астроцитах в присутствии и в отсутствие Аβ. Астроциты засевали в 12-луночный культуральный планшет по 2.5 × 105 клеток на 1 лунку. После достижения 95% конфлюентности производили замену культуральной среды на среду с Аβ в конечной концентрации 100 нМ. Через 1 сут культивирования в среде с Aβ оценивали Cx43-опосредованный транспорт в астроцитах с использованием красителя Lucifer Yellow (LY), а также иммуноцитохимически с использованием Cx43-антител.
Для оценки Cx43-опосредованной проницаемости астроциты культивировали в среде, содержащей флуоресцентный краситель LY в конечной концентрации 50 мкМ. Через 24 ч среду с LY убирали, клетки промывали раствором PBS и оценивали интенсивность флуоресценции красителя в астроцитах с помощью флуоресцентного микроскопа ZOE (Bio-Rad, США).
Иммуногистохимическое выявление белков CD38 и Cx43 в культурах in vitro. Клетки фиксировали в 4%-ном параформальдегиде в течение 15 мин, после чего отмывали раствором PBS. Наличие в клетках исследуемых маркеров определяли методом непрямой иммуногистохимии. Использовали первичные поликлональные антитела: к белкам плотных контактов ZO1 (козлиные sc-8147; Santa Cruz, США и кроличьи orb11587; Biorbyt, Великобритания); куриные к GFAP (LS-B6299; LSBio, США); мышиные к нейрон-специфичному ядерному белку NeuN (MAB377; Merck Millipore, США); кроличьи к CD38 (LS-C331640; LSBio, США), кроличьи к Cx43 (ab11370; Abcam, США). Первичные антитела использовали в рабочем разведении 1 : 300. Время инкубации составляло 18 ч при 4°С. После инкубации производили 2-х кратную отмывку клеток раствором PBS.
Вторичные поликлональные антитела от Abcam, Великобритания: ослиные, специфичные к мыши, Alexa Fluor 488 (ab150117); ослиные, специфичные к кролику, Alexa Fluor 555 (ab150077) и козьи, специфичные к курице Alexa Fluor 647 (ab150171), а также ослиные, специфичные к козе, Alexa Fluor 488 (A11055; Life Technologies, США). Антитела использовали в разведении 1 : 500, время инкубации составляло 2 ч при 37°С.
Микроскопию клеток осуществляли на флуоресцентном микроскопе ZOE (Bio-Rad, США). Наличие изучаемых маркеров изучали в клетках эндотелия (ZO1-позитивные), нейронах (NeuN-позитивные) и астроцитах (GFAP-позитивные). Каждый эксперимент проводили в 6 повторностях. В каждой экспериментальной лунке не менее, чем в пяти полях зрения, считали долю клеток (%) нейрональной, астроглиальной и эндотелиальной природы, несущих целевые молекулы .
Статистический анализ. Статистический анализ полученных результатов проводили с помощью программ GraphPad Prizm 8.0.1 (версия 8.0, США) и Stаtplus Professional (AnalystSoft Inc, США), сборка 5.9.8.5/Core v.5.9.33 и GraphPad 6.0 (США). Для оценки нормальности распределения использовали критерий Колмoгорова−Смирнова. При отсутствии условий нормальности распределения для сравнения выборок применяли U-критерий Манна–Уитни. Различия принимали значимыми при P ≤ 0.05. Результаты представлены в виде средних значений M и их стандартных отклонений SD.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Мы обнаружили, что у животных с экспериментальной БА регистрируется значимое (Р < 0.05) увеличение числа Cx43-позитивных клеток нейрональной, астроглиальной и эндотелиальной природы по сравнению с контрольной группой (табл. 1). Так, абсолютное число нейронов гиппокампа, несущих Cx43, у мышей с моделью БА оказалось выше почти в 2 раза, чем у контрольных животных, а число Cx43-позитивных астроцитов – выше в 2.5 раза. Также зарегистрировано значимое увеличение числа эндотелиоцитов, несущих Cx43, у животных с БА (табл. 1).
Таблица 1.
Число Cx43- и CD38-позитивных клеток на срезах гиппокампа мышей контрольных и с экспериментальной БА
| Условия | Число клеток, ед. (М ± SD) | ||
|---|---|---|---|
| нейроны | астроциты | эндотелиоциты | |
| Cx43+ | |||
| Контроль | 6.81 ± 1.22 | 2.94 ± 0.39 | 6.63 ± 1.05 |
| БА | 11.27 ± 1.41 | 7.36 ± 0.76 | 8.25 ± 0.93 |
| (Р = 0.0018) | (Р ≤ 0.0001) | (Р = 0.0361) | |
| CD38+ | |||
| Контроль | 8.6 ± 0.84 | 8.73 ± 0.89 | 4.2 ± 1.8 |
| БА | 4.75 ± 0.42 | 8.11 ± 0.81 | 7.1 ± 0.46 |
| (Р = 0.0003) | (Р = 0.0361) | ||
Выявленное однонаправленное увеличение числа Cx43-позитивных клеток может являться результатом подавления метаболической активности клеток нейроваскулярной единицы (гликолиза, митохондриальной дисфункции), а также быть проявлением нейровоспаления в участках аккумуляции Аβ, вызванного активированными астроцитами.
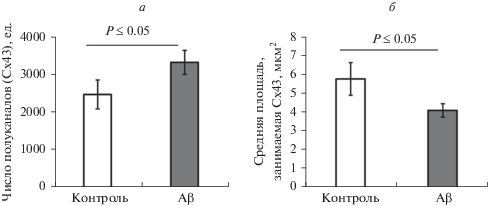
Далее мы провели иммуноцитохимическое исследование содержания Cx43 в астроцитах и изучение проницаемости коннексиновых полуканалов в астроцитах в условиях in vitro с использованием красителя LY. При исследовании содержания Cx43 в астроцитах нами установлено, что число коннексиновых полуканалов в астроцитах при культивировании в присутствии Aβ статистически значимо возрастает по сравнению с контрольной группой (Р ≤ 0.05, рис. 1а). При этом средняя площадь коннексиновых полуканалов в астроцитах экспериментальной группы оказалась в 1.5 раза меньше, чем в интактных астроцитах (Р ≤ 0.05, рис. 1б).
Рис. 1.
Содержание Cx43 в астроцитах in vitro в физиологических условиях в контроле и в присутствии Aβ (100 нМ). а – Число коннексиновых полуканалов, б – средняя площадь сигнала флуоресценции.
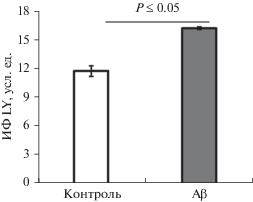
Для оценки Cx43-опосредованной проницаемости астроцитов в культуральную среду вносили флуоресцентный краситель LY и через 24 ч оценивали интенсивность флуоресценции красителя в астроцитах. Известно, что захват этого флуоресцентного красителя из внеклеточного пространства в Cx43+-клетках опосредуется именно активностью Cx43-полуканалов (Lagos-Cabré et al., 2018). В результате исследования нами было выявлено, что в астроцитах, культивированных в присутствии Aβ, интенсивность флуоресценции красителя LY была в 1.5 раза выше, чем в интактных астроцитах (Р ≤ 0.05, рис. 2).
Рис. 2.
Интенсивность захвата красителя Lucifer Yellow астроцитами из среды культивирования in vitro в контрольных условиях и в присутствии 100 нМ Aβ. ИФ – интенсивность флуоресценции.
Таким образом, астроциты гиппокампа демонстрируют увеличение числа Cx43 в ответ на токсическое действие Aβ in vitro, причем проницаемость этих полуканалов значимо увеличивается. С учетом данных о том, что метаболическое повреждение астроцитов вызывает активацию Cx43-полуканалов, эти данные могут быть интерпретированы и как доказательство нарушения процессов продукции энергии в аффектированных астроцитах в результате развития митохондриальной дисфункции, и как свидетельство их активации, необходимой для развития нейровоспаления.
Далее нами исследовано число CD38-позитивных клеток в гиппокампе животных с экспериментальной БА, т.е. клеток, несущих молекулу, потенциально сопряженную (в функциональном аспекте) с Cx43. В результате, было выявлено статистически значимое снижение CD38+-нейронов в гиппокампе головного мозга животных после интрагиппокампального введения Аβ по сравнению с ложно-оперированными животными (Р ≤ 0.05, табл. 2). В клетках эндотелиальной природы, напротив, отмечали двукратное увеличение числа CD38+-клеток в группе с экспериментальной БА относительно группы контроля (Р ≤ 0.05, табл. 1). Однако не было выявлено статистически значимых отличий при сравнении количества CD38+-астроцитов в гиппокампе животных контрольной и экспериментальной групп.
Причиной увеличения числа CD38-позитивных клеток может быть их повреждение, а также дисфункция митохондрий, косвенным подтверждением чего является увеличение содержания Cx43 как маркера метаболического ингибирования. Ранее было показано, что повреждение митохондрий вызывает открытие мегаканалов (MPT) в местах контакта их внутренней и внешней мембран, что вызывает высвобождение в цитозоль НАД+ (выступающего в качестве субстрата для CD38) и белков с проапоптотической активностью (Alano et al., 2007). В этом контексте вполне объяснимо, почему астроциты не демонстрируют значимого увеличения содержания CD38 in vivo:
1) астроциты являются более устойчивыми к повреждению в силу того, что максимально экипированы ферментами гликолиза и могут продуцировать АТФ даже при значимом подавлении активности митохондрий; 2) они имеют минимальное количество митохондрий по сравнению с нейронами и эндотелиоцитами, что позволяет им избегать выраженного окислительного стресса; 3) они имеют высокую концентрацию НАД+, причем преимущественно в цитозоле, тогда как нейроны и эндотелиоциты имеют меньшие уровни НАД+, который, в основном, аккумулируется в митохондриях; 4) они имеют высокий уровень содержания Cx43 и могут благодаря этому регулировать биодоступность НАД+ для других астроцитов в составе астроглиального синцития (Alano et al., 2007).
Далее мы проверили, насколько это справедливо для клеток НВЕ головного мозга при токсическом действии Aβ in vitro. Мы оценили количество Cx43-позитивных и CD38-позитивных клеток эндотелия, нейронов и астроцитов, культивируемых совместно в течение 24 и 48 ч в статической модели ГЭБ in vitro в присутствии и в отсутствие Аβ.
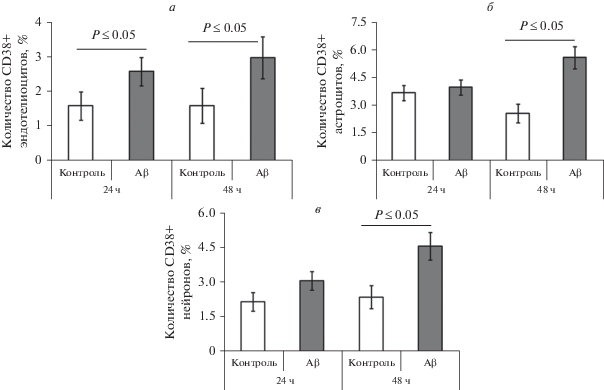
Так, при культивировании клеток ГЭБ в присутствии Aβ через 24 ч было выявлено статистически значимое увеличение количества CD38-позитивных эндотелиоцитов, которое сохранялось в течение 48 ч культивирования (Р ≤ 0.05, рис. 3а). К этому же времени (48 ч) было зафиксировано и статистически значимое увеличение в присутствии Aβ количества астроцитов и нейронов, несущих CD38 (Р ≤ 0.05, рис. 3б, в).
Рис. 3.
Количество CD38-позитивных клеток в модели ГЭБ in vitro при культивировании в контрольных условиях и в присутствии 100 нМ Aβ. а – Эндотелиоциты, б – астроциты, в – нейроны.
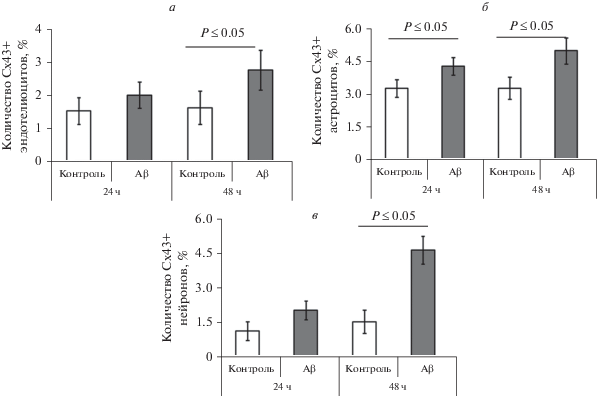
При анализе количества Cx43-позитивных клеток в составе ГЭБ при культивировании в присутствии Aβ было выявлено статистически значимое увеличение числа Cx43+-астроцитов (Р ≤ 0.05, рис. 4б), которое сохранялось на протяжении всего времени наблюдения (24 и 48 ч). Также было зафиксировано увеличение количества Cx43+-эндотелиоцитов и нейронов (48 ч культивирования с Aβ) (Р ≤ 0.05, рис. 4а, в).
Рис. 4.
Количество Cx43-позитивных клеток в модели ГЭБ in vitro при культивировании в контрольных условиях и в присутствии 100 нМ Aβ. а – Эндотелиоциты, б – астроциты, в – нейроны.
Таким образом, нами выявлено, что токсическое действие Aβ in vitro приводит к значительному увеличению количества Cx43-позитивных и CD38-позитивных клеток в составе модели ГЭБ. Это соответствует изменениям, зарегистрированным нами при исследовании действия Aβ in vivo.
Между тем известно, что выраженное увеличение экспрессии CD38 в клетках может приводить к значительному истощению уровня НАД+ (Camacho-Pereira et al., 2016), поэтому увеличение количества CD38-иммунопозитивных нейронов и эндотелиоцитов способно усугублять последствия митохондриальной дисфункции и активировать механизмы клеточной гибели. Снижение уровня НАД+ может быть одной из причин возрастных дисфункций, оказывающих влияние на процесс развития болезни Альцгеймера (Lautrup et al., 2019). Отмечается, что процесс старения сопровождается увеличением экспрессии в клетках CD38, контролирующего уровень НАД+ (Chini, 2009). Известно, что у мышей экспрессия и активность CD38 увеличиваются во время старения, а CD38 вносит значительный вклад в связанное с возрастом истощение НАД+ и митохондриальную дисфункцию, по крайней мере частично за счет ингибирования активности НАД+-зависимых сиртуинов (гистоновых деацетилаз) (Camacho-Pereira et al, 2016). Нокдаун CD38 у мышей линии APP/PS1 уменьшает аккумуляцию Aβ и уровни растворимого Aβ, а также восстанавливает пространственное обучение, возможно, за счет повышения уровня НАД+ в мозге (Blacher et al., 2015). Выявлено (Camacho-Pereira et al., 2016), что экспрессия и активность CD38 при нейродегенеративных заболеваниях возрастают, что является первым косвенным доказательством участия CD38 в нейродегенерации и нейровоспалении.
С другой стороны, в экспериментальной модели гипоксии на клеточных культурах доказано, что CD38 участвует в переносе митохондрий от астроцитов к нейронам после инсульта (Hayakawa et al., 2016), что позволяет рассматривать CD38-экспрессирующие астроциты в качестве клеток-доноров митохондрий для нейронов и эндотелиоцитов при нейродегенерации, индуцированной токсическим действием Aβ.
Также известно, что содержание Cx43 увеличивается в астроцитах, окружающих амилоидные бляшки, как в головном мозге пациентов с БА (Nagy et al., 1996), так и в головном мозге мышей с генетической моделью БА линии APP/PS1 (Yi et al., 2016), тем самым способствуя снижению жизнеспособности нейронов и развитию синаптической дисфункции. В культуре клеток Aβ не только запускает активацию Cx43-полуканалов, но и глиальных глутаматных и пуринергических рецепторов; последние, в свою очередь, высвобождают АТФ и глутамат, что способствует повреждению и гибели нейронов (Koulakoff et al., 2012). Кроме того, Cx43-опосредованное высвобождение АТФ, зарегистрированное у мышей линии APP/PS1, провоцирует дальнейшее прогрессирование нейродегенерации (Delekate et al., 2014). Интересно, что уменьшение уровня содержания Cx43 или его фармакологическое ингибирование у мышей линии APP/PS1 приводит к выраженному снижению дистрофических изменений нейритов, уменьшению митохондриального окислительного стресса, а также препятствует прогрессированию когнитивных нарушений на фоне отсутствия видимого уменьшения отложения Аβ в головном мозге (Ren et al., 2018).
С другой стороны, ряд исследований показывает, что Cx43 обладает нейропротекторным эффектом (Kozoriz et al., 2010a; Le et al., 2013). Согласно недавно полученным данным (Kajiwara et al., 2018), экспрессия Cx43 при аккумуляции Аβ в ткани головного мозга защищает клетки от альтерации. В этом контексте важно отметить, что Cx43 экспрессируется и на мембранах митохондрий, что было убедительно показано для клеток миокарда (Rodríguez-Sinovas et al., 2018), жировой ткани (Kim et al., 2017) и астроцитов (Kozoriz et al., 2010b). Выяснение, насколько митохондриальная локализация Cx43 актуальна для клеток НВЕ при экспериментальной БА, требует дальнейшего экспериментального исследования.
Таким образом, белки CD38 и Cx43 можно рассматривать в качестве перспективных мишеней для фармакологической модуляции при нейродегенерации, в том числе альцгеймеровского типа.
Список литературы
Салмина А.Б., Инжутова А.И., Моргун А.В., Окунева О.С., Малиновская Н.А., Лопатина О.Л., Петрова М.М., Таранушенко Т.Е., Фурсов А.А., Кувачева Н.В. 2012. НАД+-конвертирующие ферменты в клетках нейрональной и глиальной природы: CD38 как новая молекула-мишень для нейропротекции. Вестник Росс. акад. мед. наук. Т. 67. № 10. С. 29. (Salmina A.B., Inzhutova A.I., Morgun A.V., Okuneva O.S., Malinovskaya N.A., Lopatina O.L., Petrova M.M., Taranushenko T.E., Fursov A A.A. Kuvacheva N.V. 2012. NAD+-converting enzymes in neuronal and glial cells: CD38 as a novel target for neuroprotection. Ann. Russ. Acad. Med. Sci. V. 67. № 10. P. 29.)
Хилажева Е.Д., Бойцова Е.Б., Пожиленкова Е.А., Солончук Ю.Р., Салмина А.Б. 2015. Получение трехклеточной модели нейроваскулярной единицы in vitro. Цитология. Т. 57. № 10. С. 710. (Khilazheva E.D., Boytsova E.B., Pozhilenkova E.A., Solonchuk Yu.R., Salmina A.B. 2015. The model of neurovascular unit in vitro consisting of three cells types. Tsitologiya. V. 57. № 10. P. 710.)
Alano C.C., Tran A., Tao R., Ying W., Karliner J.S., Swanson R.A. 2007. Differences among cell types in NAD(+) compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J. Neurosci. Res. V. 85. P. 3378. https://doi.org/10.1002/jnr.21479
Blacher E., Dadali T., Bespalko A., Haupenthal V.J., Grimm M.O., Hartmann T., Lund F.E., Stein R., Levy A. 2015. Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann. Neurol. V. 78. P. 88. https://doi.org/10.1002/ana.24425
Bruzzone S., Franco L., Guida L., Zocchi E., Contini P., Bisso A., Usai C., De Flora A. 2001. A self-restricted CD38-connexin 43 cross-talk affects NAD+ and cyclic ADP-ribose metabolism and regulates intracellular calcium in 3T3 fibroblasts. J. Biol. Chem. V. 276. P. 48300. https://doi.org/10.1074/jbc.M107308200
Camacho-Pereira J., Tarragó M.G., Chini C.C.S., Nin V., Escande C., Warner G.M., Puranik A.S., Schoon R.A., Reid J.M., Galina A., Chini E.N. 2016. CD38 Dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. V. 23. P. 1127. https://doi.org/1016/j.cmet.2016.05.006
Chini E.N. 2009. CD38 as a regulator of cellular NAD: A novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. V. 15. P. 57. https://doi.org/10.2174/138161209787185788
Compagnoni G.M., Fonzo A.D., Corti S., Comi G.P. 2020. The role of mitochondria in neurodegenerative diseases: The lesson from Alzheimer’s disease and Parkinson’s disease. Mol. Neurobiol. V. 57. P. 2959. https://doi.org/10.1007/s12035-020-01926-1
Contreras J.E., Sánchez H.A., Eugenin E.A., Speidel D., Theis M., Willecke K., Bukauskas F.F., Bennett M.V., Sáez J.C. 2002. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA. V. 99. P. 495. https://doi.org/10.1073/pnas.012589799
Delekate A., Füchtemeier M., Schumacher T., Ulbrich C., Foddis M., Petzold G.C. 2014. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. V. 5. https://doi.org/10.1038/ncomms6422
Dovmark T.H., Saccomano M., Hulikova A., Alves F., Swietach P. 2017. Connexin-43 channels are a pathway for discharging lactate from glycolytic pancreatic ductal adenocarcinoma cells. Oncogene. V. 36. P. 4538. https://doi.org/10.1038/onc.2017.71
Encinas J.M., Enikolopov G. 2008. Identifying and quantitating neural stem and progenitor cells in the adult brain. Methods Cell. Biol. V. 85. P. 243. https://doi.org/10.1016/S0091-679X(08)85011-X
Epelbaum S., Youssef I., Lacor P.N., Chaurand P., Duplus E., Brugg B., Duyckaerts C., Delatour B. 2015. Acute amnestic encephalopathy in amyloid-β oligomer-injected mice is due to their widespread diffusion in vivo. Neurobiol. Aging. V. 36. P. 2043. https://doi.org/10.1016/j.neurobiolaging.2015.03.005
Franco-Iborra S., Vila M., Perier C. 2018. Mitochondrial quality control in neurodegenerative diseases: Focus on Parkinson’s disease and Huntington’s disease. Front. Neurosci. V. 12. https://doi.org/10.3389/fnins.2018.00342
Hayakawa K., Esposito E., Wang X., Terasaki Y., Liu Y., Xing C., Ji X., Lo E.H. 2016. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. V. 535. P. 551. https://doi.org/10.1038/nature18928
Johnson A.M., Roach J.P., Hu A., Stamatovic S.M., Zochowski M.R., Keep R.F., Andjelkovic A.V. 2018. Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. FASEB J. V. 32. P. 2615. https://doi.org/10.1096/fj.201700699R
Kajiwara Y., Wang E., Wang M., Sin W.C., Brennand K.J., Schadt E., Naus C.C., Buxbaum J., Zhang B. 2018. GJA1 (connexin43) is a key regulator of Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. V. 6. P. 144. https://doi.org/10.1186/s40478-018-0642-x
Kim S.N., Kwon H.J., Im S.W., Son Y.H., Akindehin S., Jung Y.S., Lee S.J., Rhyu I.J., Kim I.Y., Seong J.K., Lee J., Yoo H.C., Granneman J.G., Lee Y.H. 2017. Connexin 43 is required for the maintenance of mitochondrial integrity in brown adipose tissue. Sci. Rep. V. 7. P. 7159. https://doi.org/10.1038/s41598-017-07658-y
Koulakoff A., Mei X., Orellana J.A., Sáez J.C., Giaume C. 2012. Glial connexin expression and function in the context of Alzheimer’s disease. Biochim. Biophys. Acta. V. 1818. P. 2048. https://doi.org/10.1016/j.bbamem.2011.10.001
Kozoriz M.G., Bechberger J.F., Bechberger G.R., Suen M.W., Moreno A.P., Maass K., Willecke K., Naus C.C. 2010a. The connexin43 C-terminal region mediates neuroprotection during stroke. J. Neuropathol. Exp. Neurol. V. 69. P. 196. https://doi.org/10.1097/NEN.0b013e3181cd44df
Kozoriz M.G., Church J., Ozog M.A., Naus C.C., Krebs C. 2010b. Temporary sequestration of potassium by mitochondria in astrocytes. J. Biol. Chem. V. 285. P. 31107. https://doi.org/10.1074/jbc.M109.082073
Lagos-Cabré R., Brenet M., Díaz J., Pérez R.D., Pérez L.A., Herrera-Molina R., Quest A.F.G., Leyton L. 2018. Intracellular Ca2+ increases and connexin 43 hemichannel opening are necessary but not sufficient for Thy-1-Induced astrocyte migration. Int. J. Mol. Sci. V. 19. P. 2179. https://doi.org/10.3390/ijms19082179
Lautrup S., Sinclair D.A., Mattson M.P., Fang E.F. 2019. NAD+ in brain aging and neurodegenerative disorders. Cell Metab. V. 30. P. 630. https://doi.org/10.1016/j.cmet.2019.09.001
Le H.T., Sin W.C., Lozinsky S., Bechberger J., Vega J.L., Guo X.Q., Sáez J.C., Naus C.C. 2013. Gap junction intercellular communication mediated by connexin43 in astrocytes is essential for their resistance to oxidative stress. J. Biol. Chem. V. 289. P. 1345. https://doi.org/10.1074/jbc.M113.508390
Liu X., Sun L., Torii M., Rakic P. 2012. Connexin 43 controls the multipolar phase of neuronal migration to the cerebral cortex. Proc. Natl. Acad. Sci. USA. V. 109. P. 8280. https://doi.org/10.1073/pnas.1205880109
Liu Y., Xue Q., Tang Q., Hou M., Qi H., Chen G., Chen W., Zhang J., Chen Y., Xu X. 2013. A simple method for isolating and culturing the rat brain microvascular endothelial cells. Microvasc. Res. V. 90. P. 199. https://doi.org/10.1016/j.mvr.2013.08.004
Nagy J.I., Li W., Hertzberg E.L., Marotta C.A. 1996. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. V. 717. P. 173. https://doi.org/10.1016/0006-8993(95)01526-4
Osipova E.D., Semyachkina-Glushkovskaya O.V., Morgun A.V., Pisareva N.V., Malinovskaya N.A., Boitsova E.B., Pozhilenkova E.A., Belova O.A., Salmin V.V., Taranushenko T.E., Noda M., Salmina A.B. 2018. Gliotransmitters and cytokines in the control of blood-brain barrier permeability. Rev. Neurosci. V. 29. P. 567. https://doi.org/10.1515/revneuro-2017-0092
Ren R., Zhang L., Wang M. 2018. Specific deletion connexin43 in astrocyte ameliorates cognitive dysfunction in APP/PS1 mice. Life Sci. V. 208. P. 175. https://doi.org/10.1016/j.lfs.2018.07.033
Rodríguez-Sinovas A., Ruiz-Meana M., Denuc A., García-Dorado D. 2018. Mitochondrial Cx43, an important component of cardiac preconditioning. Biochim. Biophys. Acta Biomembr. V. 1860. P. 174. https://doi.org/10.1016/j.bbamem.2017.06.011
Sipos E., Kurunczi A., Kasza A., Horvath J., Felszeghya K., Laroche S., Toldi J., Parducz A., Penke B., Penke Z. 2007. Beta-amyloid pathology in the entorhinal cortex of rats induces memory deficits: implications for Alzheimer’s disease. Neurosci. V. 147. P. 28. https://doi.org/10.1016/j.neuroscience.2007.04.011
Song E.K., Rah S.Y., Lee Y.R., Yoo C.H., Kim Y.R., Yeom J.H., Park K.H., Kim J.S., Kim U.H., Han M.K. 2011. Connexin-43 hemichannels mediate cyclic ADP-ribose generation and its Ca2+-mobilizing activity by NAD+/cyclic ADP-ribose transport. J. Biol. Chem. V. 286. https://doi.org/10.1074/jbc.M111.307645
Swerdlow R.H. 2018. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimers Dis. V. 62. P. 1403. https://doi.org/10.3233/JAD-170585
Yi C., Mei X., Ezan P., Mato S., Matias I., Giaume C., Koulakoff A. 2016. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. V. 23. P. 1691. https://doi.org/10.1038/cdd.2016.63
Дополнительные материалы отсутствуют.