

Цитология, 2022, T. 64, № 5, стр. 457-465
Гено- и цитотоксическое действие динитрозильного комплекса железа с меркаптосукцинатом на клетки MCF-7
В. А. Тронов 1, *, Н. А. Ткачев 1, Е. И. Некрасова 2, А. Ф. Ванин 1
1 Федеральный научный центр химической физики им. Н.Н. Семенова РАН
119991 Москва, Россия
2 Институт биохимической физики им. Н.М. Эмануэля РАН
119991 Москва, Россия
* E-mail: vtronov@yandex.ru
Поступила в редакцию 29.04.2022
После доработки 31.05.2022
Принята к публикации 03.06.2022
- EDN: CGIBLT
- DOI: 10.31857/S0041377122050091
Аннотация
Цитотоксическое действие динитрозильного комплекса железа (ДНКЖ) с меркаптосукцинатом (МС) на опухолевые клетки человека MCF-7 в 2 раза выше цитотоксического эффекта смеси предшественников его синтеза (МС + Fe2+). Методом щелочных комет показано, что смесь (МС + Fe2+) индуцировала в клетках однонитевые разрывы (ОР) ДНК. Эти повреждения полностью репарировались спустя 24 ч. Хотя комплекс ДНКЖ-МС индуцировал в ДНК клеток меньше ОР, часть из них сохранялась нерепарированными в 19% клеток спустя 24 ч. Методом нейтральных комет показано, что эти разрывы являются двунитевыми (ДР) и локализованы в 17% клеток. Качественная корреляция генотоксичности по выходу нерепарированных ОР в ДНК с цитотоксичностью, определенной по МТТ-тесту, на клетках MCF-7 говорит о том, что токсичность динитрозильного комплекса по отношению к клеткам MCF-7 может быть частично ассоциирована с возникающими ДР в ДНК. Потенциально летальные ДР могут ворзникать в результате атаки клеточными эндонуклеазами однонитевых участков и АП-сайтов в ДНК, которые фомируются как интермедиаты механизма репарации NER, активируемого в ответ на появление в ДНК сложных аддуктов оснований под действием ДНКЖ-МС.
Известны заболевания, ассоциированные с дефицитом оксида азота в тканях больных. К ним относятся сахарный диабет (Giacco, Brownlee 2010; Du et al., 2001), гипертоническая болезнь (Török, 2008), глаукома (Reina-Torres et al., 2021), эндометриоз (Burgova et al., 2019), онкологические заболевания (Carmeliet, Jain, 2011; Huang, et al., 2017). В исследованиях in vitro и in vivo показано, что синтетические динитрозильные комплексы железа (ДНКЖ), обеспечивая доставку NO в ткани, открывают возможность терапии таких заболеваний и (или) снижают их патологические проявления (Wu et al., 2016; Kelsey, 2014). В экспериментах на крысах химические доноры оксида азота оказывали нейропротекторное действие при ишемическом инсульте (Godínez-Rubí, 2013). Ранее на клетках аденокарциномы человека MCF-7 мы показали, что цитотоксический эффект синтетического комплекса ДНКЖ-MС опосредован ионом нитрозония (NO+), высвобождающимся при распаде комплекса в клетках (Vanin et al., 2021). Цитотоксический эффект выражался в виде апоптоза, который регистрировали спустя 24 ч после добавления комплекса к клеткам. В этой же работе было показано, что время полу-жизни комплекса ДНКЖ-МС в водной среде при комнатной температуре составляет 2 ч. Этот факт ставит вопрос о связи цитотоксического эффекта с самим комплексом или с продуктами его распада в клетке (Vanin et al., 2021).
В настоящей работе мы, используя метод ДНК-комет, оценили гено- и цитотоксическое действие комплекса ДНКЖ-MС и смеси предшественников его синтеза – МС и ферросульфата (FeSO4) – на клетки MCF-7, а также их влияние на репарацию ДНК.
МАТЕРИАЛ И МЕТОДИКА
Синтез ДНКЖ-MС. Динитрозильный комплекс железа (ДНКЖ) с меркаптосукцинатом (MC) (ДН-КЖ-МС) синтезирован из ферросульфата и MC по разработанному нами протоколу (Vanin et al., 2021). В водном растворе ДНКЖ-MС присутствует в моноядерной (1) и биядерной (2) формах:
(1)
$\left[ {{{{\left( {{\text{R}}{{{\text{S}}}^{ - }}} \right)}}_{2}}{\text{F}}{{{\text{e}}}^{{{\text{2 + }}}}}{{{\left( {{\text{NO}}} \right)}}_{{\text{2}}}}} \right]{\text{,}}$(2)
$\left[ {{{{\left( {{\text{R}}{{{\text{S}}}^{ - }}} \right)}}_{{\text{2}}}}{\text{Fe}}_{2}^{{2 + }}{{{\left( {{\text{NO}}} \right)}}_{{\text{4}}}}} \right]{\text{,}}$(3)
${\text{HOOC}} - {\text{CH}}\left( {{\text{SH}}} \right){\text{C}}{{{\text{H}}}_{{\text{2}}}} - {\text{COOH}}{\text{.}}$Образование стабильного комплекса ДНКЖ-МС регистрировали спектрофотометрически. Концентрацию комплекса определяли по оптической плотности на длине волны 360 нм, исходя из коэффициента экстинкции 3700 М–1 см–1 на один атом железа (Vanin et al., 2011). Следует отметить, что в экспериментах с использованием эквимолярной смеси (МС + Fe2+) концентрация компонентов этой смеси не превышала 0.3 мМ. При большей их концентрации, особенно при щелочном варианте метода ДНК-комет, ионы железа в заметном количестве включались в водонерастворимые гидроокисные комплексы (FeOH)n, затруднявшие визуализацию комет и искажавшие результаты.
Культура клеток. Клетки аденокарциномы молочной железы человека линии MCF-7 были получены из коллекции лаборатории экспериментальной терапии опухолей НМИЦ онкологии им. Н.Н. Блохина (Москва). Клетки культивировали в атмосфере 5% СО2 при 37°С в среде DMEM (ThermoFisherScientific, США), содержащей 10% эмбриональной телячьей сыворотки, 2 мМ глутамина и 10 Ед/мл смеси пенициллина и стрептомицина (ПанЭко, Россия).
Оценка цитотоксичности. Цитотоксичность ДНКЖ-MC и эквимолярной смеси компонентов комплекса (MC + Fe2+) оценивали по снижению жизнеспособности клеток MCF-7 после воздействия препаратов. Жизнеспособность клеток определяли с помощью МТТ-теста. Клеточную суспензию в культуральной среде (~105 кл./0.2 мл) вносили в лунку 96-луночного планшета. Спустя 24 ч (время адаптации и прикрепления клеток), в каждую лунку добавляли раствор тестируемых соединений в стерильном фосфатно-солевом буфере (PBS) в различном разведении (не более 0.1 общего объeма в лунке). Клетки культивировали 48 ч в СО2-инкубаторе при 37°С, после чего в каждую лунку добавляли 20 мкл раствора МТТ-реагента (3-[4,5-диметилтриазол-2-ил]-2,5 дифенил тетразолия бромид) (AppliChem, Германия), до конечной концентрации в лунке 0.5 мг/мл. Клетки продолжали культивировать в присутствии МТТ-реагента 3 ч, затем отбирали среду и к оставшимся в лунках клеткам добавляли по 200 мкл ДМСО, растворяющего кристаллы формазана (37°С, 10 мин со встряхиванием). С помощью анализатора MultiscfnFC (ThermoScientific, США) измеряли оптическую плотность раствора формазана в лунке (при длине волны 570 нм), которая прямо пропорциональна количеству жизнеспособных клеток. В каждом эксперименте контрольные и экспериментальные лунки в планшетах повторялись 4-кратно.
Оценка генотоксичности методом комет. Использовали щелочной и нейтральный варианты метода комет (Olive, Banath, 2006) с некоторыми модификациями (Тронов, Некрасова, 2020). Их сочетание позволяет определять в разрывы в ДНК однонитевые (ОР), двунитевые (ДР), щелочелабильные сайты (ЩЛС) и строго их дискриминировать. Клетки (3 × × 105/0.3мл) высевали в 6-луночные плашки для культивирования в течение 24 ч при 37°С в атмосфере 5% СО2 для адгезии клеток на дно лунок. К прикрепленным клеткам добавляли тестируемые соединения, предварительно растворенные в культуральной среде до требуемой концентрации. Время совместного культивирования составляло 5 ч (время максимальной генотоксичности) и 24 ч (время эффективной репарации повреждений ДНК в клетках) По завершении совместного культивирования клетки снимали с подложки трипсинизацией, промывали холодным PBS , ресуспендировали в телячьей сыворотке, содержащей 10% ДМСО, разливали на аликвоты и замораживали в жидком азоте. Замороженные аликвоты хранили при −70°С. Размороженную суспензию клеток центрифугировали (400 g, 5 мин). Осадок суспендировали в 0.7%-ном растворе легкоплавкой агарозы (тип IV, Sigma) в PBS (ПанЭко, Россия). Из суспензии готовили слайд на предметном стекле по стандартной процедуре метода комет (Тронов и др., 2012). Стекла с застывшим гелем погружали в нейтральный лизирующий раствор: 0.5 M Na2EDTA, 2% Na-лауроилсаркозила, 0.3 мг/мл протеиназы К, pH 8, 37°С. После лизиса в течение 12–14 ч слайды либо подвергали нейтральному электрофорезу в ТАЕ-буфере рН 8 (0.8 В/см, 24 мин, 8°С), либо погружали в щелочной диссоциирующий раствор (1.2 M NaCl, 100 мМ Na2EDTA, 0.1% Na-лаурилсаркозината, 0.26 M NaOH, pH > 13) на 1 ч, при 8°С. В этих условиях хроматин диссоциирует, а ДНК расплетается в области разрывов, вызванных как тестируемыми агентами, так и под действием репарирующих ферментов клетки. Процедура завершалась погружением слайдов в электрофоретический буфер (0.03 M NaOH, 2 мМ Na2EDTA, pH ~ 12.3, 8°С, 1 ч). Электрофорез проводили в горизонтальной камере при 8°С в течение 25 мин и напряжении 0.8 В/см. После электрофореза слайды трижды по 5 мин ополаскивали нейтрализующим раствором (0.4 M Tris-HCl, pH 7.5), сушили на воздухе и дегидратировали в метаноле. Слайды окрашивали раствором ДНК-интеркалирующего красителя SYBR GreenI (Sigma), 2–4 мкг/мл в антифейде (20 мМ Tris-HCl рН 7.5, 20 мг/мл DABCO, 60% глицерина). Визуализацию комет осуществляли на флуоресцентном микроскопе МИКМЕД-2 (АО ЛОМО, Россия). Повреждение ДНК анализировали по изображениям ДНК-комет, используя программу CASP 1.2.2, оценивали повреждение по моменту хвоста комет mt (Końca et al., 2003).
Репрезентация результатов и статистика. Полученные данные представляли в виде распределений комет по параметру mt (рис. 1а, б), из которых находили усредненную поврежденность ДНК в клетках (генотоксичность, mt ± SD) и mt-цитотоксичность. Используемый в работе показатель mt-цитотоксичность определяли через 24 ч культивирования, в течение которой протекала и в основном завершалась репарация ДНК. Сохранившиеся к этому времени повреждения ДНК с большой вероятностью индуцировали в делящихся клетках апоптоз, т.е., прямо или опосредованно были связаны с жизнеспособностью клеток. Параметр mt-цитотоксичность представляет собой долю клеток/комет с высокой поврежденностью ДНК для щелочных (mt ≥ 20) и для нейтральных (mt ≥ 10) комет. Эти значения mt приняты в качестве пороговых для оценки mt-цитотоксичности, поскольку в распеделениях по mt интактных клеток более 95% комет имели значение mt ниже этих значений для щелочных и нейтральных условий электрофореза соответственно (рис. 1а, б). Эксперименты проводили независимо и повторяли трижды; результаты представлены средними значениями и их стандартными отношениями (SD); сравнение экспериментальных распределений осуществлялось с помощью непараметрической статистики Колмогорова–Смирнова, считая различия достоверными при Р < 0.05. Все статистические расчеты проведены с помощью программного обеспечения OriginPro 8.1. (Originlab Corporation, США).
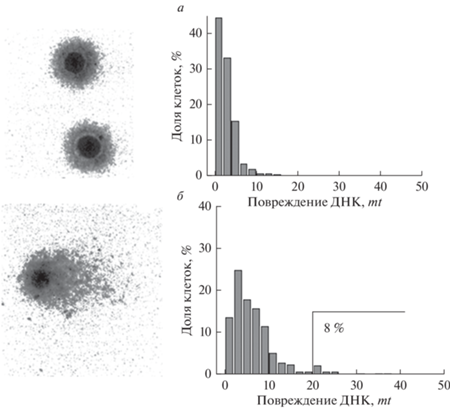
Рис. 1.
Репрезентативные микрофотографии щелочных ДНК-комет из интактных (а) и обработанных комплексом ДНКЖ-МС (б) клеток MCF-7, а также гистограммы распределения клеток по поврежденности ДНК в них. Показатель цитотоксичности (mt) представлен как доля клеток (комет) с высокой поврежденностью ДНК, для которых mt ≥ 20 (8%, б).
РЕЗУЛЬТАТЫ
Цитотоксичность комплекса ДНКЖ-МС и смеси предшественников его синтеза. Для сравнения цитотоксичностей комплекса ДНКЖ-MС и смеси предшественников его синтеза (МС + Fe2+) мы выравнивали их концентрации по Fe2+, полагая, что на 1 моль ДНКЖ-МС приходится 1 моль Fe2+, связанного с комплексом. Кроме того, в отличие от свободного Fe2+, способного вступать в реакцию Фентона, цитотоксичность МС в исследуемом диапазоне концентраций близка к нулю.
Рис. 2 демонстрирует жизнеспособность клеток MCF-7 (определенной по МТТ-тесту) после культивирования их в течение 48 ч с различными концентрациями комплекса ДНКЖ-MС и эквимолярной смеси предшественников его синтеза – (МС + Fe2+). Два факта, наблюдаемые на рисунке, обращают на себя внимание: дозозависимое увеличение жизнеспособности клеток в диапазоне концентраций комплекса ДНКЖ-MС 0–40 мкМ и более чем 2-кратное увеличение цитотоксичности комплекса по сравнению с цитотоксичностью смеси (MС + Fe2+) – значение концентрации, при которой гибнет половина клеточной популяции (IC50), составляет 0.85 мМ против 2.0 мМ соответственно.
Рис. 2.
Дозовая зависимость жизнеспособности клеток MCF-7 через 48-ч культивирования с эквимолярной смесью меркаптосукцината (МС) с Fe2+(кривая 1) и с комплексом ДНКЖ-MС (кривая 2). Жизнеспособность клеток определяли с помощью МТТ-теста. По вертикали: доля живых клеток, %.
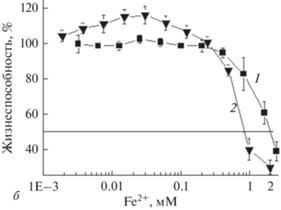
Первый факт полностью соответствует общепризнанной точке зрения, согласно которой при различных концентрациях оксида азота (его химических доноров) проявляются различные физиологические функции NO (Khan et al., 2020). На опухолевых клетках показано, что в диапазоне средних концентраций NO, создаваемых донорами NO в клетке (100–350 мкМ) наблюдается генетическая нестабильность, репарация ДНК, снижение апоптоза (Yakovlev, 2013; Huerta, 2008). Эти реакции соответствуют клеточному адаптивному ответу, который включает в себя возрастание пролиферации, что демонстрирует рис. 2 (кривая 2). В ответ на более высокие концентрации NO (500–1000 мкМ) увеличивается повреждение ДНК, активируется АТМ/ATR-сигналинг, фосфорилирующий белок р53, который тормозит пролиферацию и активирует апоптоз (Yakovlev et al., 2010). Что касается второго факта, то поскольку цито- и генотоксичность смеси (МС + Fe2+) в большой степени обусловлена свободным Fe2+, включение его в комплекс ДНКЖ уводит его из токсического эффекта комплекса.
На циркулярной ДНК плазмиды показано (Lewandowska et al., 2015), что включение железа в комплекс ДНКЖ предотвращает реакцию Фентона и тем самым защищает ДНК плазмиды от деградации под действием окислительного стресса. Полученный нами результат (рис. 2) на первый взгляд противоречит результатам этой работы. Поэтому мы оценили генотоксический эффект этих соединений в клетках MCF-7, т.е. их способность индуцировать повреждения ДНК. Эти повреждения могут возникать как под действием продуктов распада комплекса ДНКЖ (NO, NO+ и свободного Fe2+), так и в результате активации процессов репарации в ответ на повреждение ДНК (разрывы и апуриновые и апиримидиновые сайты (АП) сайты), которые в щелочных условиях лизиса клеток трансформируются в разрывы).
Влияние комплекса ДНКЖ-МС и его компонентов на ДНК и ее репарацию.
В отличие от результатов по параметру цитотоксичности с помощью МТТ-теста (рис. 2), наш результат по генотоксичности клеток MСF-7 через 5 ч совпадает с данными по повреждению ДНК in vitro, полученными в упомянутой выше работе (Lewandowska et al., 2015): mt-генотоксичность свободного железа выше таковой комплекса (рис. 3а). Однако культивирование клеток в присутствии агентов в течение 24 ч, помимо того, что снижает дозовые зависимости, но и меняет их местами: повреждения, индуцированные смесью (MS + Fe2+), практически полностью репарируются (рис. 3б); в случае с комплексом к 24 ч, как видно, сохраняются нерепарированные повреждения в субпопуляции клеток, составляющей 19%. В клетках, обработанных смесью (MС + Fe2+), объем такой субпопуляции составляет 6% от общей численности клеток в распределении (более 400 клеток). Рис. 4 наглядно демонстрирует это и достоверную разницу между mt-распределениями сравниваемых популяций клеток через 24 ч обработки агентами. Различия между распределениями достоверны при уровне значимости Р < 0.05 (статистика Колмогорова−Смирнова). Как видно, mt-цитотоксичности смеси MС+ Fe2+ 0.3 mM и комплекса ДНКЖ-MC, 1 мМ, найденные из полученных распределений, составляют 6 (рис. 4а) и 19% (рис. 4б) соответственно. Рис. 5 представляет объединенный результаты оценки цитотоксичности по МТТ-тесту и mt-распределению клеток, культивируемых с тестируемыми агентами 48 и 24 ч соответственно. Цитотоксичность комплекса, более высокая по результатам МТТ-теста, по сравнению с оценкой этого параметра по моменту хвоста комет mt. Это может быть связанo с более длительным культивированием клеток с агентами при МТТ-тестировании.
Рис. 3.
Генотоксичность через 5 ч (а) и цитотоксичность через 24 ч (б) действия смеси (MC + Fe2+) (кривая 1) и комплекса ДНКЖ-MС (кривая 2) на клетки MCF-7. Генотоксичность, mt, определяли как среднюю поврежденность ДНК в клетке через 5 ч обработки агентами. Цитотоксичность оценивали как долю клеток (%) с поврежденной ДНК через 24 ч обработки агентами.
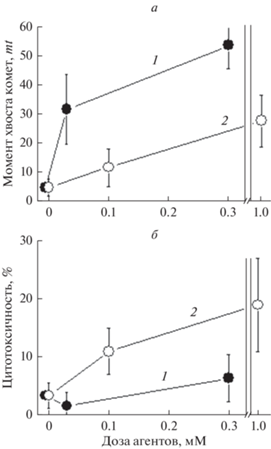
Рис. 4.
Гистограммы распределений MCF-7 клеток по повреждению ДНК (по показателю mt) после воздействия в течение 24 ч 0.3 мМ смеси (MС + Fe2+) (а) и 1 мМ комплекса ДНКЖ-MC (б). Цитотоксичность смеси и комплекса составляет 6 и 19% соответственно (число клеток в каждом распределении более 400). Гистограммы построены по результатам 3–4 экспериментов.
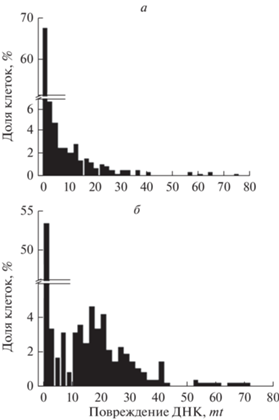
Рис. 5.
Цитотоксичность комплекса ДНКЖ-MC (1 мМ) и 0.3 мМ смеси (MC + Fe2+), оцененная по mt-распределению (черные столбики) и МТТ-тесту (серые столбики) клеток MCF-7; * различия недостоверны, **различия статистически достоверны при Р < 0.05 (cм. рис. 4).
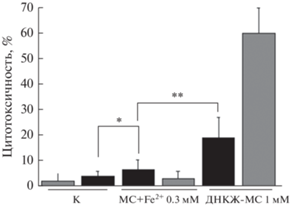
Следует подчеркнуть, что представленная на рис. 5 цитотоксичность, определенная по значениям mt (черные столбики) базируется на измерении разрывов ДНК через 24 ч, которые включают в себя ОР, ДР и ЩЛС в ДНК. Но известно, что ОР является наиболее частым и легкоустраняемым дефектом в геноме клетки, а потому сам по себе не является летальным. Стало быть, длительное его пребывание в клетке может быть связано либо c NO-инициированной инактивацией ферментов репарации, либо с тем, что он представляет собой потенциально летальный ДР, обусловливающий цитотоксический эффект (Roos, Kaina, 2012). Поэтому мы провели проверку клеток MCF7, обработанных агентами в течение 24 ч, на наличие в их ДНК двунитевых разрывов, используя для этого метод нейтральных ДНК-комет. Результат суммирован в табл. 1 в виде распределения клеток по повреждению ДНК. Каждое из 3-х представленных в таблице распределений получено путем объединения результатов 3–4-х независимых экспериментов.
Таблица 1.
Поврежденность двунитевой ДНК (днДНК) в клетках MCF-7 в ответ на воздействие 0.3 мМ смеси (MС + Fe2+) и 1 мМ комплекса ДНКЖ-МС в течение 24 ч
| Обработка клеток |
Распределение клеток по повреждению двунитевой ДНК, mt |
Параметры распределения |
||
|---|---|---|---|---|
| N | ❬mt❭ ± SD | ЦТ, % | ||
| − (Интактные) |
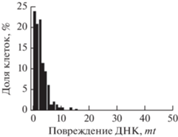 |
299 | 2.7 ± 2.4 | 2.3 |
| (MС + Fe2+) 0.3 мМ, 24 ч |
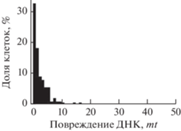 |
281 | 2.2 ± 2.5 | 1 |
| (ДНКЖ-MС) 1 мМ, 24 ч |
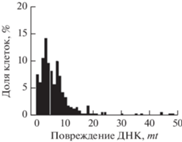 |
332 | 8.2 ± 14 | 17 |
Из табл. 1 видно, что 24-часовая обработка клеток смесью предшественников комплекса практически не оставляет в клеточной ДНК двунитевых разрывов: вид распределения и средние показатели поврежденности клеток не отличаются от таковых для интактных клеток. Комплекс ДНКЖ-МС через 24 ч оставляет существенные повреждения в ДНК в виде двунитевых разрывов. Эти разрывы локализованы в 17% клеток, и их число практически совпадает со значением цитотоксичности (19%), найденной в аналогичных экспериментах со щелочными кометами (рис. 4)
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
NO включен во многие внутри- и межклеточные сигнальные пути, регулирующие физиологические процессы клетки. В то же время, в силу высокой реакционной способности, NO повреждает ДНК в клетке, вызывая модификацию оснований (дезаминирование, окисление), фоpмирование апуриновых и апиримидиновых сайтов, разрывы цепей ДНК, внутри- и межцепочечные сшивки (см. обзоры: Szabo, Ohshima, 1997; Burney et al., 1999). Как следствие таких повреждений, продемонстрирован мутагенный эффект NO на культивируемых лимфобластах человека ТК6 по выходу мутаций в репортерных генах HPRT и ТК (Nguyen et al., 1992): частота NO-индуцированных мутаций в 15–18 раз превышала частоту спонтанного мутирования этих локусов.
В другой работе (Tamir et al., 1996) в качестве мишени для NO использовали кольцевую суперскрученную ДНК плазмиды in vitro. Оказалось, что во внеклеточной среде NO-обработанная ДНК плазмиды не подвергалась никированию, т.е. не содержала разрывов. Но после трансфекции NO-обработанной плазмиды в клетки СНО плазмида подвергалась никированию (релаксации). Другими словами, при наличии активной системы репарации в клетке в NO-поврежденной ДНК появляются разрывы. Эти результаты говорят о том, что первичным повреждением ДНК под действием NO• является безразрывное дезаминирование пуринов в ксантин и гипоксантин, которые либо подвергаются спонтанной депуринизации, либо активно удаляются гликозилазами, гидролизирующими N-гликозидную связь между химически модифицированным основанием и дезоксирибозой. В обоих случаях удаление основания оставляет безразрывный пропуск в виде апуринового и апиримидинового (АП) сайта. АП-сайт является субстратом для АП-эндонуклеазы 1, которая гиролизует в нем фосфодиэфирную связь, формируя разрыв цепи ДНК с 3'OH- и 5'dRP-концами. Далее ДНК-полимераза β удаляет 5'dRP-конец разрыва и на его место присоединяет один нуклеотид, комплементарный противолежащему нуклеотиду, тем самым заполняя пробел в АП-сайте. ДНК-лигазы I или III завершают процесс восстановления целостности ДНК по механизму BER. Следует отметить, что АП-сайт сам по себе не является разрывом в цепи ДНК, но гидролизуется при щелочном рН и потому относится к группе повреждений, называемых ЩЛС.
В случае действия смеси (MС + Fe2+) протекает реакция Фентона со свободным Fe2+, в результате которой либо окисляются основания, либо гидролизуется фосфодиэфирная связь в цепи ДНК. В обоих случаях активируется механизм BER, который удаляет и поврежденные основания, и однонитевые разрывы, как это показано на рис. 5 (различие с контролем статистически не достоверно).
В отличие от разрывов под действием смеси (MС + Fe2+), разрывы, возникшие в ответ на обработку клеток комплексом ДНКЖ-MС, репарируются медленнее, и через 24 ч наблюдается заметная доля клеток, сохраняющих высокий уровень повреждений в ДНК (рис. 5). Отсутствие репарации разрывов ДНК в NO-обработанных лимфобластах человека отмечали ранее другие авторы (Nguyen et al., 1992). Наши опыты с нейтральными ДНК-кометами показали, что эти разрывы скорее всего являются двунитевыми. В пользу этого предположения говорит тот факт, что доли клеток, имеющих в ДНК однонитевые и двунитевые разрывы, оцененные методами щелочных и нейтральных ДНК-комет, почти совпадают – 19 и 17% соответственно (рис. 4б и табл. 1). Это совпадение говорит о том, что возможно ДР появились на месте ОР в результате атаки однонитевого участка клеточной эндонуклеазой.
Такое событие вполне вероятно в ходе репарации сложных аддуктов оснований. Показано, что такие аддукты образуются в результате прямой атаки ДНК азотистым ангидридом N2O3 (Tannenbaum et al., 1994), который, в свою очередь, накапливается в клетке в результате реакции оксида азота с молекулярным кислородом (Lewis et al., 1995). Взаимодействие N2О3 с гуанином приводит к дезаминированию с образованием диазониевого катиона. Если таковые соседствуют в ДНК, то образуется межспиральный димер (cшивка) (Burney et al., 1999). Репарация таких аддуктов в клетке осуществляется механизмом NER (nucleotide excision repair). Важной особенностью NER является то, что размер выщепляемого участка ДНК, содержащего аддукт, у клеток млекопитающих составляет ~30 нуклеотидов (Shivji, 1995) (в механизме BER он ≤3 нуклеотидов в случае репарации АП-сайта и ОР, и не превышает 10 при репарации окси-модифицированных оснований).
Кроме того, обширное повреждение ДНК чревато разбалансировкой этапов репарационного процесса (Тронов, Некрасова, 2020). К рассогласованию этапов репарации может приводить и NO-индуцированная пост-трансляционная модификация белков NER, снижающая их активность и эффективность репарации в целом, (Graziewicz et al., 1996; Jaiswal et al., 2001). Именно с этим связан результат работы, в которой показано, что арсенит, экзогенный донор NO и пероксинитрита, увеличивал продукцию NO и одновременно с этим наблюдали подавление NER-эксцизии аддуктов ДНК в фибробластах человека, индуцированных УФ-излучением, цисплатином, митомицином и бензопиреном (Chien, 2004). В механизм NER у млекопитающих вовлекаются более 30 различных белков, которые являются мишенью для NO-опосредованной модификации.
Таким образом, в результате незавершенной репарации увеличивается время существования таких интермедиатов процесса NER, как АП-сайты и протяженные однонитевые участки в ДНК-дуплексе; будучи атакованы эндонуклеазами, они переходят в двунитевые разрывы.
Список литературы
Тронов В.А., Виноградова Ю.В., Поплинская В.А., Островский М.А. 2012. Механизмы радиорезистентности терминально дифференцированных клеток зрелой сетчатки глаза. Цитология. Т. 54. С. 261. (Tronov V.A., Vinogradova U.V., Loginova M.U., Poplinskaya V.A., Ostrovsky M.A. 2012. Mechanisms of radioresistance in terminally differentiated cells of mature retina. Cell Tiss. Biol. V. 6. P. 219.)
Тронов В.А., Некрасова Е.И. 2020. Повреждение ДНК и белок Р53 ограничивают пролиферацию клеток Мюллера в сетчатке мышей в ответ на действие метилнитрозомочевины. Биофизика. Т. 65. С. 543. (Tronov V.A., Nekrasova E.I. 2020. DNA Damage and p53 restrict proliferation of Muller cells in mouse retina in response to the influence of n-methyl-n-nitrosourea. Biophysics (Russ.).
Burgova E.N., Khristidis Y.I., Kurkov A.V., MikoyanV.D., Shekhter A.B., Adamyan L.V., Timashev P.S., Vanin A.F. 2019. The inhibiting effect of dinitrosyl iron complexes with thiol-containing ligands on the growth of endometrioid tumours in rats with experimental endometriosis. Cell Biochem. Biophys. V. 77. P. 69.
Burney S., Caulfield J.L., Niles J.C., Wishnok J.S., Tannenbaum S.R. 1999. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. V. 424. P. 37.
Carmeliet P., Jain R.K. 2011. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. V. 10. P. 417.
Chien Y.-H., Bau D.-T., Jan K.-Y. 2004. Nitric Oxide inhibits DNA-adducts excision in nucleotide excision repair. Free Rad. Biol. Med. V. 36. P. 1011.
Du X.L., Edelstein D., Dimmeler S., Ju Q., Sui C., Brownlee M. 2001. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Invest. V. 108. P. 1341.
Giacco F., Brownlee M. 2010. Oxidative stress and diabetic complications. Circ. Res. V.107. P. 1058.
Godínez-Rubí M., Rojas-Mayorquín A.E., Ortuño-Sahagún D. 2013. Nitric oxide donors as neuroprotective agents after an ischemic stroke-related inflammatory reaction. Oxid. Med. Cell. Long. Article ID 297357. https://doi.org/10.1155/2013/297357
Graziewicz M., Wink D.A., Laval F. 1996. Nitric oxide inhibits DNA ligase activity: Potential mechanisms for NO-mediated DNA damage. Carcinogenesis. V. 17. P. 2501.
Huang Z., Fu J., Zhang Y. 2017. Nitric oxide donor-based cancer therapy: Advances and prospects. J. Med. Chem. V. 60. P. 7617.
Huerta S., Chilka S., Bonavida B. 2008. Nitric oxide donors: novel cancer therapeutics iew) (review). Int. J. Oncol. V. 33. P. 909.
Jaiswal M., LaRusso N.F., Shapiro R.A., Billiar T.R., Gores G.J. 2001. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterol. V. 120. P. 190.
Kelsey M.S., Kwon M-Y., Chung S.W., Kim E. 2014. Coordinationtriggered NO release from a dinitrosyl iron complex leads to anti-inflammatory activity. Chem. Sci. V. 5. P. 2374.
Khan F.H., Dervan E., Bhattacharyya D.D., McAuli_J.D., Miranda K.M., Glynn S.A. 2020. The role of nitric oxide in cancer: master regulator or NOt? Int. J. Mol. Sci. V. 21. P. 9393. https://www.mdpi.com/1422-0067/21/24/9393
Końca K., Lankoff, A., Banasik, A., Lisowska, H., Kuszewski,T., Goźdź, S., Koza Z. Wojcik, A. 2003. A cross-platform public domain PC image-analysis program for the comet assay. Mutat. Res. V. 534. P. 15.
Lewandowska H., Sadło J., Męczyńska S., Stępkowski T.M., Wójciuk G., Kruszewsk M. 2015. Formation of glutathionyl dinitrosyl iron complexes protects against iron genotoxicity. Dalton Trans. V. 44. P. 12640.
Lewis R.S., Tannenbaum S.R., Deen W.M. 1995. Kinetics of N-nitrosation in oxygenated nitric oxide solutions at physiological pH: role of nitrous anhydride and effects of phosphate and chloride. J. Am. Chem. Soc. V. 117. P. 3933.
Nguyen T., Brunson D., Crespi C. L., Penman B.W., Wishnok J.S., Tannenbaum S.R. 1992. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc. Natl. Acad. Sci. USA. V. 89. P. 3030.
Olive P.L, Banath J.P. 2006. The comet assay: A method to measure DNA damage in individual cells. Nature Protocol. V. 1. P. 23.
Reina-Torres E., De Ieso M.L., Pasquale L.R., Madekurozwa M., van Batenburg-Sherwood J., Overby D.R., Stamer W.D. 2021.The vital role for nitric oxide in intraocular pressure homeostasis. Prog. Retin Eye Res. V. 83. P. 1.
Roos W.P., Kaina B. 2012. DNA damage-induced apoptosis: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Letters. V. 332. P. 237. https://doi.org/10.1016/j.canlet.2012.01.007
Shivji M.K., Podust V.N., Hubscher U., Wood R.D. Nucleotide excision repair DNA synthesis by DNA polymerase epsilon in the presence of PCNA, RFC, and RPA. 1995. Biochemistry. V. 34. P. 5011.
Szabo C., Ohshima H. 1997. DNA damage induced by peroxynitrite: Subsequent biological effects, nitric oxide: Biol. Chem. V. 1. P. 373.
Tamir S., Burney S., Tannenbaum S.R. 1996. DNA damage by nitric oxide. Chem. Res. Toxicol. V. 9. P. 821.
Tannenbaum S.R., Tamir S., deRojas-Walker T., Wishnok J.S. 1994. DNA damage and cytotoxicity by nitric oxide. In: Nitrosamines and related n-nitroso compounds. Washington: American Chemical Society. V. 10. P. 120.
Török J. 2008. Participation of nitric oxide in different models of experimental hypertension. Physiol. Res. V. 57. P. 813.
Vanin A.F., Poltorakov A.P., Mikoyan V.D., Kubrina L.N., Burbaev D.Sh. 2011. Polynuclear water-soluble dinitrosyl iron complexes with cysteine or glutathione: electron paramagnetic resonance and optical studies. Nitric Oxide – Biol. Chem. V. 23. P. 136.
Vanin A.F., Tronov V.A., Borodulin R.R. 2021. Nitrosonium cation as a cytotoxic component of dinitrosyl iron complexes with thiol-containing ligands (based on the experimental work on MCF7 human breast cancer cell culture). Cell Biochem. Biophys. V. 7. P. 93.
Wu S.C., Lu C.Y., Chen Y.L., Lo F.C., Wang T.Y., Chen Y.J., Yuan S.S., Liaw W.F., Wang Y.M. 2016. Water-soluble dinitrosyl iron complex (DNIC): A nitric oxide vehicle triggering cancer cell Death via apoptosis. Inorg. Chem. V. 55. P. 9383.
Yakovlev V.A. 2013. Nitric oxide-dependent down regulation of BRCA1 expression promotes genetic instability. Cancer Res. V. 73. P. 706.
Yakovlev V.A., Bayden A.S., Graves P.R., Kellogg G.E., Mikkelsen R.B. 2010. Nitration of the tumor suppressor protein p53 at tyrosine 327 promotesp 53 oligomerization and activation. Biochemistry. V. 49. P. 5331.
Дополнительные материалы отсутствуют.