

Цитология, 2023, T. 65, № 1, стр. 54-63
Роль MAP-киназ в индуцированном фосфорилировании гистона H2AX в трансформированных клетках
О. О. Гнедина 1, А. В. Моршнева 1, М. В. Иготти 1, *
1 Институт цитологии РАН
194064 Санкт-Петербург, Россия
* E-mail: marie.igotti@gmail.com
Поступила в редакцию 28.07.2022
После доработки 06.10.2022
Принята к публикации 16.10.2022
- EDN: GOTCEB
- DOI: 10.31857/S0041377123010030
Аннотация
Ингибиторы гистондеацетилаз (ИГД) обладают антипролиферативными свойствами и являются перспективными агентами комбинированной противоопухолевой терапии. Ранее нами было показано, что представитель семейства ИГД бутират натрия (NaBut) самостоятельно не вызывает двунитевых разрывов ДНК (ДР-ДНК), однако, подобно генотоксическим агентам, инициирует накопление фосфорилированного гистона H2AX (γH2AX), являющегося маркером ДР-ДНК. ИГД способны также снижать эффективность репарации ДНК, поврежденной при генотоксических воздействиях, в трансформированных клетках. Цель настоящей работы состояла в идентификации сигнальных путей, приводящих к накоплению γH2AX при действии ИГД в трансформированных клетках. Для этого исследовали роль киназ семейства MAPK в NaBut-индуцированном фосфорилировании гистона H2AX, а также в ингибировании репарации ДНК. Мы показали, что накопление γH2AX в трансформированных клетках в ответ на действие NaBut сопровождается снижением уровня фосфорилирования киназ ERK и PKB/Akt. Активирующее фосфорилирование киназы р38 увеличивается при действии NaBut, вследствие чего накапливается фосфатаза Wip1, что может быть одной из причин ингибирования репарации ДНК. При этом подавление активности киназы p38 отменяет NaBut-индуцированное снижение эффективности репарации. Полученные нами результаты об отмене негативного влияния NaBut на репарацию ДНК при ингибировании MAP-киназы p38, а также данные о накоплении фосфатазы Wip1 при действии NaBut позволяют предположить, что путь p38/Wip1 вовлечен в NaBut-индуцированное снижение эффективности репарации в трансформированных клетках.
Одно из первых событий в процессе узнавания и репарации двунитевых разрывов (ДР) ДНК – маркирование места повреждения, в ходе которого происходит фосфорилирование гистона H2AX в месте разрыва (Rogakou et al., 1998). В зависимости от характера воздействия, вызвавшего повреждение ДНК, в клетке активируются разные сигнальные каскады, поэтому гистон H2AX может быть фосфорилирован различными киназами. Основными киназами, ответственными за фосфорилирование H2AX, являются киназы семейства PI3K-подобных киназ (PI3K-like kinases, PIKK): ATM, ATR и ДНК-ПК (van Attikum, Gasser, 2009). При появлении двунитевых разрывов ДНК, например, после воздействия ионизирующего излучения, в фосфорилировании H2AX принимают участие киназы ATM и ДНК-ПК, тогда как ATR активируется при обнаружении длинных однонитевых участков ДНК, в ответ на репликативный стресс и ультрафиолетовое облучение (Menolfi, Zha, 2020).
Ранее нами было показано, что ингибиторы гистондеацетилаз (ИГД), подобно генотоксическим агентам, вызывают накопление фосфорилированной формы гистона H2AX (γH2AX) в онкоген-трансформированных фибробластах (Abramova et al., 2011), что, однако, не сопровождается появлением двунитевых разрывов ДНК (ДР-ДНК) (Abramova et al., 2011; Gnedina et al., 2022). Накопление γH2AX при действии ИГД было показано и в других типах опухолевых и трансформированных клеток (Gaymes et al., 2006; Wang et al., 2012), но ни в одной из опубликованных работ не было доказано наличие ДР-ДНК методом нейтральных комет, специфически выявляющим именно двунитевые разрывы.
ИГД способны повышать чувствительность ряда опухолевых клеток к ионизирующему излучению (Konsoula et al., 2011) и к действию генотоксических агентов (Abramova et al., 2011; Thurn et al., 2013; Wu et al., 2017), что сопровождается увеличением количества γH2AX при совместном действии этих факторов. Многочисленные экспериментальные данные указывают на способность ИГД модулировать клеточный ответ на повреждение ДНК (DNA damage response, DDR) через изменение экспрессии генов, кодирующих белки распознавания ДНК-повреждений и репарации, таких как ATM, BRCA1/2 (Konsoula et al., 2011; Thurn et al., 2013), Ku70/Ku80, Rad51, DNA-PKcs и Mre11 (Adimoolam et al., 2007; Lee et al., 2010; Munshi et al., 2005; Wu et al., 2017), в раковых, но не в нормальных клетках (Lee et al., 2010; Munshi et al., 2005). Способность ИГД сенсибилизировать раковые клетки к терапевтическим агентам, нацеленным на повреждение ДНК, и их низкая токсичность для нормальных медленно делящихся клеток позволяет рассматривать эти вещества как перспективные агенты противораковой терапии.
Представленная работа направлена на выявление вероятных мишеней, вовлеченных в сенсибилизацию раковых клеток с помощью ИГД. Такими мишенями могут быть митоген-активируемые протеинкиназы (MAP-киназы). MAP-киназы представляют собой семейство серинтреониновых киназ, передающих сигнал от внешних стимулов (факторов роста, цитокинов, гормонов) к нижележащим сигнальным белкам, регулирующим различные клеточные процессы – клеточный рост, пролиферацию, дифференцировку и пр. MAP-киназы принимают участие в регулировании ответа клетки на повреждение ДНК (Sanchez-Prieto et al., 2000; Wu et al., 2006; Picco, Pagès, 2013), в частности в фосфорилировании гистона H2AX (Kasibhatla et al., 1998; Lu et al., 2008; Phong et al., 2010) и регуляции активности белков системы репарации ДНК (Wu et al., 2006; Golding et al., 2007; Wei et al., 2011; Wang et al., 2013). Для выявления сигнальных путей, регулирующих фосфорилирование H2AX в отсутствие ДР-ДНК в ответ на действие ИГД, мы исследовали вклад MAP-киназ в накопление γH2AX, а также ингибирование репарации ДНК при действии ИГД в трансформированных клетках.
МАТЕРИАЛ И МЕТОДИКА
Клеточные линии и воздействия. В работе использовали линию трансформированных клеток mERas, полученную из первичных эмбриональных фибробластов мыши с помощью Ca-фосфатной трансфекции онкогеном cHa-ras, несущим активирующие точечные мутации в кодонах 12 и 61, и ранним районом Е1А генома аденовируса человека 5-го типа (Pospelova et al., 1999). Эмбриональные фибробласты мыши с дезактивирующими мутациями киназы p38 (p38Y182F/Y182F (p38ki/ki), клеточная линия mERas-p38−/−)), а также с нокаутом киназ JNK1/2 (mERas-JNK−/−) были любезно предоставлены доктором Дмитрием Булавиным (Институт исследований рака и старения Ниццы (IRCAN), INSERM, Университет Лазурного Берега, CNRS, Ницца, Франция). Клетки культивировали при 37°C и 5% CO2 в среде DMEM (Биолот, Россия), дополненной 10% сыворотки FCS (Gibco, США), 2 мМ L-глутамина (Биолот, Россия) и содержащей 40 мкг/мл гентамицина (Биолот, Россия). Клетки обрабатывали 4 мМ бутирата натрия (NaBut) в течение короткого (1–4 ч) или длительного времени (24–72 ч) и (или) ингибиторами киназ МЕК (50 мкМ PD98059), p38 (20 мкМ SB203580), JNK (10 мкМ SP600125), PI3K (20 мкМ LY294002 или 10 мкМ вортманнина (WM)) в течение 24 ч.
Вестерн-блотинг белков. Клетки лизировали в буфере PBS, содержащем 1% NP-40, 0.5% дезоксихолата натрия, 0.1% додецилсульфата натрия (SDS), ингибиторы протеаз и фосфатаз, а также 1 мМ PMSF. Концентрацию белка измеряли спектрофотометрически при длине волны 595 нм по методу Бредфорда (Bradford, 1976). Клеточные экстракты (20–40 мкг) прогревали в буфере для проб (60 мМ Tris-HCl, pH 6.8, 2% SDS, 10% глицерина, 5% β-меркаптоэтанола, 0.01% бромфенолового синего) при 95°C в течение 5 мин. Белки разделяли электрофоретически в 10%-ном полиакриламидном геле в присутствии 0.1% SDS, переносили на мембрану PVDF (Millipore). Мембраны блокировали в течение 1.5 ч при комнатной температуре в блокирующем буфере (5%-ное обезжиренное сухое молоко в PBST (PBS с 0.5% Tween 20)) и инкубировали с соответствующими первичными антителами, разведенными в 1–5%-ном BSA/PBST, в течение 1 ч при комнатной температуре или в течение ночи при 4°C. Мембраны отмывали в PBST и инкубировали 1 ч при комнатной температуре в PBST с 5%-ным молоком, содержащем вторые антитела, коньюгированные с пероксидазой хрена. Белки на мембранах выявляли методом усиления хемилюминесценции (Thermo Sci., США) и визуализировали на Syngene PXi6 Access. В качестве первичных использовали антитела к следующим белкам: киназе ERK (no. sc-94), p38 (no. sc-535) (Santa Cruz Biotechnology, Inc. США), белку репарации Mre11 (no. NB100–142; Novus Biologicals, LLC, США), к фосфорилированной (р) форме pERK (no. 4377), pp38 (no. 9211), SAPK/JNK (no. 9252), pJNK (no. 9255), Akt (no. 9272), pAkt (no. 4058), фосфогистону H2AX (γH2AX; no. 2577), фосфатазе Wip1 (no. 11901), белку pp53 (no. 12571), GAPDH (no. 2118) (Cell Signaling, США).
Оценка эффективности репарации ДР методом реактивации транскрипции. Метод реактивации транскрипции (Host Cell Reactivation assay, HCR) для изучения эффективности восстановления как ДР-ДНК, так и повреждений ДНК другого характера широко используется в биологических исследованиях (Johnson, Latimer, 2005; Thoms et al., 2007; Sears, Turchi, 2012). В репортерный вектор pGL3basic-luc вносят ДР в область, кодирующую люциферазу, с помощью эндонуклеазы рестрикции, что делает невозможным транскрипцию поврежденного гена до его репарации в клетке-реципиенте. Клетки, рассеянные на 96-луночные планшеты (в среде DMEM с 10% FCS без антибиотика) в плотности 10 × 103 кл./лунку, трансфицировали интактным или линеаризованным вектором pGL3basic-luc с помощью трансфицирующего агента Lipofectamine 2000 (Invitrogen, США) в соответствии с инструкцией производителя. Для контроля эффективности трансфекции клетки одновременно трансфицировали контрольным вектором pRL-TK, экспрессирующим люциферазу Renilla.
Через 16 ч после трансфекции клетки оставляли необработанными или обрабатывали специфическими ингибиторами киназ отдельно или совместно с NaBut в течение 24 ч, после чего клетки лизировали и измеряли в лизатах активность люциферазы с использованием набора для репортерного анализа с двумя люциферазами – Firefly и Renilla (Promega, США) на люминометре TD-20/20 (Turner Design, США) в соответствии с инструкцией производителя. Результаты вычисляли в изменениях активности люциферазы Firefly относительно активности люциферазы Renilla (в разах). По активности люциферазы, синтезированной после восстановления поврежденной плазмидной ДНК, оценивали эффективность репарации ДР. Люциферазная активность в клетках с поврежденной ДНК, не обработанных агентами, принята за 1 ед. Было проведено не менее трех независимых экспериментов по совместной трансфекции. На графиках представлены средние значения из трех независимых экспериментов и ошибки среднего.
РЕЗУЛЬТАТЫ
Индуцированное NaBut накопление γH2AX сопровождается изменением активности МАР-киназ. ИГД вызывают накопление фосфорилированной формы гистона H2AX (γH2AX) (Gaymes et al., 2006; Abramova et al., 2011; Wang et al., 2012). Однако в отличие от генотоксических агентов, также индуцирующих накопление γH2AX, действие ИГД не приводит к ДР-ДНК (Abramova et al., 2011; Gnedina et al., 2022). Данные вестерн-блот-анализа (рис. 1) показывают, что индуцированное NaBut накопление фосфорилированной формы H2AX сопровождается изменением активности белков семейства MAP-киназ. Установлено, что NaBut вызывает накопление фосфорилированых форм киназ p38 и JNK1 в клетках mERas (рис. 1). Увеличение содержания γH2AX и активированных (фосфорилированных) форм киназ p38 и JNK1 обнаруживается через 24 ч после добавления NaBut к клеткам и продолжает увеличиваться до 72 ч действия агента. Стоит отметить, что NaBut не изменяет фосфорилирования киназы JNK2, являющейся положительным регулятором белка p53, в отличие от JNK1 (Fogarty et al., 2003; Tafolla et al., 2005). Активирующее фосфорилирование MAP-киназы ERK, а также киназы PKB/Akt, наоборот, уменьшается при действии NaBut с течением времени (рис. 1). По данным вестерн-блотинга с антителами против фосфорилированных форм киназ видно, что NaBut приводит практически к полной инактивации ERK и PKB/Akt в клетках mERas.
Рис. 1.
Вестерн-блоты белков клеток mERas, необработанных или обработанных ингибитором HDAC бутиратом натрия (NaBut) в течение 1–72 ч, с антителами к фосфорилированной форме гистона H2AX (γH2AX), а также к тотальным и фосфорилированным формам киназ ERK, p38, JNK и Akt. Gapdh использован в качестве контроля нагрузки.

Таким образом, индуцированное NaBut фосфорилирование гистона H2AX сопровождается активацией киназ p38 и JNK1 и снижением активности ERK и PKB/Akt.
Роль МАР-киназ в регулировании базального и индуцированного фосфорилирования гистона H2AX. Для изучения вовлеченности МАР-киназ в фосфорилированиe гистона H2AX анализировали изменение содержания γH2AX при подавлении активности соответствующих киназ, как с помощью специфических химических ингибиторов, так и на генетическом уровне. Результаты вестерн-блотинга показывают, что подавление активности киназ ERK и JNK специфическими ингибиторами PD98059 и SP600125 соответственно снижает базальный уровень фосфорилированности H2AX (рис. 2а). Ингибитор киназы p38, вещество SB203580, увеличивает экспрессию γH2AX (рис. 2а). Также по результатам вестерн-блотинга видно, что содержание γH2AX повышено в клетках mERas с генетически инактивированной киназой p38 (mERas(p38–/–)) по-сравнению с нативными клетками mERas (рис. 2в), что подтверждает негативную роль киназы p38 в фосфорилировании H2AX.
Рис. 2.
Вестерн-блоты с антителами к фосфорилированной форме гистона H2AX (γH2AX) белков клеток mERas с нормальными (а, б) или инактивированными (в, г) формами киназ JNK (mERas(JNK–/–) или p38 (mERas(p38–/–). а, б – Клетки mERas не обработаны (−) или обработаны ингибиторами (инг) МАР-киназ или киназ PI3, ATM, Chk1 или ДНК-ПК отдельно, или в комбинации с NaBut в течение 24 ч. в – Базальный уровень γH2AX в контрольных клетках mERas и в клетках с инактивированной киназой p38 (mERas(p38–/–)). г – Клетки не обработаны или обработаны NaBut в течение 24–72 ч. Gapdh использован в качестве контроля нагрузки. Обозначения: PD – PD98059 (ингибитор киназы ERK); SB – SB203580 (ингибитор киназы p38); SP – SP600125 (ингибитор киназы JNK1/2); WM и LY – соответственно вортманн и LY294002 (ингибиторы киназы PI3).
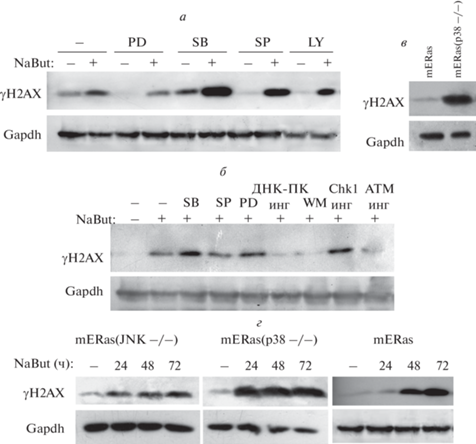
Индуцированное NaBut фосфорилирование H2AX не снижается при подавлении MAP-киназ p38, JNK или ERK специфическими химическими ингибиторами (рис. 2а, б). Только ингибитор PI3-киназы WM или специфические ингибиторы PI3-подобных киназ ДНК-ПК и АТМ отменяют NaBut-индуцированное накопление γН2АХ (рис. 2б). Соответствующие результаты получены при использовании клеток, в которых активность киназ p38 и JNK подавлена с помощью генетических модификаций. Данные вестерн-блотинга (рис. 2г) показывают, что в клетках mERas с инактивированными киназами р38 или JNK (mERas(p38–/–) и mERas(JNK–/–) соответственно) NaBut вызывает накопление γН2АХ с течением времени (рис. 2г), как и в контрольных трансформантах mERas.
При подавлении активности киназы p38 усиливается индуцированная NaBut экспрессия γН2АX (рис. 2). На рисунке 2г видно, что при сниженной активности киназы p38 в клетках mERas(p38–/–) γH2AX накапливается быстрее при действии NaBut, достигая максимального уровня уже через 24 ч действия агента. При этом уровень γH2AX, накопленный в результате действия NaBut, значительно превышает количество γH2AX в случае интактной p38 (рис. 2а, г).
Таким образом, подавление активности MAP-киназ не отменяет индуцированное NaBut накопление γH2AX, а ингибирование MAP-киназы p38, наоборот, приводит к увеличению количества γH2AX, как базального, так и индуцированного NaBut.
Роль MAP-киназ в индуцированном ИГД подавлении репарации в трансформированных клетках. Ранее было показано, что ИГД пролонгируют существование фокусов γH2AX, индуцированных генотоксическим воздействием (облучением, ингибиторами топоизомеразы II), благодаря снижению эффективности репарации ДНК в трансформированных клетках (Abramova et al., 2011; Gnedina et al., 2022). Для идентификации сигнальных путей, приводящих к ИГД-зависимому ингибированию репарации, анализировали влияние MAP-киназ на репарацию ДР-ДНК методом реактивации транскрипции поврежденной ДНК в клетке-реципиенте. Данные экспериментов по реактивации транскрипции показывают, что фармакологическое ингибирование PI3-киназы или PI3-подобных киназ ДНК-ПК и ATM снижает эффективность репарации ДР (рис. 3а, светлые столбцы), тогда как ингибирование киназ ERK, JNK и p38 не влияет на эффективность базальной репарации (рис. 3а).
Рис. 3.
Реактивация клетками-хозяевами транскрипции поврежденного эндонуклеазой гена люциферазы вектора pGL3-luc в контрольных клетках mERas (а) и в клетках mERas с инактивированными формами киназы JNK (JNK–/–) или p38 (p38‒/‒) (б). Клетки трансфицировали интактным или поврежденным вектором pGL3-luc и инкубировали без NaBut (серые столбики) или в присутствии NaBut (темные столбики) отдельно или в комбинации с ингибиторами (инг) MAP-киназ p38 (SB), ERK (PD, PD03), JNK (SP) или киназ ДНК-ПК, ATM/ATR или PI3K (WM, LY) в течение 24 ч. Относительную активность люциферазы, соответствующую эффективности репарации ДНК при действии химических веществ, рассчитывали по отношению к эффективности восстановления транскрипции гена люциферазы в ничем не обработанных клетках mERas. PD03 – PD0325901, другие обозначения те же, что и на рис. 2.
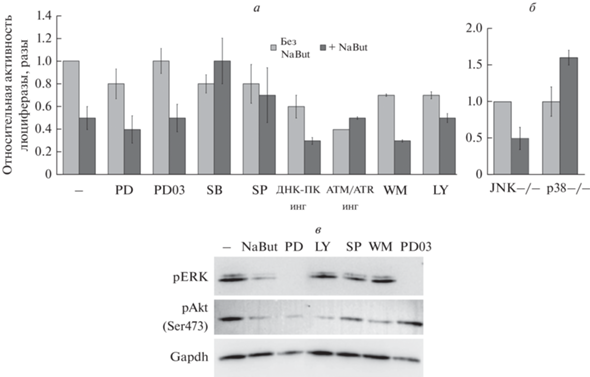
Индуцированное NaBut ингибирование репарации ДНК (рис. 3а, б, темные столбцы) отменялось только при подавлении активности киназы p38 веществом SB203580. Подобные результаты, свидетельствующие об определяющей роли киназы p38 в ИГД-зависимом ингибировании репарации, были получены при использовании клеток с генетически инактивированными киназами (рис. 3б). В клетках с инактивированной киназой JNK ДНК-репарация снижается в присутствии NaBut, как и в контрольных клетках mERas (рис. 3а, б). Однако в клетках, экспрессирующих p38 с инактивирующими мутациями, NaBut не снижает эффективность восстановления ДР. Полученные результаты позволяют рассматривать киназу p38, как один из возможных определяющих факторов в ИГД-индуцированной задержке репарации ДНК в трансформированных клетках.
Вестерн-блот, представленный на рис. 3в, демонстрирует эффективность работы ингибиторов киназ, использованных нами. Вещество PD98059 снижает содержание фосфорилированных форм киназ ERK1/2 и PKB/Akt (рис. 3в), а его ингибирующая активность в отношении вышележащих киназ в каскадах MEK/ERK и PI3K/Akt описана в литературе (Ye et al., 2018). Более специфичный ингибитор для MEK/ERK-пути вещество PD0325901 снижает содержание только pERK1/2. Ингибиторы PI3-киназы вещества LY294002 и WM уменьшают уровень фосфорилированности PKB/Akt. Ингибиторы киназ p38 и JNK не влияют на фосфорилированность этих киназ, а изменяют только их активность. Вещество SB203580 ингибирует каталитическую активность MAP-киназы p38 через связывание с АТФ-связывающим карманом киназы (Young et al., 1997) и не ингибирует фосфорилирование p38 (Kumar et al., 1999). Вещество SP600125 ингибирует активность киназ JNK за счет конкурирования за сайт связывания с АТФ (Bennett et al., 2001) и также не влияет на фосфорилирование самих киназ семейства JNK.
Таким образом, мы показали, что индуцированное NaBut ингибирование репарации ДНК отменялось только при подавлении активности МАР-киназы p38, как при использовании специфического химического ингибитора p38, так и при ее генетической инактивации.
Ингибитор гистон-деацетилаз бутират натрия активирует фосфатазу Wip1. Для выяснения возможной роли MAP-киназы p38 в ИГД-зависимой задержке репарации ДНК и накоплении γH2AX в трансформированных клетках исследовали влияние NaBut на изменение нижележащей мишени p38 фосфатазы Wip1 (Lowe et al., 2012). Фосфатаза Wip1 является членом семейства серин-треониновых фосфатаз PP2C, которая способна как самостоятельно дефосфорилировать гистон H2AX (Cha et al., 2010; Moon et al., 2010), так и через инактивирование киназы ATM (Shreeram et al., 2006) снижать количество γH2AX, в результате чего нарушается рекрутирование важных факторов репарации ДНК к поврежденным участкам и задерживается репарация ДР ДНК.
Результаты вестерн-блоттинга показывают, что NaBut параллельно с накоплением активированной формы киназы p38 вызывает накопление онкосупрессорного белка p53 и, как следствие, увеличение содержания фосфатазы Wip1 в трансформированных клетках mERas (рис. 4а).
Рис. 4.
Вестерн-блоты белков из клеток mERas, обработанных только NaBut в течение 24 ч (а) и NaBut отдельно или совместно с ингибитором фосфатаз окадаиковой кислотой (ОК) в течение 6 ч (б). ОК отменяет индуцированное NaBut появление гипофосфорилированной формы (Гипо-р) репарационного белка Mre11; Гипер-р и Укороч – гиперфосфорилированная и укороченная форма Mre11 соответственно.

Ранее нами было показано, что ИГД-индуцированное нарушение репарации ДНК в трансформированных клетках связано с дерегуляцией ключевых репарационных белков, а именно с уменьшением пула фосфорилированного белка репарации ДР Mre11 и накоплением его гипофосфорилированной и укороченной неактивной формы (Gnedina et al., 2022). Для изучения возможной роли фосфатазы Wip1 в дефосфорилировании и инактивировании белка Mre11 клетки mERas были обработаны NaBut отдельно или совместно с ингибитором фосфатаз окадаиковой кислотой. Результаты вестерн-блот-анализа показывают, что окадаиковая кислота предотвращает индуцированное NaBut появление гипофосфорилированного белка Mre11, а также его укороченной неактивной формы (рис. 4б).
ОБСУЖДЕНИЕ
Несмотря на наличие новых терапевтических стратегий, распространенной тактикой противоопухолевой терапии остается применение генотоксических агентов. Генотоксические агенты вызывают разрывы ДНК, которые сопровождаются накоплением маркера ДР-ДНК фосфорилированной формы гистона H2AX (γH2AX) (Rogakou et al., 1998; van Attikum, Gasser, 2009). ИГД самостоятельно не вызывают ДР-ДНК, однако, подобно генотоксическим агентам, вызывают накопление γH2AX (Gaymes et al., 2006; Abramova et al., 2011; Wang et al., 2012), а также продлевают существование фокусов γH2AX, индуцированных генотоксическими воздействиями (Abramova et al., 2011), что позволяет предположить такую реорганизацию клеточного ответа на повреждение ДНК при действии ИГД в трансформированных клетках, которая приводит к нарушению или невозможности восстановления повреждений ДНК, вызванных генотоксическими агентами. Для идентификации сигнальных путей, приводящих к накоплению γH2AX при действии ИГД в трансформированных клетках, нами исследована роль представителей семейства MAP-киназ.
Мы показали, что индуцированное NaBut фосфорилирование гистона H2AX сопровождается активацией киназ p38 и JNK1 и снижением активности ERK и PKB/Akt (рис. 1). Подавление активности киназ ERK и JNK уменьшало базальный уровень γH2AX, но не отменяло NaBut-индуцированного фосфорилирования H2AX и не влияло на задержку репарации ДР-ДНК, обусловленную действием NaBut (рис. 3). Ингибирование MAP-киназы p38 увеличивало как базальное, так и индуцированное NaBut фосфорилирование H2AX (рис. 2). Индуцированное NaBut ингибирование репарации ДНК отменялось при снижении активности p38 как при использовании специфического химического ингибитора p38, так и при генетической инактивации (рис. 3).
В литературе широко представлены данные о роли MAP-киназ в клеточном ответе на повреждения ДНК. Сигнальный путь MEK/ERK участвует в регулировании клеточного ответа на ДНК-повреждения, способствуя остановке клеточного цикла (Wu et al., 2006; Yan et al., 2007) для предотвращения деления с поврежденной ДНК и инициирования репарации. Также ERK может осуществлять регулирование фосфорилирования гистона H2AX через модуляцию активности киназ ATM и ATR (Wu et al., 2006; Golding et al., 2007).
Киназы JNK вовлечены в регулирование ответа клетки на повреждение ДНК и репарации через транскрипционный фактор p53 (Picco, Pagès, 2013), изменяя его стабильность и активность. Киназы JNK могут также непосредственно фосфорилировать гистон H2AX по положению Ser139 в ответ на генотоксические воздействия (Lu et al., 2006; Sluss, Davis, 2006).
МАР-киназы семейства p38 активируются в ответ на повреждение ДНК, накапливаются преимущественно в ядре, где стабилизируют и активируют транскрипционный фактор p53 (Sanchez-Prieto et al., 2000), следствием чего является пролиферативный блок клеточного цикла G1/S, репарация ДНК и (или) запуск апоптоза. Существуют противоречивые данные о влиянии киназ p38 на фосфорилирование гистона H2AX. С одной стороны, показано, что ингибирование активности p38 не отменяет экспрессию γH2AX, вызванную генотоксическим воздействием, в клетках HeLa, и активирует апоптоз (Phong et al., 2010). С другой стороны, ингибирование p38 снижает фосфорилирование H2AX и апоптоз, индуцированные сывороточным голоданием в трансформированных кератиноцитах (Lu et al., 2008). Более того, p38 способны непосредственно фосфорилировать H2AX, что необходимо для индукции апоптоза, вызванного сывороточным голоданием (Lu et al., 2008).
Показанная нами активация MAP-киназы p38 при действии ИГД приводит к p53-зависимому накоплению фосфатазы Wip1 (рис. 4). Серин-треониновая фосфатаза Wip1 вовлечена в передачу сигнала о повреждении ДНК (Cha et al., 2010; Moon et al., 2010) и способна как самостоятельно дефосфорилировать гистон γH2AX (Cha et al., 2010; Moon et al., 2010), так и через инактивирование киназы ATM (Shreeram et al., 2006) снижать количество γH2AX, в результате чего нарушается рекрутирование важных факторов репарации ДНК к поврежденным участкам и задерживается репарация ДР-ДНК (Moon et al., 2010). Полученные результаты о накоплении Wip1 при действии NaBut позволяют сделать предположение о ее роли в NaBut-зависимом ингибировании репарации.
Ранее нами было показано, что снижение эффективности восстановления ДР-ДНК при действии NaBut в трансформированных клетках связано с нарушением стабильности одного из компонентов репарационного комплекса MRN (Nbs1/Rad50/Mre11) белка Mre11 (Gnedina et al., 2022). NaBut вызывает накопление гипофосфорилированной формы Mre11, а также его неактивной укороченной формы (Gnedina et al., 2022). Известно, что дефосфорилирование белка Mre11 является инициирующим событием для его дальнейшего протеолиза до укороченной формы, лишенной ДНК-связывающей и нуклеазной активности (Nicholson et al., 2017). Инактивацию комплекса MRN связывают с повышением чувствительности опухолевых клеток к генотоксической терапии за счет снижения эффективности репарации повреждений ДНК (Bian et al., 2019).
Мы предположили участие фосфатазы Wip1 в дестабилизации репарационного белка Mre11 и показали, что ингибитор фосфатаз окадаиковая кислота, подавляющая активность Wip1 (Fiscella et al., 1997), отменяет NaBut-индуцированное накопление гипофосфорилированной формы белка Mre11 и его неактивной укороченной формы в трансформированных клетках (рис. 4б). Таким образом, индуцированная NaBut инактивация белка Mre11, опосредованная фосфатазой Wip1, предотвращает последующую сборку комплекса репарации и восстановление поврежденной ДНК, что и может объяснять ИГД-зависимое ингибирование репарации ДР-ДНК и персистирование фокусов γH2AX. Действие генотоксических агентов на клетки с модифицированной подобным образом системой ответа на ДНК-повреждения и репарации приводит к накоплению ДР-ДНК, репарация которых затруднена. Следовательно, можно предположить, что ИГД, самостоятельно не вызывая ДР, модифицируют систему ответа клетки на ДНК-повреждения таким образом, что сенсибилизируют клетки к генотоксическому стрессу, препятствуя репарации вносимых ими повреждений ДНК.
Фосфатаза Wip1 в настоящее время активно изучается как потенциальная мишень комбинированной противоопухолевой терапии в сочетании с генотоксическими агентами. Однако четкого видения, в каком направлении необходимо изменение активности Wip1, пока нет. Существуют данные о повышении чувствительности опухолевых клеток к генотоксическим агентам при ингибировании Wip1 (Belova et al., 2005; Pechackova et al., 2016). С другой стороны, увеличение апоптотической гибели р53-негативных опухолевых клеток при действии генотоксических агентов происходило при повышении экспрессии фосфатазы Wip1 через активацию транскрипции гена проапоптотического белка Bax (Goloudina et al., 2012; Eren et al., 2021).
Предложенная нами модель сенсибилизации трансформированных клеток к ДНК-повреждающим воздействиям с помощью фосфатазы Wip1 через дефосфорилирование и инактивирование репарационного белка Mre11 еще нуждается в дополнительном обосновании в исследованиях с применением более специфических ингибиторов фосфатазы и методов генетической инактивации wip1. Однако полученные результаты позволяют сделать предположение об участии p38/Wip1-пути в ИГД-индуцированном накоплении γH2AX и ингибировании репарации ДНК в трансформированных клетках.
Список литературы
Abramova M.V., Svetlikova S.B., Kukushkin A.N., Aksenov N.D., Pospelova T.V., Pospelov V.A. 2011. HDAC inhibitor sodium butyrate sensitizes E1A + Ras-transformed cells to DNA damaging agents by facilitating formation and persistence of γH2AX foci. Cancer Biol. Ther. V. 12. P. 1069.
Adimoolam S., Sirisawad M., Chen J., Thiemann P., Ford J.M., Buggy J.J. 2007. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA. V. 104. P. 19482.
Belova G.I., Demidov O., Fornace A.J., Bulavin D.V. 2005. Chemical inhibition of Wip1 phosphatase contributes to suppression of tumorigenesis. Cancer Biol. Ther. V. 4. P. 1154.
Bennett B.L., Sasaki D.T., Murray B.W., O’Leary E.C., Sakata S.T., Xu W., Leisten J.C., Motiwala A., Pierce S., Satoh Y., Bhagwat S.S., Manning A.M., Anderson D.W. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA. V. 98. P. 13681.
Bian L., Meng Y., Zhang M., Li D. 2019. MRE11-RAD50-NBS1 complex alterations and DNA damage response: implications for cancer treatment. Mol. Cancer. V. 18. P. 169.
Bradford M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. V. 72. P. 248.
Cha H., Lowe J.M., Li H., Lee J.-S., Belova G.I., Bulavin D.V., Fornace A.J. 2010. Wip1 directly dephosphorylates gamma-H2AX and attenuates the DNA damage response. Cancer Res. V. 70. P. 4112.
Eren M.K., Kartal N.B., Pilevneli H. 2021. Oncogenic WIP1 phosphatase attenuates the DNA damage response and sensitizes p53 mutant Jurkat cells to apoptosis. Oncol. Lett. V. 21. P. 479.
Fiscella M., Zhang H., Fan S., Sakaguchi K., Shen S., Mercer W.E., Vande Woude G.F., O’Connor P.M., Appella E. 1997. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA. V. 94. P. 6048.
Fogarty M.P., Downer E.J., Campbell V. 2003. A role for c-Jun N-terminal kinase 1 (JNK1), but not JNK2, in the beta-amyloid-mediated stabilization of protein p53 and induction of the apoptotic cascade in cultured cortical neurons. Biochem. J. V. 371. P. 789.
Gaymes T.J., Padua R.A., Pla M., Orr S., Omidvar N., Chomienne C., Mufti G.J., Rassool F.V. 2006. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: a mechanism for leukemia-specific HDI-dependent apoptosis? Mol. Cancer Res. MCR. V. 4. P. 563.
Gnedina O.O., Morshneva A.V., Skvortsova E.V., Igotti M.V. 2022. HDAC inhibitor sodium butyrate attenuates the DNA repair in transformed but not in normal fibroblasts. Int. J. Mol. Sci. V. 23. P. 3517.
Golding S., Rosenberg E., Neill S., Dent P., Povirk L., Valerie K. 2007. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res. V. 67. P. 1046.
Goloudina A.R., Tanoue K., Hammann A., Fourmaux E., Le Guezennec X., Bulavin D.V., Mazur S.J., Appella E., Garrido C., Demidov O.N. 2012. Wip1 promotes RUNX2-dependent apoptosis in p53-negative tumors and protects normal tissues during treatment with anticancer agents. Proc. Natl. Acad. Sci. V. 109. P. E68.
Kasibhatla S., Brunner T., Genestier L., Echeverri F., Mahboubi A., Green D.R. 1998. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-κB and AP-1. Mol. Cell. V. 1. P. 543.
Konsoula Z., Cao H., Velena A., Jung M. 2011. Adamantanyl-histone deacetylase inhibitor H6CAHA exhibits favorable pharmacokinetics and augments prostate cancer radiation sensitivity. Int. J. Radiat. Oncol. Biol. Phys. V. 79. P. 1541.
Kumar S., Jiang M.S., Adams J.L., Lee J.C. 1999. Pyridinylimidazole compound SB 203580 inhibits the activity but not the activation of p38 mitogen-activated protein kinase. Biochem. Biophys. Res. Commun. V. 263. P. 825.
Lee J.H., Choy M.L., Ngo L., Foster S.S., Marks P.A. 2010. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA. V. 107. P. 14639.
Lowe J., Cha H., Lee M.-O., Mazur S.J., Appella E., Fornace A.J. 2012. Regulation of the Wip1 phosphatase and its effects on the stress response. Front. Biosci. J. Virtual Libr. V. 17. P. 1480.
Lu C., Zhu F., Cho Y.-Y., Tang F., Zykova T., Ma W., Bode A.M., Dong Z. 2006. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell. V. 23. P. 121.
Lu C., Shi Y., Wang Z., Song Z., Zhu M., Cai Q., Chen T. 2008. Serum starvation induces H2AX phosphorylation to regulate apoptosis via p38 MAPK pathway. FEBS Lett. V. 582. P. 2703.
Menolfi D., Zha S. 2020. ATM, ATR and DNA-PKcs kinases–the lessons from the mouse models: inhibition ≠ deletion. Cell Biosci. V. 10. Article number 8.
Moon S.-H., Lin L., Zhang X., Nguyen T.-A., Darlington Y., Waldman A.S., Lu X., Donehower L.A. 2010. Wild-type p53-induced phosphatase 1 dephosphorylates histone variant γ-H2AX and suppresses DNA double strand break repair. J. Biol. Chem. V. 285. P. 12935.
Munshi A., Kurland J.F., Nishikawa T., Tanaka T., Hobbs M.L., Tucker S.L., Ismail S., Stevens C., Meyn R.E. 2005. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res. V. 11. P. 4912.
Nicholson J., Jevons S.J., Groselj B., Ellermann S., Konietzny R., Kerr M., Kessler B.M., Kiltie A.E. 2017. E3 Ligase cIAP2 mediates downregulation of MRE11 and radiosensitization in response to HDAC inhibition in bladder cancer. Cancer Res. V. 77. P. 3027.
Pechackova S., Burdova K., Benada J., Kleiblova P., Jenikova G., Macurek L. 2016. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget. V. 7. P. 14458.
Phong M.S., Van Horn R.D., Li S., Tucker-Kellogg G., Surana U., Ye X.S. 2010. P38 mitogen-activated protein kinase promotes cell survival in response to DNA damage but is not required for the G2 DNA damage checkpoint in human cancer cells. Mol. Cell. Biol. V. 30. P. 3816.
Picco V., Pagès G. 2013. Linking JNK activity to the DNA damage response. Genes Cancer. V. 4. P. 360.
Pospelova T.V., Medvedev A.V., Kukushkin A.N., Svetlikova S.B., van der Eb A.J., Dorsman J.C., Pospelov V.A. 1999. E1A + cHa-ras transformed rat embryo fibroblast cells are characterized by high and constitutive DNA binding activities of AP-1 dimers with significantly altered composition. Gene Expr. V. 8. P. 19.
Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. V. 273. P. 5858.
Sanchez-Prieto R., Rojas J.M., Taya Y., Gutkind J.S. 2000. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. V. 60. P. 2464.
Shreeram S., Demidov O.N., Hee W.K., Yamaguchi H., Onishi N., Kek C., Timofeev O.N., Dudgeon C., Fornace A.J., Anderson C.W., Minami Y., Appella E., Bulavin D.V. 2006. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol. Cell. V. 23. P. 757.
Sluss H.K., Davis R.J. 2006. H2AX is a target of the JNK signaling pathway that is required for apoptotic DNA fragmentation. Mol. Cell. V. 23. P. 152.
Tafolla E., Wang S., Wong B., Leong J., Kapila Y.L. 2005. JNK1 and JNK2 oppositely regulate p53 in signaling linked to apoptosis triggered by an altered fibronectin matrix: JNK links FAK and p53. J. Biol. Chem. V. 280. P. 19992.
Thurn K.T., Thomas S., Raha P., Qureshi I., Munster P.N. 2013. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol. Cancer Ther. V. 12. P. 1535.https://doi.org/10.1158/1535
Van Attikum H., Gasser S.M. 2009. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. V. 19. P. 207.
Wang D., Zhao M., Chen G., Cheng X., Han X., Lin S., Zhang X., Yu X. 2013. The histone deacetylase inhibitor vorinostat prevents TNFα-induced necroptosis by regulating multiple signaling pathways. Apoptosis Int. J. Program. Cell Death. V. 18. P. 1348.
Wang H., Zhou W., Zheng Z., Zhang P., Tu B., He Q., Zhu W.-G. 2012. The HDAC inhibitor depsipeptide transactivates the p53/p21 pathway by inducing DNA damage. DNA Repair. V. 11. P. 146.
Wei F., Yan J., Tang D. 2011. Extracellular signal-regulated kinases modulate DNA damage response – a contributing factor to using MEK inhibitors in cancer therapy. Curr. Med. Chem. V. 18. P. 5476.
Wu D., Chen B., Parihar K., He L., Fan C., Zhang J., Liu L., Gillis A., Bruce A., Kapoor A., Tang D. 2006. ERK activity facilitates activation of the S-phase DNA damage checkpoint by modulating ATR function. Oncogene. V. 25. P. 1153.
Wu Y.-H., Hong C.-W., Wang Y.-C., Huang W.-J., Yeh Y.-L., Wang B.-J., Wang Y.-J., Chiu H.-W. 2017. A novel histone deacetylase inhibitor TMU-35435 enhances etoposide cytotoxicity through the proteasomal degradation of DNA-PKcs in triple-negative breast cancer. Cancer Lett. V. 400. P. 79.
Yan Y., Black C.P., Cowan K.H. 2007. Irradiation-induced G2/M checkpoint response requires ERK1/2 activation. Oncogene. V. 26. P. 4689.
Ye M., Zhang Y., Gao H., Xu Y., Jing P., Wu J., Zhang X., Xiong J., Dong C., Yao L., Zhang J., Zhang J. 2018. Activation of the aryl hydrocarbon receptor leads to resistance to EGFR TKIs in non-small cell lung cancer by activating Src-mediated bypass signaling. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. V. 24. P. 1227.
Young P.R., McLaughlin M.M., Kumar S., Kassis S., Doyle M.L., McNulty D., Gallagher T.F., Fisher S., McDonnell P.C., Carr S.A., Huddleston M.J., Seibel G., Porter T.G., Livi G.P., Adams J.L. et al. 1997. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. J. Biol. Chem. V. 272. P. 12116.
Дополнительные материалы отсутствуют.