

Цитология, 2022, T. 64, № 3, стр. 208-215
Роль опухолевого супрессора RB в развитии локализованного и кастрационно-резистентного рака предстательной железы
В. М. Рябов 1, М. А. Воскресенский 2, Б. В. Попов 1, *
1 Институт цитологии РАН
194064 Санкт-Петербург, Россия
2 Городская многопрофильная больница № 2 МЗ РФ
194354 Санкт-Петербург, Россия
* E-mail: borisvp478@gmail.com
Поступила в редакцию 25.02.2022
После доработки 11.03.2022
Принята к публикации 12.03.2022
- EDN: CCROUK
- DOI: 10.31857/S0041377122030099
Аннотация
Рак предстательной железы (РПЖ) занимает в настоящее время в западных странах лидирующее положение по заболеваемости и смертности среди всех опухолевых заболеваний. РПЖ возникает в форме аденокарциномы (локализованный РПЖ), которая может находиться неопределенно долгое время в дремлющей форме, не угрожающей жизни пациента, или трансформироваться в агрессивный рак, нечувствительный к андроген-депривационной терапии – кастрационно-резистентный рак простаты, протекающий с метастазированием и быстро наступающим летальным исходом (КР-РПЖ). РПЖ возникает из эпителия предстательной железы, формирование и функционирование которого происходит под действием андрогенов. Андрогены, в основном дигидротестостерон, активируют сигнальный путь андрогенного рецептора (AR), который регулирует рост и деление эпителия простаты в нормальных условиях и в случае возникновения локализованного РПЖ. Андроген-депривационная терапия, например, с помощью ингибиторов андрогенного рецептора (энзалутамид, абиратерон), тормозит развитие локализованного РПЖ в течение 1.5–2-х лет, но затем теряет эффективность и неизбежно приводит к переходу заболевания в агрессивный КР-РПЖ. Инактивация опухолевого супрессора RB вносит вклад в развитие рака любой тканевой специфичности вследствие мутаций, потери гена или посттрансляционной модификации его продукта. Оценка состояния RB и его продукта показывает, что ген RB изменяется менее чем у 1% пациентов при локализованном РПЖ, но его потеря является причиной возникновения КР-РПЖ у 17–33% пациентов. В настоящем кратком обзоре данных литературы рассматривается роль сигнального пути pRb в патогенезе локализованного и кастрационно-резистентного РПЖ.
Ген чувствительности к возникновению ретинобластомы (RB) был первым идентифицированным опухолевым супрессором. Его существование было изначально предположено Knudson (1971), который изучал истории болезней пациентов, страдающих редкой формой опухоли сетчатки глаза у детей – ретинобластомой. Автор предположил, что ретинобластома вызывается двумя мутациями одного гена. В доминантно наследуемой форме одна мутация такого гена наследуется через зародышевые клетки, а вторая возникает в соматических клетках сетчатой оболочки глаза (Knudson, 1971). Следуя этой гипотезе, ген RB успешно клонировали в 1986–1987 гг. (Friend et al., 1986; Lee et al., 1987). С этого времени RB и его продукт (pRb) постепенно заняли центральное место в литературе, описывающей основные функции клеток. Было найдено, что pRb играет центральную роль в биологии эмбриональных и тканеспецифических стволовых клеток зрелого организма, тканеспецифических дифференцированных клеток различных тканей. Мутации или утрата гена RB, инактивация его продукта служат причиной возникновения различных заболеваний, а поиск новых подходов и лекарств для лечения разнообразных болезней требует оценки статуса RB и его продукта (Sage, 2012).
pRb регулирует основные функции клеток во всех многоклеточных организмах, в том числе у растений и животных. Область активности pRb включает регуляцию способности к самоподдержанию, поддержание состояния покоя, выбор клеточной судьбы и тканеспецифической дифференцировке в эмбриональных и тканеспецифических стволовых клетках зрелого организма (Sage, 2012). В дифференцированных клетках различной тканевой специфичности pRb выполняет роль убиквитарного регулятора клеточного цикла, дифференцировки, метаболизма, апоптоза, клеточного старения, аутофагии, клеточной памяти, ответа на повреждение ДНК и стабильности генома (рис. 1) (Weinberg, 1995; Sherr, 1996; Dyson, 1998; Knudsen, Knudsen, 2008; Dick et al., 2018; Baryshev et al., 2020; Popov et al., 2020).
Рис. 1.
Молекулярно-клеточные механизмы регуляции основных клеточных функций pRb – продуктом гена RB. pRb регулирует функции клетки через канонический и неканонический сигнальные пути. Канонический сигнальный путь включает регуляцию клеточного цикла, неканонический – регуляцию репарации ДНК, обмена веществ, ответа на повреждение ДНК, митохондриальный биогенез, митохондриальную транскрипцию и апоптоз, индуцированный фактором некроза опухолей α (TNFα). E2F – транскрипционные факторы семейства E2F, Ezh2 – метилтрансфераза, триметилирующая сайт Н3К27, Bax – белок семейства Bcl-2, регулирующий проницаемость внешней мембраны митохондрий.
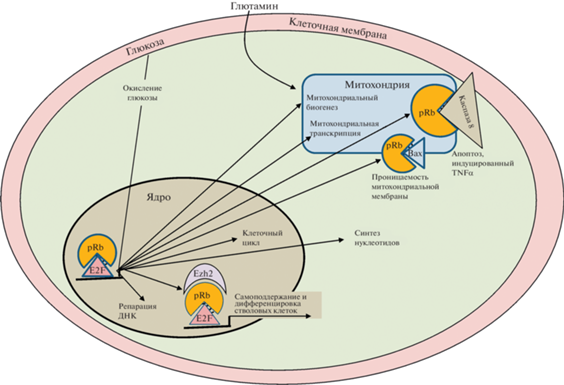
Более 200 белков выполняют свои специфические функции путем взаимодействия с pRb, создавая, таким образом, биохимическую основу для его многофункциональности. Основными партнерами pRb являются транскрипционные факторы семейства E2F, которые опосредуют влияние pRb на транскрипцию множества генов, регулирующих базальные функции клеток. Транскрипционные факторы E2F предоставляют pRb возможность находить мишени и регулировать транскрипцию путем прямого торможения активности E2F или с помощью привлечения других регуляторных молекул, например, метилтрансферазы Ezh2, которая метилирует K27 гистона H3 (H3K27me3), вызывая сайленсинг генов. Формирование троичного комплекса, включающего pRb, E2F1 и Ezh2, играет ключевую роль в регуляции самоподдержания, прогрессии клеточного цикла, дифференцировки и стабильности генома (Dick et al., 2018). С другой стороны, потеря, мутации, метилирование промотора RB или посттрансляционная модификация его продукта приводит к клеточным катастрофам, лежащим в основе различных заболеваний, наиболее разрушительным из которых является рак, включая РПЖ (Mandigo et al., 2021).
В настоящем кратком обзоре литературы рассматривается роль сигнального пути pRb−E2F1 в патогенезе локализованного и кастрационно-резистентного РПЖ. Опубликованные результаты свидетельствуют, что при локализованном РПЖ pRb сохраняет активность, но она используется опухолевыми клетками для создания конкурентных ростовых преимуществ. Потеря RB с полной утратой регуляторных функций сигнального пути pRb−E2F1 приводит и доминирует в патогенезе кастрационно-резистентного РПЖ.
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕГУЛЯЦИИ ОСНОВНЫХ КЛЕТОЧНЫХ ФУНКЦИЙ ПРОДУКТОМ ГЕНА RB
Белок pRb физически взаимодействует более чем с 200 регуляторными молекулами, однако основными его партнерами являются белки семейства E2F. Весь спектр регуляторного влияния pRb осуществляется через канонический и неканонический сигнальные пути. Наиболее хорошо изученным в настоящее время является канонический сигнальный путь, опосредующий регуляторное влияние pRb на деление клетки. Неканонический сигнальный путь аккумулирует механизмы онкосупрессорной активности pRb, которые в значительной степени опосредуются его взаимодействием с одним из факторов семейства E2F–E2F1.
КАНОНИЧЕСКИЙ СИГНАЛЬНЫЙ ПУТЬ pRb: РЕГУЛЯЦИЯ КЛЕТОЧНОГО ЦИКЛА
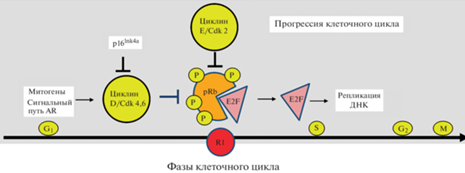
pRb тормозит деление клетки в рестрикционной точке R1 путем связывания и временной инактивации активаторных E2F (рис. 2). Под действием митогенов в клетке образуются активные киназы циклин D/Cdk2 и циклин E/Cdk2, которые последовательно фосфорилируют pRb, способствуя освобождению активаторных E2F. Наконец, активаторные E2F транскрипционно вызывают синтез белков, необходимых для репликации ДНК и дальнейшей прогрессии клеточного цикла. Активность комплексов циклин D/Cdk4,6 тормозится белком p16Ink4a. Гиперметилирование промотора гена р16Ink4a (при раке пищевода, головы и шеи, немелкоклеточной карциномы легких, раке молочной железы, толстого кишечника) или потеря гена р16Ink4a (в случаях глиомы, рака головы и шеи, яичников, мочевого пузыря, эндометрия) сопровождается гиперактивацией циклина D/Cdk4 и гиперфосфорилированием pRb. В таких условиях отменяется рестрикционная точка R1, что способствует постоянному и автономному делению клетки. Подобная ситуация возникает и при амплификации гена циклина D1 (при раке пищевода, головы и шеи, молочной железы, эндометрия и меланоме) (Mandigo et al., 2021). При инактивации pRb, опосредованной гиперактивацией циклина D/Cdk4,6, в клинике используются ингибиторы Cdk4,6 (пальбоклиб, рибоциклиб), эффективность действия которых зависит от присутствия функционального pRb. Таким образом, pRb может использоваться как эффективный прогностический маркер для выбора таргетной терапии для лечения рака (Mandigo et al., 2021).
Рис. 2.
Роль фактора pRb в регуляции клеточного цикла. pRb контролирует прохождение клеткой рестрикционной точки R1 в конце фазы G1 путем связывания и временной инактивации активаторных транскрипционных факторов E2F. G1, S, G2, M – фазы клеточного цикла; R1 – рестрикционная точка фазы G1, в которой регулируется переход G1/S; AR – андрогенный рецептор; Cdk 2,4,6 – циклин-зависимые киназы.
НЕКАНОНИЧЕСКИЙ СИГНАЛЬНЫЙ ПУТЬ pRb
Клинические и экспериментальные исследования роли pRb показали, что механизм его действия выходит за пределы канонического сигнального пути. Было установлено, что pRb локализуется не только в промоторной, но и в инхансерной области генов, а также в повторяющихся последовательностях ДНК (Kareta et al., 2015; Ishak et al., 2016). Гиперфосфорилированный pRb был найден в связанном с хроматином виде в фазе S клеточного цикла (Avni et al., 2008), хотя в соответствии с гипотезой о канонической роли pRb его гиперфосфорилирование в рестрикционной точке R1 способствует диссоциации комплекса pRb/E2F и лишает pRb способности связывать хроматин (рис. 2). Клинические работы показали, что инактивация pRb часто коррелирует с повышением чувствительности к химеотерапии (Knudsen, Knudsen, 2008). Было установлено, что pRb регулирует не только клеточный цикл, но также потребление и окисление глюкозы, синтез нуклеотидов, обмен глютамина, цикл трикарбоновых кислот, митохондриальную транскрипцию, митохондриальный апоптоз, индуцированный TNFα, репарацию ДНК, самоподдержание и дифференцировку эмбриональных стволовых клеток (ЭСК) и соматических стволовых клеток (СК), ответ клетки на повреждение ДНК, стабильность генома (рис. 1) (Dick et al., 2018). Вновь найденные объекты регуляторного влияния pRb отнесены к неканоническому сигнальному пути, механизмы участия pRb в некоторых из них рассмотрены ниже.
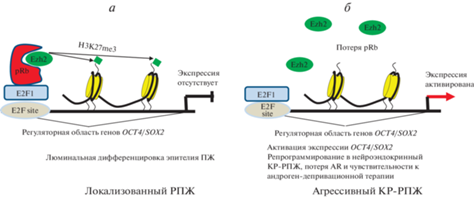
Роль pRb в регуляции самоподдержания и дифференцировки СК. Изучение роли белков семейства Polycomb (PcG) показало, что как в эмбриональных (ЭСК), так и в соматических СК промоторы генов, поддерживающих мультипотентность и линейную дифференцировку, содержат “бивалентные” домены, включающие активную (Н3К4ме3) и репрессивную метку (Н3К27ме3) (Sharma et al., 2010; Попов, 2019). Эти модификации катализируются соответственно белками семейств Trithorax (TrxG) и PcG (Piunti, Shilatifard, 2016). В активно делящихся самоподдерживающихся ЭСК гены линейной дифференцировки не транскрибируются, но их репрессия отменяется при индукции линейной дифференцировки, что связано с утратой репрессивной метки Н3К27ме3. Активация экспрессии дифференцировочных генов сочетается с торможением транскрипции генов плюрипотентности (OCT4, SOX2, NANOG) в ЭСК путем повышения уровня репрессивной метки Н3К27ме3 в их промоторах. Такая регуляция позволяет дифференцировку СК в один клеточный тип, но предотвращает де- и трансдифференцировку. Современные исследования установили, что уровень метилирования промоторов генов плюрипотентности регулируется путем формирования комплексов pRb/E2F1/Ezh2, которые триметилируют сайт Н3К27. Экспериментальная инактивация pRb сопровождается снижением уровня метилирования и репрограммированием СК (Kareta et al., 2015). В клинических условиях, потеря RB и репрограммирование люминального фенотипа в нейроэндокринный найдены у 15–25% пациентов с КР-РПЖ (Vlachostergios et al., 2017).
Роль pRb в регуляции метаболизма. Было установлено, что сигнальный путь pRb−E2F1 регулирует множество метаболических процессов, включая синтез нуклеотидов, окисление глюкозы и функцию митохондрий (рис. 1). Прямыми мишенями E2F1 являются гены ферментов синтеза нуклеотидов: тимидинкиназы (ТК) и дегидрофолатредуктазы (DHFR) (Anderson et al., 1996; Jensen et al., 1997); дегидрогеназы 4 пируваткиназы (PDK4), тормозящей вход пирувата в цикл трикарбоновых кислот (Hsieh et al., 2008); коактиватора 1α PPARgamma (PCG-1al-pha), способствующего митохондриальной транскрипции, биогенезу и потреблению кислорода (Da-li-Youcef et al., 2007); гены белков, участвующих в транспорте электронов и окислительном фосфорилировании, например, субъединицы АТФ-синтетазы, цитохром-с-оксидазы, убихинол-цитохром-с-редуктазы, и сукцинатдегидрогеназы (Dali-Youcef et al., 2007; Blanchet et al., 2011). Экспрессия транспортера глютамина ASCT2 и глютаминазы (GLS1) также регулируется сигналами pRb−E2F1 (Reynolds et al., 2014).
Клеточный ответ на повреждение ДНК. Ответ клетки на повреждение ДНК включает репарацию ДНК, гомологичную рекомбинацию (HR), и соединение негомологичных концов ДНК (NHEJ). Репарация ДНК регулируется pRb−E2F1 путем влияния на экспрессию генов, кодирующих факторы репарации MSH2, BRCA1, PCNA (Polager et al., 2002). В контексте регуляции негомологичной рекомбинации pRb взаимодействует с белками Ku70, Ku80, что необходимо для привлечения модификаторов хроматина (Cook et al., 2015). При HR вслед за двухнитевыми разрывами ДНК происходит фосфорилирование E2F1 киназами ATM/ATR с последующим привлечением к месту повреждения ДНК pRb и BRG1, инициирующего резекцию концов ДНК и последующую репарацию (Velez-Cruz et al., 2016).
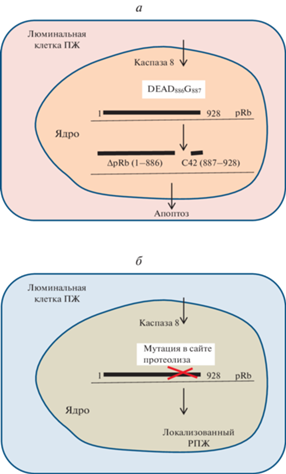
Проницаемость мембраны митохондрий и митохондриальный апоптоз. В нормальных условиях pRb опосредует индуцированный TNFα апоптоз в митохондриях. В ходе возникновения апоптоза TNFα индуцирует образование каспазы 8, которая вызывает протеолиз pRb в С-концевом домене, что необходимо для активации последующего каспазного каскада (рис. 3а) (Hilgendorf et al., 2013). Мутация RB в сайте протеолиза или взаимодействие его продукта с β-катенином вызывает устойчивость pRb к действию каспазы 8 и тормозит апоптоз (рис. 3б) (Han et al., 2013). Кроме того, pRb физически взаимодействует с белком Bax в мембране митохондрий, что увеличивает ее проницаемость, способствуя апоптозу (Wang et al., 2017).
Рис. 3.
Схематическое изображение роли pRb в торможении индуцированного TNFα митохондриального апоптоза при локализованном РПЖ. а – в нормальных условиях в ходе апоптоза, индуцированного TNFα, фактор pRb протеолизируется в митохондриях под действием каспазы 8 с образованием двух фрагментов, что необходимо для последующего каспазного каскада; б – при локализованном РПЖ pRb может приобретать устойчивость к действию каспазы 8 в результате мутации или взаимодействия с β-катенином. DEAD886−C887 – сайт pRb, в котором в нормальных условиях происходит его протеолиз каспазой 8 при апоптозе, индуцированном TNFα; ΔpRb (1−886), C42 (887−928) – фрагменты pRb, образующиеся при его протеолизе каспазой 8.
Роль сигнального пути pRb−E2F1−AR в формировании и функционировании эпителия предстательной железы. Предстательная железа представляет собой небольшой вспомогательный орган в мужской репродуктивной системе, который показывает самую высокую чувствительность к онкогенной трансформации среди других органов у мужчин. РПЖ диагностируется в течение жизни у каждого восьмого лица мужского пола. В настоящее время заболеваемость и смертность от РПЖ занимают лидирующие позиции среди всех онкологических заболеваний: в западных странах и в США в 2021 г. они достигли соответственно 26 и 11% от всех опухолевых болезней у мужчин (Siegel et al., 2021).
Предстательная железа формируется и функционирует в эмбриональной жизни и после рождения путем взаимодействия мезенхимных и эпителиальных клеток, экспрессирующих андрогенный рецептор (AR) (Cunha, 1994). Предстательная железа возникает из эпителиальных клеток, продуцирующих AR, который также является критическим эффектором возникновения, прогрессии РПЖ и эффективности его терапии (Balk, Knudsen, 2008). Удаление AR в начале применения противоопухолевой терапии является эффективным, но неизбежно сопровождается рецидивом опухоли вследствие неконтролируемого восстановления сигнального пути AR (Feldman, Feldman, 2001). AR способствует делению клетки путем индукции экспрессии циклина D1, образования активной киназы циклин D/Cdk4,6 и последующего гиперфосфорилирования (инактивации) pRb (Xu et al., 2006). В начале фазы G1 pRb находится в гипофосфорилированном активном состоянии и индуцирует остановку клеточного цикла в рестрикционной точке R1 перед началом репликации путем связывания и секвестрирования активаторных транскрипционных факторов E2Fs, включая E2F1 (рис. 2). Фосфорилирование pRb киназами циклин D/Cdk4,6 и циклин Е/Cdk2 в результате активации сигнального пути AR освобождает связанные E2Fs, которые способствуют последовательному формирования различных киназ циклин/Cdks и делению клетки (Dyson, 1988). Утрата RB играет ключевую роль в патогенезе и снижении эффективности медикаментозного лечения РПЖ, направленного на модуляцию сигнального пути AR. Прогрессирование РПЖ включает две отдельные стадии: локализованный РПЖ и КР-РПЖ. В первой стадии болезнь может протекать неопределенно долго в дремлющей форме, не угрожающей жизни, или внезапно трансформироваться в агрессивный КР-РПЖ, сопровождающийся быстрым летальным исходом.
Локализованный РПЖ. Прогностические маркеры, свидетельствующие о трансформации локализованного РПЖ в КР-РПЖ не найдены. Опубликованные данные дают возможность предположить, что клинически локализованный РПЖ, при котором повышается уровень белка Ezh2, характеризуется плохим прогнозом (Varambally et al., 2002). В нормальных тканях продукция Ezh2 тормозится сигнальным путем pRb−E2F1, инактивация pRb в эксперименте и клинике повышает уровень Ezh2 (Bra-cken, Helin, 2009). Подобный механизм лежит также в основе активации сети плюрипотентности в дифференцированных клетках и трансдифференцировки люминального эпителия предстательной железы в нейроэпителий (рис. 3) (Kareta et al., 2015). Оценка внутриклеточного уровня pRb показывает, что утрата гена RB может быть причиной перехода локализованного РПЖ во вторую стадию болезни, а также доминировать в ходе КР-РПЖ, однако уровень белка не снижается в первой стадии при локализованном РПЖ (Sharma et al., 2010). Экспериментальное удаление RB ведет к повышению уровня E2F1, обогащению его связывания с промотором AR, повышению продукции AR. Потеря RB изменяет рекрутирование партнерских белков к AR в нелигированном и лигированном состояниях, отменяет транскрипционный ответ AR к андрогенам (Sharma et al., 2010).
КР-РПЖ. В начальной стадии развития рака клетки различных опухолей сохраняют функциональный pRb, чтобы получить конкурентные преимущества для роста и деления. Потеря RB сопровождается индукцией аутофагии путем накопления реактивных молекул кислорода, активацией апоптоза транскрипционным фактором E2F1 в условиях потери супрессорного влияния на него pRb. В нормальных условиях TNFα вызывает в митохондриях синтез каспазы 8, протеолизирующей pRb, что необходимо для активации последующего каспазного каскада (рис. 3) (Hilgendorf et al., 2013). В некоторых типах рака толстого кишечника локус RB амплифицируется, и его продукт предотвращает опосредованное E2F1 транскрипционное торможение синтеза β-катенина, а также участвует в формировании комплекса pRb/β-катенин, в котором pRb становится нечувствительным к протеолизу, вызываемому каспазой 8, и, таким образом, тормозит митохондриальный апоптоз, способствуя росту опухоли (Viatour, Sage, 2011; Han et al., 2013). Продукция pRb позволяет клеткам опухоли легких, молочной и поджелудочной желез приобретать резистентность к химеотерапии, RB подвергается мутациям менее чем в 1% случаев локализованного РПЖ, что свидетельствует о его способности поддерживать рост опухоли в этой стадии (Knudsen, Knudsen, 2008).
Андроген-депривационная терапия неизбежно способствует переходу локализованного РПЖ в стадию КР-РПЖ (Knudsen, Scher, 2009). Утрата RB выявляется в 17–33% КР-РПЖ, в этих случаях в клетках полностью отменяется функция pRb в противоположность другим опухолям, в клетках которых активность pRb ослабляется путем фосфорилирования комплексом циклин D/Cdk4,6 (Mandigo et al., 2021). Утрата RB при КР-РПЖ ведет к нарушению сигнального пути pRb−E2F1, повышению способности E2F1 связывать хроматин и расширению цистрома E2F1. Новые связывающие сайты E2F1 локализуются в промоторных, внутригенных и дистальных межгенных областях (McNair et al., 2018). Расширение области связывания E2F1 происходит вследствие увеличения числа связывающих его сайтов с низким аффинитетом и последующей экспрессии генов, активирующих автономное деление клеток опухоли. Потеря RB может вызывать формирование КР-РПЖ с нейроэндокринным фенотипом (НЭ-РПЖ), который возникает в 10–20% случаев КР-РПЖ (Vlachostergios et al., 2017). При НЭ-РПЖ клетки не продуцируют AR и его мишень – простат-специфический антиген. С механистической точки зрения возникновение такого фенотипа может быть следствием активации сигнального пути pRb−E2F1−Ezh2 и сети плюрипотентности (рис. 4). Идентификация специфических генов, нарушение регуляции которых происходит при потере RB и КР-РПЖ может обеспечить понимание патогенеза и разработку новых подходов для лечения этой смертельной в настоящее время болезни.
Рис. 4.
Комплекс pRb/E2F1/Ezh2 тормозит экспрессию генов плюрипотентности в дифференцирванных клетках эпителия предстательной железы (ПЖ). а – В нормальных условиях и при локализованном РПЖ экспрессия генов OCT4 и SOX2 тормозится комплексом pRb/E2F1/Ezh2, который триметилирует сайт Н3К27, вызывающий сайленсинг этих генов; б – потеря pRb нарушает образование комплексов pRb/E2F1/Ezh2 на промоторах OCT4 и SOX2 в клетках эпителия ПЖ, что сопровождается активацией их экспрессии, репрограммированием в нейроэпителий и формированием НЭ-РПЖ. OCT4, SOX2 – гены сети плюрипотентности, остальные обозначения те же, что и на рис. 1.
ЗАКЛЮЧЕНИЕ
Ген RB, определяющий чувствительность к возникновению опухоли сетчатки глаз у детей (ретинобластомы), был первым идентифицированным опухолевым супрессором. Изучение RB показало, что он вносит существенный вклад в регуляцию основных клеточных функций. Мутации, потеря RB, инактивация его продукта pRb играют патогенетическую роль в возникновении и развитии каждой опухоли. pRb влияет на транскрипцию множества генов путем физического взаимодействия более чем с 200 регуляторными белками. Основным партнером pRb является транскрипционный фактор E2F1, имеющий сайты связывания в промоторных областях множества генов. Сигнальный путь pRb−E2F1 регулирует продукцию AR, под влиянием которого происходит формирование, развитие и функционирование предстательной железы. Нарушение передачи сигналов pRb−E2F1 являются причиной возникновения и прогрессирования РПЖ, который в настоящее время занимает лидирующие позиции по заболеваемости и смертности от всех онкологических заболеваний у мужчин в западных странах. В своем развитии РПЖ проходит две стадии: локализованный РПЖ, который может неопределенно долго находиться в дремлющей форме, не угрожающей жизни пациента, и КР-РПЖ, неизбежно и быстро приводящий к смерти больного. Одной из причин перехода локализованного РПЖ в КР-РПЖ является применение андроген-депривационной терапии, которая вначале эффективно тормозит передачу сигналов pRb−E2F1−AR, но неизбежно приводит к реактивации сигнального пути AR по коллатеральным путям. При локализованном РПЖ, клетки опухоли используют активность pRb для создания ростовых преимуществ по сравнению с нормальными клетками, в частности путем его участия в торможении передачи сигналов, необходимых для активации митохондриального апоптоза. Полная утрата функций pRb может быть причиной развития КР-РПЖ. В таких случаях происходит расширение цистрома транскрипционного фактора E2F1, который активирует экспрессию множества генов, способствующих автономному росту опухоли. Идентификация таких генов для разработки эффективной противоопухолевой терапии является приоритетной задачей в лечении КР-РПЖ.
Список литературы
Попов Б.В. 2019. Семейство POLYCOMB: стволовые, опухолевые стволовые клетки и рак предстательной железы. Цитология Т. 61. С. 769. (Popov B.V. 2019. POLYCOMB family: stem cells, cancer stem cells and prostate cancer. Tsitologiya. V. 61. P. 769).
Anderson M.M., Chen J., Cole C.N., Conrad S.E. 1996. Activation of the human thymidine kinase (TK) promoter by simian virus 40 large T antigen requires both the T antigen pRb family-binding domain and TK promoter sequences resembling E2F-binding sites. J. Virol. V. 70. P. 6304.
Avni D., Yang H., Martelli F., Hofmann F., ElShamy W.M., Ganesan S., Scully R., Livingston D.M. 2008. Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol. Cell. V. 12. P. 735.
Balk S.P., Knudsen K.E. 2008. AR, the cell cycle, and prostate cancer. Nucl. Recept. Signal. V. 6. P. e001. https://doi.org/10.1621/nrs.06001
Baryshev M., Petrov N., Ryabov V., Popov B. 2020. Transient expression of inactive RB in mesenchymal stem cells impairs their adipogenic potential and is associated with hypermethylation of the PPARγ2 promoter. Genes Dis. V. 7. P. 165.
Blanchet E, Annicotte J.S., Lagarrigue S., Aguilar V., Clapé C., Chavey C., Fritz V., Casas F., Apparailly F., Auwerx J., Fajas L. 2011. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol. V. 13. P. 1146.
Bracken A.P., Helin K. 2009. Polycomb group proteins: navigators of lineage pathways in cancer. Nat. Rev. Cancer. V. 9. P. 773.
Cook R., Zoumpoulidou G., Luczynski M.T., Rieger S., Moquet J., Spanswick V.J., Hartley J.A., Rothkamm K., Huang P.H., Mittnacht S. 2015. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep. V. 10. P. 2006.
Cunha G.R. 1994. Role of mesenchymal-epithelial interactions in normal and abnormal development of the mammary gland and prostate. Cancer. V. 74. P. 1030.
Dick F.A., Goodrich D.W., Sage J., Dyson N.J. 2018. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer. V. 18. P. 442.
Dali-Youcef N., Mataki C., Coste A., Messaddeq N., Giroud S., Blanc S., Koehl C., Champy M.F., Chambon P., Fajas L., Metzger D., Schoonjans K., Auwerx J. 2007. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc. Natl. Acad. Sci. USA. V. 104. P. 10703.
Dyson N. 1998. The regulation of E2F by pRB-family proteins. Genes Dev. V 12. P. 2245.
Feldman B.J., Feldman D. 2001. The development of androgen-independent prostate cancer. Nat. Rev. Cancer. V. 1. P. 34.
Friend S.H., Bernards R., Rogelj S., Weinberg R.A., Rapaport J.M., Albert D.M., Dryja T.P. 1986. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. V. 323. P. 643.
Han J., Soletti R.C., Sadarangani A., Sridevi P., Ramirez M.E., Eckmann L., Borges H.L., Wang J.Y. 2013. Nuclear expression of β-catenin promotes RB stability and resistance to TNF-induced apoptosis in colon cancer cells. Mol. Cancer Res. V. 11. P. 207.
Hilgendorf K.I., Leshchiner E.S., Nedelcu S., Maynard M.A., Calo E., Ianari A., Walensky L.D., Lees J.A. 2013. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev. V. 27. P. 1003.
Hsieh M.C.F., Das D., Sambandam N., Zhang M.Q., Nahlé Z. 2008. Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J. Biol. Chem. V. 283. P. 27410.
Ishak C.A., Marshall A.E., Passos D.T., White C.R., Kim S.J., Cecchini M.J., Ferwati S., MacDonald W.A., Howlett C.J., Welch I.D., Rubin S.M., Mann M.R.W., Dick F.A. 2016. An RB-EZH2 Complex mediates silencing of repetitive DNA sequences. Mol. Cell. V. 4. P. 1074.
Jensen D.E., Black A.R., Swick A.G., Azizkhan J.C. 1997. Distinct roles for Sp1 and E2F sites in the growth/cell cycle regulation of the DHFR promoter. J. Cell Biochem. V 67. P. 24.
Kareta M.S., Gorges L.L., Hafeez S., Benayoun B.A., Marro S., Zmoos A.F., Cecchini M.J., Spacek D., Batista L.F., O’Brien M., Ng Y.H., Ang C.E., Vaka D., Artandi S.E., Dick F.A. et al. 2015. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell. V. 16. P. 39.
Knudson A.G. Jr. 1971. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA. V. 68. P. 820.
Knudsen E.S., Knudsen K.E. 2008.Tailoring to RB: tumour suppressor status and therapeutic response. Nat. Rev. Cancer. V. 8. P. 714.
Knudsen K.E., Scher H.I. 2009. Starving the addiction: New opportunities for durable suppression of AR signaling in prostate cancer. Clin. Cancer Res. V. 15. P. 4792.
Lee W.H., Bookstein R., Hong F., Young L.J., Shew J.Y., Lee E.Y. 1987. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. V. 235. P. 1394.
Mandigo A.C., Tomlins S.A., Kelly W.K., Knudsen K.E. 2021. Relevance of pRB Loss in Human Malignancies. Clin. Cancer Res. V. 28. P. 255.
McNair C., Xu K., Mandigo A.C., Benelli M., Leiby B., Rod-rigues D., Lindberg J., Gronberg H., Crespo M., De Laere B., Dirix L., Visakorpi T., Li F., Feng F.Y., de Bono J., Demichelis F., Rubin M.A., Brown M., Knudsen K.E.. 2018. Differential impact of RB status on E2F1 reprogramming in human cancer. J. Clin. Invest. V. 128. P. 341.
Piunti A., Shilatifard A. 2016. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science. V. 352. P. aad9780.
Polager S., Kalma Y., Berkovich E., Ginsberg D. 2002. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene. V. 21. P. 437.
Popov B., Petrov N., Ryabov V., Evsyukov I. 2020. p130 And pRb in the maintenance of transient quiescence of mesenchymal stem cells. Stem Cells Int.https://doi.org/10.1155/2020/8883436
Reynolds M.R., Lane A.N., Robertson B., Kemp S., Liu Y., Hill B.G., Dean D.C., Clem B.F. 2014. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. V. 33. P. 556.
Sage J. 2012. The retinoblastoma tumor suppressor and stem cell biology. Genes Dev. V. 26. P. 1409.
Sharma A., Yeow W.S., Ertel A., Coleman I., Clegg N., Thangavel C., Morrissey C., Zhang X., Comstock C.E., Witkiewicz A.K., Gomella L., Knudsen E.S., Nelson P.S., Knudsen K.E. 2010. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Invest. V. 120. P. 4478.
Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. 2021. Cancer Statistics, 2021. CA Cancer J. Clin. V. 71. P. 7.
Sherr C.J. 1996. Cancer cell cycles. Science. V. 274. P. 1672.
Varambally S., Dhanasekaran S.M., Zhou M., Barrette T.R., Kumar-Sinha C., Sanda M.G., Ghosh D., Pienta K.J., Sewalt R.G., Otte A.P., Rubin M.A., Chinnaiyan A.M. 2002. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. V. 419. P. 624.
Vélez-Cruz R., Manickavinayaham S., Biswas A.K., Clary R.W., Premkumar T., Cole F., Johnson D.G. 2016. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. V. 30. P. 2500.
Viatour P., Sage J. 2011. Newly identified aspects of tumor suppression by RB. Dis. Model Mech. V. 4. P. 581.
Vlachostergios P.J., Puca L., Beltran H. 2017. Emerging variants of castration-resistant prostate cancer. Curr. Oncol. Rep. V. 19. P. 32.
Wang C.Y., Xu Z.B., Wang J.P., Jiao Y., Zhang B. 2017. Rb deficiency accelerates progression of carcinoma of the urinary bladder in vivo and in vitro through inhibiting autophagy and apoptosis. Int. J Oncol. V. 50. P. 1221.
Weinberg R.A. 1995. The retinoblastoma protein and cell cycle control. Cell. V. 81. P. 323.
Xu Y., Chen S.Y., Ross K.N., Balk S.P. 2006. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. V. 66. P. 7783.
Дополнительные материалы отсутствуют.