

Цитология, 2023, T. 65, № 1, стр. 64-81
Нарушение уровня транспортеров лактата в клетках головного мозга при остром токсическом действии бета-амилоида in vitro и in vivo
Я. В. Горина 1, *, Е. В. Харитонова 1, Е. Д. Хилажева 1, А. А. Семенова 1, А. В. Моргун 2, Ю. К. Комлева 1, 3, О. Л. Лопатина 2, А. Б. Салмина 1, 4
1 Научно-исследовательский институт молекулярной медицины и патобиохимии Красноярского государственного медицинского университета им. профессора В.Ф. Войно-Ясенецкого Министерства здравоохранения РФ (КрасГМУ)
660022 Красноярск, Россия
2 Кафедра поликлинической педиатрии и пропедевтики детских болезней с курсом ПО КрасГМУ
660022 Красноярск, Россия
3 Центр коллективного пользования Молекулярные и клеточные технологии, КрасГМУ
660022 Красноярск, Россия
4 Лаборатория нейробиологии и тканевой инженерии Института мозга Научного центра неврологии
125367 Москва, Россия
* E-mail: yana_20@bk.ru
Поступила в редакцию 12.08.2022
После доработки 22.09.2022
Принята к публикации 07.10.2022
- EDN: GOTNXZ
- DOI: 10.31857/S0041377123010042
Аннотация
Снижение энергетического метаболизма головного мозга коррелирует с когнитивными нарушениями при болезни Альцгеймера. Накапливающиеся экспериментальные данные указывают на то, что переносчики лактата и монокарбоксилатные транспортеры (МСТ) принимают непосредственное участие в церебральном энергетическом метаболизме. Однако в настоящее время изменения уровня лактата и МСТ при болезни Альцгеймера остаются неясными. Цель исследования заключалась в изучении содержания лактата и уровня его транспортеров MCT1 и MCT2 в клетках нейрональной, астроглиальной и эндотелиальной природы при остром токсическом действии бета-амилоида (Aβ1–42) in vitro и in vivo. Показано, что в условиях острого токсического действия Aβ1–42 in vivo значимо (P ≤ 0.05) уменьшается уровень лактата в ткани гиппокампа и повышается в диализате на фоне низкого уровня MCT1 и MCT2. In vitro выявлена высокая (P ≤ 0.05) продукция лактата астроцитами, сопряженная с низким (P ≤ 0.05) уровнем MCT2 на нейронах. Таким образом, Aβ1–42 вызывает снижение уровня лактата в ткани гиппокампа и повышение его уровня в диализате in vivo, что коррелирует с нарушением уровня MCT1 и MCT2. Это указывает на нарушение энергетического метаболизма за счет острого токсического действия Aβ1–42. При этом выявленное повышение продукции лактата астроцитами in vitro может свидетельствовать о включении компенсаторного механизма, направленного на поддержание астроцитарно-нейронального взаимодействия.
Болезнь Альцгеймера (БА) – хроническое нейродегенеративное заболевание, характеризующееся наличием клубков гиперфосфорилированного тау-белка, отложением пептида бета-амилоида (Aβ1–42) и потерей нейронов (Forlenza et al., 2010). Характерной особенностью пациентов с БА является неспособность консолидировать долговременную память, что приводит к прогрессирующему ухудшению памяти по мере развития заболевания (Bondi et al., 2017).
Важно отметить, что энергетические потребности головного мозга очень высокие, и в этом контексте действуют жесткие регулирующие механизмы, обеспечивающие своевременную доставку необходимых энергетических субстратов в соответствии с активностью нейронов, для формирования и консолидации памяти (Falkowska et al., 2015; Shin et al., 2018). Ряд исследований убедительно продемонстрировали, что снижение энергетического метаболизма головного мозга может не только предшествовать, но и способствовать развитию БА с выраженным проявлением митохондриальной и когнитивной дисфункции (Mosconi et al., 2009; Croteau et al., 2018). Глюкоза является одним из ключевых компонентов сигнальных путей для поддержания функции нейронов. При этом в условиях покоя астроциты потребляют больше всего глюкозы (около 85%) и выделяют лактат, в то время как нейроны вносят минимальный вклад в потребление этого субстрата в головном мозге (Bolaños et al., 2010).
Как показывают последние экспериментальные данные, нарушение церебрального метаболизма глюкозы, обусловленное снижением нейронального поглощения глюкозы, дисфункцией митохондрий и увеличением продукции активных форм кислорода, обнаруживается у лиц с семейной формой БА еще до накопления бляшек Aβ1–42 (Ding et al., 2013; Wang et al., 2020).
Многие исследования также продемонстрировали, что переносчики глюкозы в значительной мере уменьшаются в головном мозге при БА, что, в свою очередь, может способствовать нарушению памяти (Correia et al., 2012; Jin et al., 2013; Bartolotti, Lazarov, 2019). Это прямо указывает на существование тесной корреляции между гипометаболизмом глюкозы и дефицитом памяти при БА (Cunnane et al., 2011).
Несмотря на ряд свидетельств того, что лактат является важным энергетическим субстратом для организма, особенно на ранних этапах онтогенеза, его присутствие в головном мозге интерпретируется как признак церебрального повреждения. Кроме того, долгое время лактат считался потенциально токсичным продуктом метаболизма, однако теперь он признан не только ключевым участником нейрон-астроглиального метаболического сопряжения, но даже предпочтительным источником энергии при определенных обстоятельствах (Tang, 2018; Muraleedharan et al., 2020).
Известно, что в физиологических условиях активация анаэробного гликолиза является обычным способом адаптации при дефиците энергии в головном мозге (Koenig, 2008).
Необходимо отметить и установленные доказательства того, что аэробный гликолиз и связанное с ним производство и транспорт лактата от астроцитов к нейронам важен для поддержания синаптической пластичности и долговременной памяти (Newman et al., 2011; Suzuki et al., 2011). Было выявлено, что интрагиппокампальные инфузии лактата в значительной степени улучшают память у крыс, при этом эффект выраженно подавляется фармакологическим ингибированием транспорта лактата в нейроны (Newman et al., 2011).
Стоит отметить, что транспорту лактата в нейроны ЦНС способствуют монокарбоксилатные транспортеры (МСТ). Известно, что MCT1 во взрослом головном мозге экспрессируется, в основном, астроцитами и клетками церебрального эндотелия, а MCT2 – преимущественно нейронами (Salmina et al., 2015), но экспрессия этих транспортеров может быть зарегистрирована и на других клетках, например, транспортеры MCT1 при гипоксии активно экспрессируются на нейронах, тогда как экспрессия MCT2 на нейронах в данных условиях резко снижается. Интересно, что PGC-1 (стимулятор митохондриального биогенеза) увеличивает уровень MCT1 в разных клетках (Bergersen, 2015).
Примечательно, что MCT1 может работать и на импорт, и на экспорт лактата, в зависимости от энергетических потребностей клеток, тогда как MCT2, в основном, используется для транспорта лактата внутрь клеток, однако некоторые авторы считают, что MCT1 и MCT2 в большей степени предназначены для импорта лактата, а MCT4 – для высвобождения лактата из клеток (Bergersen, 2015).
Ряд исследований показал, что содержание лактата и уровня МСТ изменяются при патологических заболеваниях, таких как опухоль и ишемия головного мозга (Moreira et al., 2009; Pinheiro et al., 2012). Сопутствующие исследования продемонстрировали, что уровни лактата в значительной мере повышены в спинномозговой жидкости у пациентов с БА на фоне сопровождающегося гипометаболизма глюкозы (Liguori et al., 2016). Однако исследования влияния уровня лактата и содержания MCT на нейрональную и астроглиальную дисфункцию при БА весьма ограничены.
Таким образом, цель настоящего исследования – изучение содержания лактата и содержания его транспортеров – MCT1 и MCT2 в клетках нейрональной, астроглиальной и эндотелиальной природы при остром токсическом действии Aβ1–42 in vitro и in vivo.
МАТЕРИАЛ И МЕТОДИКА
Для проведения исследований использовали моделирование БА в условиях in vivo и in vitro. Использование в нашем исследовании мышиной модели БА обеспечивает не только лучшее понимание поведенческих изменений, ассоциированных с нарушением экспрессии транспортеров лактата в процессе развития заболевания, но также учитывает биологическую сложность живого организма. Однако модель in vivo ограничивает доступность к интересующей ткани, затрудняет мониторинг в реальном времени и измерение уровня целевых маркеров при развитии заболевания, нивелируя влияние побочных факторов. Напротив, модель БА in vitro с использованием клеточных культур (нейронов, астроцитов и эндотелиоцитов) – модель гемато-энцефалического барьера (ГЭБ) с добавлением Aβ1–42 – позволяет провести исследования на живых клетках, а также дает возможность осуществлять непрерывное наблюдение за процессами, протекающими на клеточном уровне (Shin et al., 2019). Иными словами – получить подробную информацию о молекулярных механизмах в интересующих нервных клетках определенного региона головного мозга. При этом возможное влияние сопутствующих факторов, наличие которых вероятно в условиях in vivo, исключается. Таким образом, модель БА in vitro выступает в качестве значимого дополнительного инструмента для преодоления ограничений модели заболевания на животных.
В нашем случае модель БА in vitro использовалась для изучения уровня лактата и содержания его транспортеров, то есть возможных метаболических изменений, происходящих в клетках нейрональной, астроглиальной и эндотелиальной природы (в модели ГЭБ) при культивировании в присутствии Aβ1–42. Блок-схема эксперимента in vivo представлена на рис. 1.
Рис. 1.
Блок-схема эксперимента по определению уровня лактата и его транспортеров in vivo. ИК – интрагиппокампальное, ПО – послеоперационный, СФМ – спектрофотометрия, ИГХ –иммуногистохимия.

Животные и эксперименты с ними. В работе использовали 20 самцов мышей линии C57BL/6 в возрасте 4 мес. Экспериментальная группа животных (C57BL/6 + Aβ1–42) – мыши после введения в зону гиппокампа CA1 амилоида Aβ1–42 по 1 мкл билатерально согласно стереотаксическим координатам (ML ± 1.3 мм, в AP – 2.0 мм. DV – 1.9 мм) (n = 10). Контрольная группа мышей (C57BL/6 + PBS) – ложно-оперированные животные после введения фосфатно-солевого буферного раствора (PBS), растворителя для Aβ1–42 (n = 10).
Животные во всех вариантах экспериментов находились в клетках со свободным доступом к воде и корму при температуре 21 ± 1°С и регулярном световом цикле 12 ч день/12 ч ночь. Планирование и проведение экспериментов на животных осуществляли при соблюдении “принципа 3R”: Replacement – использование альтернативных методов (культуры клеток); Reduction – использование минимального количества животных в эксперименте; Refinement – не подвергать животных безосновательному стрессу до, во время и после проведения эксперимента. Рандомизации не было при распределении объектов исследования. Размер выборки не рассчитывался.
Все исследования на животных проводили в соответствии с соблюдением принципов гуманности, изложенных в Директиве Европейского сообщества (2010/63/ЕС). Исследования выполняли после утверждения заявки и протокола на использование лабораторных животных для исследования на заседании биоэтической комиссии по работе с животными при локальном этическом комитете Красноярского государственного медицинского университета имени профессора В.Ф. Войно-Ясенецкого Министерства здравоохранения РФ (выписка из протокола № 3 от 29.10.2019 г.).
Экспериментальное моделирование нейродегенерации in vivo. Моделирование БА проводили путем интрагиппокампального введения Aβ1–42 (Sigma-Aldrich, США) по стереотаксическим координатам мозга в зону СА1. Aβ1–42 предварительно растворяли в ДМСО (Sigma-Aldrich, США) до концентрации 4 мМ, затем брали аликвоту и разбавляли в PBS (Sigma-Aldrich, США) до концентрации 50 мкM с последующей агрегацией в термостате при 37°С в течение 7 сут. 1 мкл Aβ1–42 вводили с каждой стороны гиппокампа в зону СА1 (Epelbaum et al., 2015). Контрольной группе животных вводили PBS, содержащий ДМСО в том же количестве, что и при растворении Aβ1–42, с использованием той же процедуры.
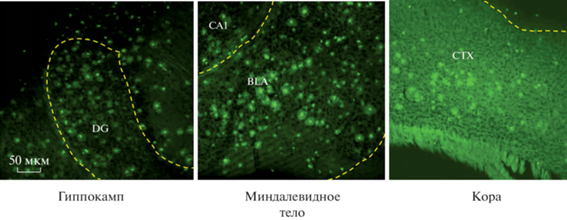
Признаки БА выявляли, начиная с 10 сут (Sipos et al., 2007) после оперативного вмешательства. Верификацию модели БА проводили с помощью окрашивания срезов головного мозга тиофлавином S (Sigma-Aldrich, США) (Комлева и др., 2015). После введения Aβ1–42 в ткани головного мозга амилоидные бляшки флуоресцировали зеленым цветом (рис. 2).
Рис. 2.
Визуализация бляшек бета-амилоида (Aβ1−42) в областях головного мозга экспериментальных мышей. DG – зубчатая извилина гиппокампа, CA1 – область гиппокампа, BLA – базолатеральное миндалевидное тело, CTX – кора.
Оценку когнитивных функций у исследуемых групп животных проводили с использованием нейроповеденческого теста Fear conditioning, результаты по которому были представлены нами ранее (Горина и др., 2017).
Микродиализ ткани головного мозга экспериментальных животных. Сбор межклеточной жидкости (диализата) осуществляли с использованием установки для микродиализа (Eicom Corporation, США). Перед проведением микродиализа экспериментальным животным в левый желудочек головного мозга с помощью стереотаксического аппарата устанавливали направляющую канюлю по координатам ML 1 мм, в AP – 0.46 мм, DV 2.2 мм, которую затем фиксировали стоматологическим цементом.
После операции животное помещали в клетку на 5 сут для полного восстановления со свободным доступом к воде и корму при постоянной температуре 21 ± 1°С и регулярном световом цикле 12 ч день/12 ч ночь.
Сбор целевых компонентов межклеточной жидкости головного мозга осуществляли с использованием искусственной спинномозговой жидкости, приготовление которой осуществляли следующим образом: в деионизованной воде (5 мл) растворяли 0.36525 г NaCl (Sigma-Aldrich, США), 0.0093 г KCl (Sigma-Aldrich, США) и 0.0075 г NaH2PO4 (Sigma-Aldrich, США). К 0.5 мл полученного раствора добавляли 0.01092 г NaHCO3 (Sigma-Aldrich, США), 0.009 г кристаллической глюкозы (Sigma-Aldrich, США), 0.01 г CaCl2 (Sigma-Aldrich, США), 0.0025 г MgCl2 (Sigma-Aldrich, США) и доводили общий объем аликвоты до 5 мл. Полученный раствор фильтровали.
Сбор диализата проводился в течение 24 ч у свободнодвижущегося животного, подключенного через капилляры к направляющей канюле. Мышь находилась в прозрачном боксе со свободным доступом к воде и корму. Диализат (V = 20 мкл) собирался со скоростью 0.3 мкл/мин в микропланшет, расположенный в коллекторе с системой охлаждения до 2°C.
Иммуногистохимия ткани головного мозга. Через 60 мин после проведения микродиализа осуществляли транскардиальную перфузию 4%-ным параформальдегидом (Sigma-Aldrich, США) с последующим забором головного мозга. Мозг фиксировали в 10%-ном нейтральном забуференном формалине рН 7.4 (Sigma-Aldrich, США), после чего погружали в 20%-ный раствор сахарозы (Sigma-Aldrich, США). С помощью микротома (Thermo Scientific Microm HM 650, США) изготавливали срезы толщиной 50 мкм. Изучали наличие и уровень целевых маркеров методом непрямой иммуногистохимии для свободно плавающих срезов (Encinas, Enikolopov, 2008).
После промывки в PBS срезы блокировали 3%-ным бычьим сывороточным альбумином (BSA, Sigma-Aldrich, США) в PBS и 1%-ном Тритоном X-100 (Sigma-Aldrich, США) в течение 1 ч при комнатной температуре с последующим инкубированием в течение ночи с первичными антителами с 3%-ным BSA в PBS и 0.2%-ным Тритоном Х-100 при 4°С. После инкубации с первичными антителами срезы промывали и инкубировали со вторичными антителами в течение 2 ч при комнатной температуре. Наименование первичных и вторичных антител представлены в табл. 1. Изображения получали с помощью конфокального микроскопа (Olympus FV 10i, Япония).
Таблица 1.
Антитела, используемые в иммуногистохимическом исследовании in vivo и in vitro
| Антитела in vivo (разбавление 1 : 1000) | |
|---|---|
| Первичные | Вторичные |
| Против CD31 моноклональные мыши (MBS532307; MyBioSource, США) | Козлиные антимышиные, Alexa Fluor 488 (ab 150157; Abcam, Великобритания) |
| Против NSE (нейрон-специфическая энолаза) поликлональные цыпленка (GTX85462; GeneTex, США) | Козлиные антицыплячьи, Alexa Fluor 488 (ab 150169; Abcam, Великобритания) |
| Против GFAP (глиальный фибриллярный кислый белок) моноклональные мыши (MBS8241552; MyBioSource, США) | Козлиные антимышиные, Alexa Fluor 488 (ab 150113; Abcam, Великобритания) |
| Против MCT1 поликлональные кролика (TA321556; OriGene Technologies, США) | Ослиные антикроличьи, Alexa Fluor 555 (ab 150074; Abcam, Великобритания) |
| Против MCT2 поликлональные кролика (bs-3995R; Bioss Inc., США) | Ослиные антикроличьи, Alexa Fluor 555 (ab 150074; Abcam, Великобритания) |
| Антитела in vitro | |
| Первичные (разбавление 1 : 300) | Вторичные (разбавление 1 : 500) |
| Против ZO1 поликлональные козла (sc-8147; Santa Cruz; США) | Ослиные антикозьи, Alexa Fluor 488 (A11055; Thermo Fisher Scientific, США) |
| Против NeuN моноклональные мыши (MAB377; Merck Millipore, США) | Козлиные антимышиные, Alexa Fluor 488 (ab150117; Abcam, Великобритания) |
| Против GFAP поликлональные цыпленка (LS-B6299; LSBio, США) | Козлиные антицыплячьи, Alexa Fluor 555 (A-21437; Thermo Fisher Scientific, США) |
| Против MCT1 поликлональные кролика (LS-B14628; LSBio, США) | Козлиные антикроличьи, Alexa Fluor 555 (ab 150078; Abcam, Великобритания) |
| Против MCT2 поликлональные кролика (ab224627; Abcam, Великобритания) | Ослиные антикроличьи, Alexa Fluor 555 (ab 150074, Abcam, Великобритания) |
В иммуногистохимическом исследовании в экспериментальной и контрольной группах было по 5 животных. От каждого животного отбирали по 3 среза головного мозга. Подсчет клеток, экспрессирующих MCT1 и MCT2 в клетках нейрональной, астроглиальной и эндотелиальной природы, в каждом срезе в области гиппокампа проводили в трех полях зрения (100 × 100 мкм). Регистрировали среднее значение по трем полям зрения. В графическом изображении представлены среднее значение по трем полям зрения с каждого среза (число срезов n = 15). При обработке полученных результатов принимали во внимание долю клеток, которые несли целевую метку и выражали в процентах от общего числа клеток.
Определение уровня лактата в ткани гиппокампа и диализате in vivo. Уровень лактата определяли в группах по 5 животных. Подготовку ткани гиппокампа проводили следующим образом: образцы тканей (около 10 мг ткани) промывали в холодном PBS, ресуспендировали ткань в 6-кратном объеме буфера для анализа лактата (Lactate Assay Buffer, Abcam, Великобритания) на льду с помощью гомогенизатора Даунса (WK Life Sciences GmbH, Германия), затем центрифугировали (центрифуга с охлаждением Hitachi Koki, Япония) при 11000 g в течение 5 мин при 4°C, после чего супернатанты собирали и переносили в чистые пробирки. Уровень лактата определяли спектрофотометриечски с помощью микропланшетного фотометра (Anthos 2010; Biochrom, Великобритания) согласно протоколу производителя, представленного в наборе L-Lactate Assay Kit (Colorimetric/Fluorometric) (Abcam, Великобритания).
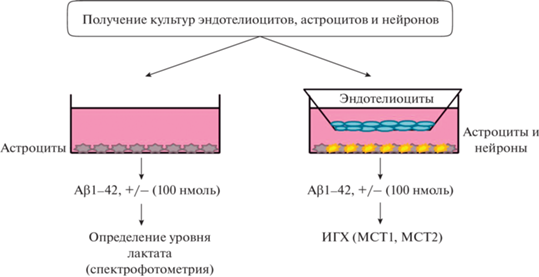
Получение первичных культур клеток и моделирование нейроваскулярной единицы/ГЭБ. Блок-схема эксперимента in vitro представлена на рис. 3. В работе использовали первичные культуры клеток церебрального эндотелия, астроцитов и нейронов, выделенные из головного мозга эмбрионов мыши линии C57BL/6 (общее число животных 32).
Рис. 3.
Схема-дизайн эксперимента по определению уровня лактата и его транспортеров in vitro. ИГХ – иммуногистохимия.
Выделение церебральных эндотелиоцитов проводили по модифицированному протоколу (Liu et al., 2013). Кору головного мозга выделяли в холодном растворе Хенкса (ПанЭко, Россия), мелко измельчали и центрифугировали при 150 g и комнатной температуре в течение 3 мин. Затем к осадку добавляли двукратный объем 25%-ного раствора фетальной бычьей сыворотки (FBS, HyClone, Южная Америка), тритурировали 25 раз пипеткой 5 мл с последующим центрифугированием гомогената при 600 g и комнатной температуре в течение 10 мин. Забирали нижний слой и переносили в коническую пробирку. Повторяли этапы тритурирования и центрифугирования 3 раза. Затем проводили ферментативную обработку пеллета в двукратном объеме (по отношению к объему пеллета) 0.1%-ного раствора коллагеназы II (ПанЭко, Россия) при температуре 37°С в течение 35 мин с периодическим перемешиванием. Затем проводили ресуспензирование осадка с последующим центрифугированием при 150 g в течение 5 мин.
Культивирование фрагментов и отдельных эндотелиальных клеток осуществляли при температуре 37°С и 5% CO2 в культуральных флаконах, предварительно покрытых желатином (Gelatin Solution0.1%. Biological Industries, США) в культуральной среде DMEM (ПанЭко, Россия), содержащей 20% раствора FBS (ПанЭко, Россия), 3 мг/мл глюкозы (Sigma-Aldrich, США), 0.58 мг/мл глутамина (Sigma-Aldrich, США), 100 Ед./мл пенициллина, 100 мг/мл стрептомицина (HyClone, Южная Америка); смену среды проводили каждые 3 сут. Клетки во флаконе промывали 2 раза раствором Хенкса (ПанЭко, Россия) и обрабатывали 0.25%-ным раствором трипсина и ЭДТА (ПанЭко, Россия). Смену среды осуществляли каждые 2–3 сут.
Выделение и культивирование нейросфер. Головной мозг помещали в ледяной 2%-ный раствор глюкозы в PBS, затем выделяли интересующие области (гиппокамп, стенки боковых желудочков) и иссекали до размеров 1 мм3. Далее переносили в свежий 2%-ный раствор глюкозы в PBS на 1 мин. После осаждения ткани супернатант удаляли. Оставшуюся ткань ресуспендировали в 1 мл среды NeuroCult NS-A Proliferation (StemCell, США). Тритурацию ткани проводили (около 30 раз) стерильным пластиковым наконечником до получения однородной суспензии клеток. Затем добавляли 1 мл свежей среды NeuroCult NS-A Proliferation. Через 2 мин после осаждения неразделенных кусочков ткани собирали супернатант и переносили его в другую пробирку. Собранный супернатант центрифугировали при 150 g в течение 5 мин, после чего удаляли супернатант и добавляли 1 мл свежей среды NeuroCult NS-A Proliferation с последующей тритурацией.
С помощью цитометра Scepter Cell Counter (Millipore, США) проводили подсчет клеток. Клетки (1.5 × 106 кл./мл) сеяли в культуральные флаконы T-75 см2 с 25 мл среды NeuroCult NS-A Proliferation и культивировали в условиях инкубатора при 37°C и 5% CO2. Образование нейросфер наблюдали через 24–48 ч.
Получение культуры астроцитов. Поводили направленную дифференцировку нейросфер в среде Astrocyte Medium (ScienCell, США) следующего состава: базальная среда (Basal Medium, ScienCell, США), 10% FBS (HyClone, США), AGS (ScienCell, США), раствора пенициллина-стрептомицина (ScienCell, США) в конечной концентрации 50 Ед./мл. Через 7–9 сут наблюдали образование монослоя астроцитов.
Получение сокультуры астроцитов и нейронов. Сокультуру получали из нейросфер путем направленной дифференцировки в астроциты и нейроны (Моргун и др., 2013). В качестве культуральной среды использовали NeuroCult NS-A Proliferation Kit, с добавлением гепарина, основного фактора роста фибробластов (βFGF) и эпидермального фактора роста (EGF). Полученные клетки (1.5 × 106 кл./мл) засевали в культуральные флаконы T-75 см2 с 25 мл среды. Далее проводили дифференцировку нейросфер в астроциты и нейроны в культуральной среде NeuroCult NS-A Differentiation Kit.
Получение модели ГЭБ in vitro. Смесь астроцитов и нейронов помещали на дно культурального планшета, после устанавливали культуральную вставку (Corning-Costar, США), на которую помещали эндотелиоциты. Для получения модели нейроваскулярной единицы эндотелиоциты культивировали совместно с астроцитами и нейронами (при соотношении 1 : 2 : 1 соответственно) в культуральном планшете в среде DMEM с FBS, глутамином и смесью антибиотиков при 37°C и 5% CO2.
Моделирование острого токсического действия Aβ1–42 in vitro. Для исследования действия Аβ1–42 на культуру изолированных астроцитов клетки экспериментальной группы культивировали в среде, содержащей Аβ1–42 в конечной концентрации 100 нмоль. Через 24 ч производили забор культуральной среды для последующего определения в ней концентрации лактата. В экспериментальной и контрольной группах было по 6 лунок.
Для исследования острого токсического действия Aβ1–42 в модели ГЭБ к клеткам добавляли Аβ1–42 в той же конечной концентрации (100 нмоль) в питательной среде. Через 24 ч и 48 ч оценивали изменение уровня исследуемых молекул на каждом типе клеток (астроцитах нейронах, эндотелиоцитах). В обеих группах было по 10.
Оценка уровня лактата в среде культивирования астроцитов в присутствии и отсутствие Аβ1–42. Определение уровня лактата (Colorimetric/Fluorometric) проводили по стандартному протоколу фирмы-изготовителя (Abcam, Великобритания) в среде, в которой культивировались астроциты с добавлением Aβ1–42 (в конечной концентрации 100 нмоль) и без его добавления (контроль). Для этого смешивали 50 мкл образца с 50 мкл реакционной смеси (46 мкл Lactate Assay Buffer, 2 мкл Lactate Probe, 2 мкл Enzyme Mix), инкубировали при комнатной температуре в защищенном от света месте в течение 30 мин. Оптическую плотность определяли с помощью микропланшетного ридера CLARIOstar Plus (Германия) при длине волны 492 нм. Полученные результаты выражали в нмоль на 1 лунку по данным калибровочной кривой, построенной по стандартным растворам различной концентрации (0, 2, 4, 6, 8 и 10 нмоль/мкл), приготовленным из стандарта, прилагаемого в наборе.
Иммуногистохимическое исследование in vitro. Регистрацию уровня MCT1 и MCT2 в клетках эндотелия, нейронах и астроцитах, культивируемых совместно в течение 24 и 48 ч, осуществляли в модели ГЭБ после добавления 100 нмоль Aβ1–42 и без его добавления (контроль). Первичные антитела для иммуногистохимического исследования (см. табл. 2 ) использовали в рабочем разведении 1 : 300. Время инкубации составляло 18 ч при 4°С. Вторичные антитела использовали в разведении 1 : 500, время инкубации составляло 2 ч при 37°С (табл. 1).
Микроскопию клеток осуществляли на флуоресцентном микроскопе ZOE (Bio-Rad, США). Каждый эксперимент повторяли 10 раз. Считали клетки, позитивные по каждому виду антигена в образце (не менее 5 полей зрения). При обработке полученных результатов принимали во внимание долю клеток, которые несли целевую метку и выражали ее в процентах от общего числа клеток (100%).
Статистический анализ. Статистический анализ полученных результатов проводили с помощью программ GraphPad Prizm 8.0.1 (версия 8.0, США) и Stаtplus Professional (AnalystSoft Inc, США), сборка 5.9.8.5/Core v.5.9.33 и GraphPad 6.0 (США). В связи с отсутствием нормальности распределения сравнение двух групп проводили с использованием непараметрического U-критерий Манна–Уитни. Проверку статистических гипотез выполняли при критическом уровне значимости P = 0.05. Результаты представлены в виде Me [Q1; Q3]), где Ме – медиана, Q1 – нижний квартиль, Q3 – верхний квартиль.
Реактивы. Использовали Aβ1−42, ДМСО, PBS, тиофлавин S, глутамин, NaCl, KCl, NaH2PO4, NaH-CO3, CaCl2, MgCl2, параформальдегид, сахарозу, BSA и Triton X-100 от Sigma-Aldrich (США); лактат (Lactate Assay Buffer), L-Lactate Assay Kit (Colorimetric/Fluorometric) (Abcam, Великобритания); культуральную среду DMEM, раствор Хенкса, раствор трипсина и коллагеназу II, ЭДТА (ПанЭко, Россия); фетальную бычью сыворотку (FBS), пенициллин, стрептомицина (HyClone, Южная Америка), NeuroCult NS-A Proliferation (StemCell, США), среду Astrocyte Medium (ScienCell, США).
РЕЗУЛЬТАТЫ
Мы оценили уровни лактата в ткани и во внеклеточном пространстве (диализате) головного мозга в норме и при остром токсическом действии Aβ1–42 in vivo. В ходе исследований установлено, что уровень лактата в гиппокампе животных опытной группы значимо (P = 0.0449) снижается по сравнению с ложно-оперированными животными. Интересно, что во внеклеточном пространстве головного мозга наблюдается противоположная картина, а именно высокий уровень лактата в условиях острого действия Aβ1–42 (P = 0.0358) по сравнению с ложно-оперированными животными (рис. 4).
Рис. 4.
Уровни лактата в ткани головного мозга при остром токсическом действии Aβ1–42 in vivo. Концентрация лактата в ткани гиппокампа (а) и в диализате головного мозга (б) животных с интрагиппокампальным введением Aβ1–42 (C57BL/6 + Aβ1–42) и ложно-оперированных животных (C57BL/6 + PBS). Данные показаны в виде Me [Q1; Q3], где Ме – медиана, Q1 – нижний квартиль, Q3 – верхний квартиль, P – уровень значимости.

Для выяснения вклада астроцитов в продукцию лактата при токсическом действии Aβ1–42 мы проанализировали уровень лактата в среде культивирования клеток. При оценке концентрации лактата в среде выявлено, что в астроцитах, культивированных в присутствии Aβ1–42 (100 нмоль), уровень лактата в среде значимо выше, чем в астроцитах контрольной группы (P = 0.0014) (рис. 5).
Рис. 5.
Уровень лактата в среде культивирования астроцитов in vitro в физиологических условиях в отсутствие (контроль, К) и в присутствии Aβ1–42 (100 нмоль). Данные показаны в виде Me [Q1; Q3], P – уровень значимости (U-критерий Манна–Уитни).

Определение уровня MCT1 в экспериментальных группах in vivo показало статистически значимое (Р = 0.0308) снижение уровня MCT1 в нейронах в гиппокампе головного мозга у животных с интрагиппокампальным введением Аβ1–42 по сравнению с ложно-оперированными животными (рис. 6а, 7). Аналогичную ситуацию наблюдали и при исследовании содержания MCT1 на астроцитах и эндотелиоцитах в гиппокампе, а именно, выраженное снижение (P = 0.0492 и Р = 0.0284 соответственно) у животных экспериментальной группы по сравнению с контролем (рис. 6б–в, 8, 9).
Рис. 6.
Уровень MCT1 в нейронах (а), астроцитах (б) и эндотелиоцитах (в) в гиппокампе головного мозга in vivo у животных с интрагиппокампальным введением Aβ1–42 (C57BL/6 + Aβ1–42) и ложно-оперированных животных (C57BL/6 + PBS). Данные показаны в виде Me [Q1; Q3], P – уровень значимости (U-критерий Манна–Уитни).

Кроме того, нами установлено, что при остром токсическом действии Aβ1–42 значимо снижается и уровень MCT2 как в нейронах (рис. 10, 11), так и в астроцитах и эндотелиоцитах по сравнению с контрольной группой (рис. 10, 12, 13).
Рис. 7.
Уровень MCT1 в нейронах в гиппокампе головного мозга животных in vivo. Маркер MCT1 – красный, NSE (нейрон-специфическая энолаза) – зеленый, ядра окрашены DAPI (синий цвет). GCL – гранулярный слой, SGZ – субгранулярная зона. Стрелками на совмещенных изображениях (Merge) показаны клетки, несущие целевую метку. Увел. об.: 10×.

Рис. 8.
MCT1 в астроцитах в гиппокампе головного мозга животных in vivo. а – MCT1 (красный цвет), GFAP (глиальный фибриллярный кислый белок; зеленый) и ядра, окрашенные DAPI (синий); стрелками на совмещенных изображениях (Merge) показаны клетки, несущие целевую метку. Увел. об.: 10×. б – Саггитальный срез гиппокампа мыши с обозначением полей зрения в зубчатой извилине (зеленый цвет – белок GFAP. Увел. об.: 1×.

Рис. 9.
MCT1 в эндотелиоцитах в гиппокампе головного мозга животных in vivo. CD31 показан зеленым цветом, ядра – синим (DAPI). GCL – гранулярный слой, SGZ – субгранулярная зона. Стрелками показаны клетки, несущие целевую метку. Увел. об.: 10×.

Рис. 10.
Уровень MCT2 в гиппокампе головного мозга животных in vivo на нейронах (а), астроцитах (б) и эндотелиоцитах (в) у животных с интрагиппокампальным введением Aβ1–42 (C57BL/6 + Ab1–42) и ложно-оперированных животных (C57BL/6 + PBS). Доля положительных клеток представлена в процентах от общего числа клеток в поле зрения. Данные показаны в виде Me [Q1; Q3], P – уровень значимости (U-критерий Манна–Уитни).
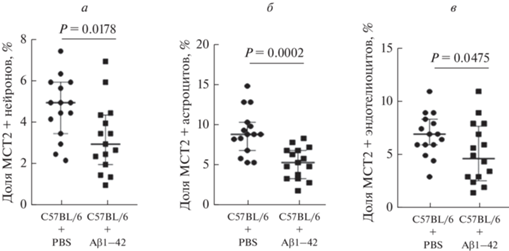
Рис. 11.
Уровень MCT2 в нейронах в гиппокампе головного мозга животных in vivo. Маркеры: MCT2 – красный, NSE (нейрон-специфическая энолаза) – зеленый, ядра – синий (DAPI). Стрелками на совмещенных изображениях (Merge) показаны клетки, несущие целевую метку. Увел. об.: 10×.

Рис. 12.
MCT2 в астроцитах в гиппокампе головного мозга животных экспериментальных групп in vivo. Маркеры: MCT2 – красный, GFAP (глиальный фибриллярный кислый белок – зеленый, ядра – синий (DAPI). Стрелками обозначены клетки, несущие целевую метку. Увел. об.: 10×.
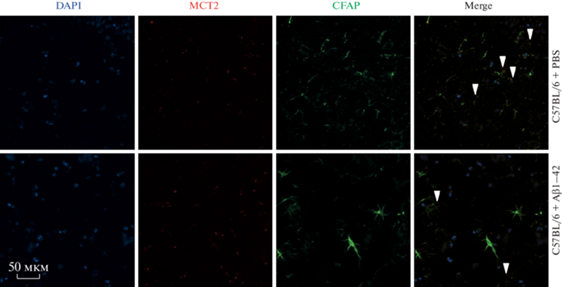
Рис. 13.
MCT2 в эндотелиоцитах в гиппокампе головного мозга животных in vivo. Маркер MCT2 – красный, CD31 – зеленый, ядра – синий (DAPI). GCL – гранулярный слой, SGZ – субгранулярная зона. Стрелки показывают клетки, несущие целевую метку. Увел. об.: 10×.

При анализе уровня МСТ1 в клетках нейроваскулярной единицы/ГЭБ как в физиологических условиях, так и в присутствии Aβ1–42 статистически значимых отличий выявлено не было (P > 0.05). Однако наблюдали тенденцию к снижению количества МСТ1-позитивных астроцитов и нейронов (рис. 14).
Рис. 14.
Уровень МСТ1 в клетках нейроваскулярной единицы/ГЭБ при их культивировании в течение 24 и 48 ч в отсутствие (контроль, К) и в присутствии Aβ1–42 (100 нМ). а – Эндотелиоциты, б – астроциты, в – нейроны (P > 0.05 – сравнение с контрольной группой, U-критерий Манна–Уитни). Доли положительных клеток представлены в % от общего числа клеток в поле зрения. Данные показаны в виде Me [Q1; Q3], P – уровень значимости.

При культивировании клеток нейроваскулярной единицы/ГЭБ в присутствии Aβ1–42 в течение 48 ч произошло значимое снижение уровня МСТ2 в нейронах (P ≤ 0.05; рис. 15в). Также была выявлена тенденция к снижению МСТ2-иммунопозитивных эндотелиоцитов и астроцитов (рис. 15а, б).
Рис. 15.
Уровень МСТ2 в клетках нейроваскулярной единицы/ГЭБ при культивировании in vitro в отсутствие (контроль, К) и в присутствии Aβ1–42 (100 нМ). а – Эндотелиоциты, б – астроциты, в – нейроны (P > 0.05 – сравнение с контрольной группой, U-критерий Манна–Уитни). Доля положительных клеток представлена в процентах от общего числа клеток в поле зрения. Данные показаны в виде Me [Q1; Q3], P – уровень значимости.

ОБСУЖДЕНИЕ
Как установлено в одной из работ, высокий уровень церебрального лактата во фронтальной коре мышей с генетической моделью БА (линия APP/PS1), обусловленный нарушением обработки лактата, коррелировал со снижением пространственной памяти (Harris, 2017).
Согласно результатам клинических исследований, у пациентов с БА увеличение уровня лактата в переднем отделе поясной извилины находится в тесной взаимосвязи со степенью выраженности когнитивных нарушений (Лобзин и др., 2013).
Интересно, что лечение L-лактатом после ишемического инсульта оказывало нейропротекторный эффект и снижало неврологический дефицит у мышей (Berthet et al., 2012). Примечательно, что этот эффект лактат оказывает через активацию транскрипции экспрессии нейротрофического фактора мозга (BDNF), как было показано на астроцитах человека, а также на клеточной линии SH-SY5Y (Coco et al., 2013). При этом важно отметить, что BDNF является не только необходимым фактором выживания нервных клеток в ЦНС, но и играет существенную роль в реализации долговременной памяти (Bekinschtein et al., 2008).
Более того, экзогенный лактат, как было показано, увеличивает экспрессию мРНК и белков – как MCT1, так и цитохром-с-оксидазы в мышечных клетках (Hashimoto et al., 2007). Таким образом, лактат может вызывать ряд событий, ведущих к активации факторов транскрипции, участвующих в обеспечении его транспорта и возможном процессинге через митохондрии. Кроме того, в нескольких исследованиях сообщалось, что лактат увеличивает вазодилатацию (Yamanishi et al., 2006; Gordon et al., 2016), а непрерывное производство лактата в активированном головном мозге может служить сигнальным механизмом для увеличения кровотока и доставки лактата в мозг.
Установленное нами значительное увеличение уровня лактата в диализате животных экспериментальной группы соответствует данным из литературы о высокой концентрации лактата в спинномозговой жидкости пациентов с БА, что связывают с токсическим действием тау-белка на митохондрии (Liguori et al., 2015). Более того, обратная зависимость между локальным низким метаболизмом глюкозы в ткани головного мозга при БА и высоким уровнем лактата в ликворе интерпретируется некоторыми авторами как признак нейродегенерации при БА (Liguori et al., 2016).
Также показано, что Aβ1–42 увеличивает поглощение глюкозы и гликолиз, производство перекиси водорода и высвобождение глутатиона в культивируемых астроцитах. Aβ-индуцированные изменения метаболизма глюкозы связаны с агрегацией, интернализацией и клиренсом Aβ1–42 в астроцитах (Nielsen et al., 2009; Mohamed, Posse de Chaves, 2011). Следствием внутриклеточного накопления Aβ1–42 является изменение метаболизма астроцитарной глюкозы, что приводит к снижению глутатиона и лактата, а это, в свою очередь, способствует повышенной уязвимости соседних нейронов (Allaman et al., 2010).
Необходимо отметить и тот установленный факт, что транспортеры семейства MCT играют важную роль в метаболических взаимодействиях между клетками головного мозга (Pierre, Pellerin, 2005; Cunnane et al., 2011). Так, установлено, что нарушение уровня транспортеров MCT1 и MCT4 в астроцитах вызывает амнезию, которая, как и нарушение долговременной потенциации, предотвращается введением экзогенного лактата. Нарушение содержания MCT2 в нейронах также приводит к амнезии, однако введение лактата не оказывает значимого влияние, что позволяет предположить, что транспорт лактата из атроцитов в нейроны необходим для формирования долговременной памяти и синаптической пластичности (Suzuki et al., 2011). В другой работе показано, что лактат, транспортируемый из астроцитов в нейроны с помощью MCT1–МСТ4, необходим для поддержания синаптической передачи в возбуждающих синапсах даже при условии наличия достаточного запаса глюкозы и внутриклеточного АТФ (Nagase et al., 2014). Кроме того, MCT1 может стимулировать митохондриальный метаболизм, тем самым облегчая использование лактата в качестве метаболического субстрата для цикла трикарбоновых кислот и окислительного фосфорилирования (Harris, Attwell, 2012; Hui et al., 2017). С другой стороны, дисфункция MCT1 может привести к аберрантному переносу лактата, что, в свою очередь, приведет к нарушению энергетического обмена и когнитивному дефициту (Newman et al., 2011; Suzuki et al., 2011; Boury-Jamot et al., 2016).
Так, в ходе одного из исследований у мышей (линия C57BL/6) в гиппокампе выявлены изменения экспрессии генов, вызванные обучением, которые участвуют в метаболическом взаимодействии астроцитов и нейронов, а именно: через 24 ч после тренировки в тесте ингибирующего избегания наблюдали повышенную экспрессию генов, кодирующих транспортеры MCT1 и MCT4, альфа2-субъединицу Na/K-ATФaзы и транспортер глюкозы GLUT1. Примечательно, что при оценке функциональной роли одного из этих генов, участвующих в обучении (гена MCT1), обнаружено нарушение долговременной памяти у MCT1-нокаутных мышей (Tadi et al., 2015).
В другом исследовании продемонстрировано, что подавление MCT1 и MCT4 в глиальных клетках и MCT2 в нейронах гиппокампа крыс вызывает нарушение процесса обучения и дисфункцию памяти (Pérez-Escuredo et al., 2016). Снижение содержания лактата, сопровождающееся подавлением экспрессии гена MCT в коре головного мозга и гиппокампе, способствовало нарушению пространственного обучения и памяти у крыс с моделью БА (Lu et al., 2015). В целом, это убедительно доказывает значимое влияние лактата и MCT на энергетический метаболизм, формирование гиппокамп-зависимой памяти и активность нейронов гиппокампа, что существенным образом нарушается при развитии БА.
Таким образом, установленное нами снижение уровня лактата в ткани головного мозга и увеличение уровня лактата в диализате при остром токсическом действии Aβ1–42 in vivo являются признаками нарушения энергетического метаболизма вследствие токсического действия Aβ1–42 на нервные клетки.
В дополнение к этому, нами выявлено, что клетки астроглии под влиянием Aβ1-42 in vitro характеризуются повышенной продукцией лактата, что, в совокупности с представленными выше результатами дает право предполагать, что они вносят важный вклад в увеличение уровня лактата во внеклеточном пространстве, сохраняя свою гликолитическую активность.
Это сопряжено с зарегистрированным нами при остром токсическом действии Aβ1–42 in vivo и in vitro снижением уровня импорт-ориентированных изоформ MCT в нейронах, астроглии и церебральном эндотелии. С учетом полученных нами ранее данных о гипометаболизме глюкозы при развитии БА (Горина и др., 2017), логично предположить, что такие изменения уровня MCT1 и MCT2 маркируют энергодефицит в ткани головного мозга. Это соответствует результатам, полученным у животных с генетической моделью БА (линия APP/PS1), что приводит к лактат-дефициту в нейронах, в норме зависящих от транспорта лактата из гликолитически активных астроцитов (Zhang et al., 2018). Более того, согласно ранее полученным нами результатам (Горина и др., 2017), при остром токсическом действии Aβ1–42 у мышей выявлено нарушение ассоциативного обучения и эмоциональной памяти в тесте “Fear conditioning” (условного рефлекса страха), о чем указывало отсутствие формирования условного рефлекса на протяжении всего периода тестирования в ответ на неприятный стимул. Это позволяет сделать предположение о том, что установленные патологические изменения поведения сопряжены с нарушением уровня транспортеров лактата в клетках головного мозга в условиях острого токсического действия Aβ1–42.
Таким образом, установлено, что в условиях острого токсического действия Aβ1–42 у экспериментальных животных наблюдается снижение уровня лактата в ткани гиппокампа и повышение его уровня в диализате in vivo, которое сопряжено с аберрантной экспрессией транспортеров лактата MCT1 и MCT2 в нейронах, астроглии и церебральном эндотелии. В свою очередь, это может препятствовать транспорту лактата от глиальных клеток до нейронов, вызывая выраженный дефицит лактата в нейронах. В совокупности это указывает о нарушении энергетического метаболизма вследствие Aβ-индуцированного патологического изменения церебрального метаболизма глюкозы. При этом зафиксированное нами выраженное увеличение продукции лактата астроцитами in vitro может свидетельствовать о включении компенсаторного механизма, направленного на поддержание межклеточного метаболического сопряжения в поврежденных регионах головного мозга.
Список литературы
Горина Я.В., Комлева Ю.К., Лопатина О.Л., Черных А.И., Салмина А.Б. 2017. Влияние инсулинорезистентности на нарушение метаболизма глюкозы в миндалине головного мозга при экспериментальной болезни Альцгеймера. Бюлл. сибирской мед. № 6. С. 1. (Gorina Ya.V., Komleva Yu.K., Lopatina O.L., Chernykh A.I., Salmina A.B. 2017. Influence of insulin resistance on increased risk of brain amygdala development in experimental Alzheimer’s disease. Bulletin of Siberian medicine. № 6. Р. 1.)
Комлева Ю.К., Малиновская Н.А., Горина Я.В., Лопатина О.Л., Волкова В.В., Салмина А.Б. 2015. Экспрессия молекул CD38 и CD157 в ольфакторных луковицах головного мозга при экспериментальной болезни Альцгеймера. Сибирское медицинское обозрение. № 5. С. 45. (Komleva Yu.K., Malinovskaya N.A., Gorina Ya.V., Lopatina O.L., Volkova V.V., Salmina A.B. 2015. Expression of CD38 and CD157 molecules in the olfactory bulbs of the brain in experimental Alzheimer’s disease. Siberian Med. Rev. № 5. Р. 45.)
Лобзин В.Ю., Одинак М.М., Фокин В.А., Воробьев С.В., Емелин А.Ю., Лупанов И.А., Кудяшева А.В., Соколов А.В. 2013. Метаболические изменения головного мозга при болезни Альцгеймера, сосудистой и смешанной деменции. Биомед. журнал медлайн.ру. С. 1085. (Lobzin V.Yu., Odinak M.M., Fokin V.A., Vorobyov S.V., Emelin A.Yu., Lupanov I.A., Kudyasheva A.V., Sokolov A.V. 2013. Brain metabolic changes in Alzheimer’s disease, vascular and mixed dementia. Biomed. J. Medline.ru. P. 1085.)
Моргун А.В., Кувачева Н.В., Комлева Ю.К., Кутищева И.А., Окунева О.С., Дробушевская А.И., Хилажева Е.Д., Черепанов С.М., Салмина А.Б. 2013. Дифференцировка эмбриональных прогениторных клеток мозга крыс в астроциты и нейроны. Сибирское мед. обозрение. № 6. С. 9. (Morgun A.V., Kuvacheva N.V., Komleva Yu.K., Kutishcheva I.A., Okuneva O.S., Drobushevskaya A.I., Khilazheva E.D., Cherepanov S.M., Salmina A.B. 2013. Differentiation of rat brain embryonic progenitor cells into astrocytes and neurons. Siberian Med. Rev. № 6. Р. 9.)
Allaman I., Gavillet M., Bélanger M., Laroche T., Viertl D., Lashuel H.A., Magistretti P.J. 2010. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J. Neurosci. V. 30. P. 3326. https://doi.org/10.1523/JNEUROSCI.5098-09.2010
Bartolotti N., Lazarov O. 2019. CREB signals as PBMC-based biomarkers of cognitive dysfunction: a novel perspective of the brain-immune axisBrain Behav. Immun. V. 78. P. 9. https://doi.org/10.1016/j.bbi.2019.01.004
Bekinschtein P., Cammarota M., Katche C., Slipczuk L., Rossato J.I., Goldin A., Izquierdo I., Medina J.H. 2008. BDNF is essential to promote persistence of long-term memory storage. Proc. Natl. Acad. Sci. USA. V. 105. P. 2711. https://doi.org/10.1073/pnas.0711863105
Bergersen L.H. 2015. Lactate transport and signaling in the brain: potential therapeutic targets and roles in body-brain interaction. J. Cereb. Blood Flow. Metab. V. 35. P. 176. https://doi.org/10.1038/jcbfm.2014.206
Berthet C., Castillo X., Magistretti P.J., Hirt L. 2012. New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc Dis. V. 34. P. 329. https://doi.org/10.1159/000343657
Bolaños J.P., Almeida A., Moncada S. 2010. Glycolysis: a bioenergetic or a survival pathway? Trends Biochem. Sci. V. 35. P. 145. https://doi.org/10.1016/j.tibs.2009.10.006
Bondi M.W., Edmonds E.C., Salmon D.P. 2017. Alzheimer’s disease: past, present, and future. J. Int. Neuropsychol. Soc. V. 23. P. 818. https://doi.org/10.1017/S135561771700100X
Boury-Jamot B., Carrard A., Martin J.L., Halfon O., Magistretti P.J., Boutrel B. 2016. Disrupting astrocyte-neuron lactate transfer persistently reduces conditioned responses to cocaine. Mol. Psychiatry. V. 21. P. 1070. https://doi.org/10.1038/mp.2015.157
Coco M., Caggia S., Musumeci G., Perciavalle V., Graziano A.C.E., Pannuzzo G., Cardile V. 2013. Sodium L-lactate differently affects brain-derived neurothrophic factor, inducible nitric oxide synthase, and heat shock protein 70 kDa production in human astrocytes and SH-SY5Y cultures. J. Neurosci. Res. V. 91. P. 313. https://doi.org/10.1002/jnr.23154
Correia S.C., Santos R.X., Carvalho C., Cardoso S., Candeias E., Santos M.S., Oliveira C.R., Moreira P.I. 2012. Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer’s disease and diabetes interrelation. Brain Res. V. 1441. P. 64. https://doi.org/10.1016/j.brainres.2011.12.063
Croteau E., Castellano C.A., Fortier M., Bocti C., Fulop T., Paquet N., Cunnane S.C. 2018. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. V.107. P.18. https://doi.org/10.1016/j.exger.2017.07.004
Cunnane S., Nugent S., Roy M., Courchesne-Loyer A., Croteau E., Tremblay S., Castellano A., Pifferi F., Bocti C., Paquet N., Begdouri H., Bentourkia M., Turcotte E., Allard M., Barberger-Gateau P., Fulop T., Rapoport S.I. 2011. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition. V. 27. P. 3. https://doi.org/10.1016/j.nut.2010.07.021
Ding F., Yao J., Rettberg J.R., Chen S., Brinton R.D. 2013. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One. V. 8. P. e79977. https://doi.org/10.1371/journal.pone.0079977
Encinas J.M., Enikolopov G. 2008. Identifying and quantitating neural stem and progenitor cells in the adult brain. Methods Cell Biol. V. 85. P. 243. https://doi.org/10.1016/S0091-679X(08)85011-X
Epelbaum S., Youssef I., Lacor P.N., Chaurand P., Duplus E., Brugg B., Duyckaerts C., Delatour B. 2015. Acute amnestic encephalopathy in amyloid-β oligomer-injected mice is due to their widespread diffusion in vivo. Neurobiol Aging. V. 36. P. 2043. https://doi.org/10.1016/j.neurobiolaging.2015.03.005
Falkowska A., Gutowska I., Goschorska M., Nowacki P., Chlubek D., Baranowska-Bosiacka I. 2015. Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int. J. Mol. Sci. V. 16. P. 25959. https://doi.org/10.3390/ijms161125939
Forlenza O.V., Diniz B.S., Gattaz W.F. 2010. Diagnosis and biomarkers of predementia in Alzheimer’s disease. BMC Med. V. 8. P. 89. https://doi.org/10.1186/1741-7015-8-89
Gordon G.R., Howarth C., MacVicar B.A. 2016. Bidirectional control of blood flow by astrocytes: a role for tissue oxygen and other metabolic factors. Adv. Exp. Med. Biol. V. 903. P. 209. https://doi.org/10.1007/978-1-4899-7678-9_15
Harris J.J., Attwell D. 2012. The energetics of CNS white matter. J. Neurosci. V. 32. P. 356. https://doi.org/10.1523/JNEUROSCI.3430-11.2012
Harris R.A. 2017. Cerebral lactate metabolism and memory: Implications for Alzheimer’s disease. Electronic Thesis and Dissertation Repository. P. 4529.
Hashimoto T., Hussien R., Oommen S., Gohil K., Brooks G.A. 2007. Lactate sensitive transcription factor network in L6 cells: activation of MCT1 and mitochondrial biogenesis. FASEB J. V. 21. P. 2602. https://doi.org/10.1096/fj.07-8174com
Hui S., Ghergurovich J.M., Morscher R.J., Jang C., Teng X., Lu W., Esparza L.A., Reya T., Le Z., Yanxiang Guo J., White E., Rabinowitz J.D. 2017. Glucose feeds the TCA cycle via circulating lactateNature. V. 551. P. 115. https://doi.org/10.1038/nature24057
Jin N., Qian W., Yin X., Zhang L., Iqbal K., Grundke-Iqbal I., Gong C.-X., Liu F. 2013. CREB regulates the expression of neuronal glucose transporter 3: a possible mechanism related to impaired brain glucose uptake in Alzheimer’s disease. Nucleic Acids Res. V. 41. P. 3240. https://doi.org/10.1093/nar/gks1227
Koenig M.K. 2008. Presentation and diagnosis of mitochondrial disorders in children. Pediatr. Neurol. V. 38. P. 305. https://doi.org/10.1016/j.pediatrneurol
Liguori C., Chiaravalloti A., Sancesario G., Stefani A., Sancesario G.M., Mercuri N.B., Schillaci O., Pierantozzi M. 2016. Cerebrospinal fluid lactate levels and brain [18F]FDG PET hypometabolism within the default mode network in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging. V. 43. P. 2040. https://doi.org/10.1007/s00259-016-3417-2
Liguori C., Stefani A., Sancesario G., Sancesario G.M., Marciani M.G., Pierantozzi M. 2015. CSF lactate levels, τ proteins, cognitive decline: a dynamic relationship in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry. V. 86. P. 655. https://doi.org/10.1136/jnnp-2014-308577
Liu Y., Xue Q., Tang Q., Hou M., Qi H., Chen G., Chen W., Zhang J., Chen Y., Xu X. 2013. A simple method for isolating and culturing the rat brain microvascular endothelial cells. Microvasc. Res. V. 90. P. 199. https://doi.org/10.1016/j.mvr.2013.08.004
Lu W., Huang J., Sun S., Huang S., Gan S., Xu J., Yang M., Xu S., Jiang X. 2015. Changes in lactate content and monocarboxylate transporter 2 expression in Aβ25−35-treated rat model of Alzheimer’s disease. Neurol Sci. V. 36. P. 871. https://doi.org/10.1007/s10072-015-2087-3
Mohamed A., Posse de Chaves E. 2011. Aβ internalization by neurons and glia. Int. J. Alzheimers Dis. P. 127984. https://doi.org/10.4061/2011/127984
Moreira T.J., Pierre K., Maekawa F., Repond C., Cebere A., Liljequist S., Pellerin L. 2009. Enhanced cerebral expression of MCT1 and MCT2 in a rat ischemia model occurs in activated microglial cells. J. Cereb. Blood Flow Metab. V. 29. P. 1273. https://doi.org/10.1038/jcbfm.2009.50
Mosconi L., Mistur R., Switalski R., Tsui W.H., Glodzik L., Li Y., Pirraglia E., De Santi S., Reisberg B., Wisniewski T., de Leon M. 2009. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. J. Eur. J. Nucl. Med. Mol. Imaging. V. 36. P. 811. https://doi.org/10.1007/s00259-008-1039-z
Muraleedharan R., Gawali M.V., Tiwari D., Sukumaran A., Oatman N., Anderson J., Nardini D., Bhuiyan M.A.N., Tkáč I., Ward A.L., Kundu M., Waclaw R., Chow L.M., Gross C., Rao R., Schirmeier S., Dasgupta B. 2020. AMPK-Regulated Astrocytic lactate shuttle plays a non-cell-autonomous role in neuronal survival. Cell Rep. V. 32. P. 108092. https://doi.org/10.1016/j.celrep.2020.108092
Nagase M., Takahashi Y., Watabe A.M., Kubo Y., Kato F. 2014. On-site energy supply at synapses through monocarboxylate transporters maintains excitatory synaptic transmission. J. Neurosci. V. 34. P. 2605. https://doi.org/10.1523/JNEUROSCI.4687-12.2014
Newman L.A., Korol D.L., Gold P.E. 2011. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One. V. 6. P. e28427. https://doi.org/0.1371/journal.pone.0028427
Nielsen H.M., Veerhuis R., Holmqvist B., Janciauskiene S. 2009. Binding and uptake of A beta1-42 by primary human astrocytes in vitro. Glia. V. 57. P. 978. https://doi.org/10.1002/glia.20822
Pérez-Escuredo J., Van Hée V.F., Sboarina M., Falces J., Payen V.L., Pellerin L., Sonveaux P. 2016. Monocarboxylate transporters in the brain and in cancer Biochim. Biophys. Acta. V. 1863. P. 2481. https://doi.org/10.1016/j.bbamcr.2016.03.013
Pierre K., Pellerin L. 2005. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J. Neurochem. V. 94. P. 1. https://doi.org/10.1111/j.1471-4159.2005.03168.x
Pinheiro C., Longatto-Filho A., Azevedo-Silva J., Casal M., Schmitt F.C., Baltazar F. 2012. Role of monocarboxylate transporters in human cancers: state of the art. J. Bioenerg. Biomembr. V. 44. P. 127–139. https://doi.org/10.1007/s10863-012-9428-1
Salmina A.B., Kuvacheva N.V., Morgun A.V., Komleva Y.K., Pozhilenkova E.A., Lopatina O.L., Gorina Y.V., Taranushenko T.E., Petrova L.L. 2015. Glycolysis-mediated control of blood-brain barrier development and function. Int. J. Biochem. Cell Biol. V. 64. P. 174. https://doi.org/10.1016/j.biocel.2015.04.005
Shin B.K., Kang S., Kim D.S., Park S. 2018. Intermittent fasting protects against the deterioration of cognitive function, energy metabolism and dyslipidemia in Alzheimer’s disease-induced estrogen deficient rats. Exp. Biol. Med. (Maywood). V. 243. P. 334. https://doi.org/10.1177/1535370217751610
Shin Y., Choi S.H., Kim E., Bylykbashi E., Kim J.A., Chung S., Kim D.Y., Kamm R.D., Tanzi R.E. 2019. Blood-brain barrier dysfunction in a 3D in vitro model of Alzheimer’s disease. Adv. Sci. V. 6. P.1900962. https://doi.org/10.1002/advs.201900962
Sipos E., Kurunczi A., Kasza A., Horvath J., Felszeghy K., Laroche S., Toldi J., Parducz A., Penke B., Penke Z. 2007. Beta-amyloid pathology in the entorhinal cortex of rats induces memory deficits: implications for Alzheimer’s disease. Neurosci. V. 147. P. 28. https://doi.org/10.1016/j.neuroscience.2007.04.011
Suzuki A., Stern S.A., Bozdagi O., Huntley G.W., Walker R.H., Magistretti P.J., Alberini C.M. 2011. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. V. 144. P. 810. https://doi.org/10.1016/j.cell.2011.02.018
Tadi M., Allaman I., Lengacher S., Grenningloh G., Magistretti P.J. 2015. Learning-induced gene expression in the hippocampus reveals a role of neuron-astrocyte metabolic coupling in long term memory. PLoS One. 2015. V. 10. P. e0141568. https://doi.org/10.1371/journal.pone.0141568
Tang B.L. 2018. Brain activity-induced neuronal glucose uptake/glycolysis: is the lactate shuttle not required? Brain Res. Bull. V. 137. P. 225. https://doi.org/10.1016/j.brainresbull.2017.12.010
Wang Y., Shang Y., Mishra A., Bacon E., Yin F., Brinton R. 2020. Midlife chronological and endocrinological transitions in brain metabolism: system biology basis for increased Alzheimer’s risk in female brain. Sci. Rep. V. 10. P. 8528. https://doi.org/10.1038/s41598-020-65402-5
Yamanishi S., Katsumura K., Kobayashi T., Puro D.G. 2006. Extracellular lactate as a dynamic vasoactive signal in the rat retinal microvasculature. Am. J. Physiol. Heart Circ. Physiol. V. 290. P. 925H. https://doi.org/10.1152/ajpheart.01012.2005
Zhang M., Cheng X., Dang R., Zhang W., Zhang J., Yao Z. 2018. Lactate deficit in an Alzheimer disease mouse model: the relationship with neuronal damage. J. Neuropathol. Exp. Neurol. V. 77. P. 1163. https://doi.org/10.1093/jnen/nly102
Дополнительные материалы отсутствуют.