

Журнал физической химии, 2019, T. 93, № 12, стр. 1810-1823
Сравнительный анализ водородной, ван-дер-ваальсовой и галогенной связей в комплексах аммиака с молекулами HCl и ClF
a Российская академия наук, Институт органической химии им. Н.Д. Зелинского
119991 Москва, Россия
* E-mail: isaevaln@ioc.ac.ru
Поступила в редакцию 29.01.2019
После доработки 29.01.2019
Принята к публикации 20.02.2019
Аннотация
Методом MP2 теории возмущений Меллера–Плессета второго порядка в корреляционно-согласованном базисе Даннинга aug-cc-pVTZ, дополненном диффузными функциями, проведены квантово-химические расчеты молекулярных комплексов, образованных аммиакам и метанидом (донор электронной пары) c молекулами HCl и ClF (акцептор) при различных вариантах их взаимной ориентации. Во всех комплексах отмечается удлинение ковалентной связи акцептора и красный сдвиг соответствующей полосы в ИК-спектре. Выполнен NBO-анализ и анализ топологии электронной плотности, построены карты сдвига электронной плотности при образовании комплексов. Рассчитана структура переходного состояния для взаимопревращения комплексов NH3···HCl и NH3···ClH, отвечающих минимумам на поверхности потенциальной энергии. Переход между конфигурациями сопровождается эволюцией межмолекулярного связевого пути с заменой критической точки контакта N···H критической точкой контакта N···Cl. Данные расчетов, дополненные сравнительным анализом потенциальных кривых взаимодействия, указывают на сходную природу межмолекулярного связывания в комплексах H3N и HCl с различной ориентацией мономеров.
В теории орбиталей натуральных связей (NBO) классическая трехцентровая четырехэлектронная (3с-4e) водородная (Н-) связь Y···H–X определяется как донорно-акцепторное взаимодействие nY → $\sigma _{{{\text{X}}--{\text{H}}}}^{*}$ между центром Y с неподеленной электронной парой и анти-связью X–H акцептора электронов [1]. В этом определении предполагается, что при образовании Н-связи электронная пара Y (основание Льюиса) ориентирована в направлении протона акцептора (кислота Льюиса). В то же время, очевидно, что существуют четыре возможных варианта взаимной ориентации неподеленной электронной пары и акцептора, которые представлены на рис. 1.
Рис. 1.
Возможные варианты взаимной ориентации неподеленной электронной пары атома Y и ковалентной связи X–H акцептора. Обозначения см. текст.

Вариант а на рис. 1 отвечает традиционному водородному связыванию, рассматриваемому в литературе. Из общих соображений понятно, что именно при такой ориентации мономеров (“голова к голове”, А-комплексы) должно происходить наиболее эффективное связывание донора электронной пары с акцептором. Анализ свойств молекулярных комплексов с ориентацией мономеров “хвост к голове” (вариант б на рис. 1, Б‑комплексы) показал их сходство с комплексами А-типа и позволил определить межмолекулярное взаимодействие Y···H–X в этих комплексах как водородную связь, более слабую по сравнению с Н-связью в А-комплексах [2–4].
Можно предположить, что связывание мономеров по вариантам в и г (рис. 1) также возможно при определенных условиях; природа межмолекулярного взаимодействия в таких комплексах представляет немалый интерес для теоретической химии. Поиск подобных молекулярных систем показал, что молекулы NH3 и HСl могут образовывать комплексы всех четырех типов ориентации мономеров. Эти комплексы представляют исключительно удобный объект для сравнительного анализа нековалентных взаимодействий N···H–Cl и N···Cl–H, который выполнен в настоящей работе. Рассмотрены также А- и Б-комплексы NH3 с ClF с целью сравнения Н-связи с галогенной связью, которая интенсивно изучается в последние годы. В частности, А-комплекс H3N···ClF с галогенной связью рассматривался ранее как в экспериментальных, так и в теоретических работах [5–10].
В ряде исследований показано, что сила нековалентного взаимодействия и ориентация взаимодействующих молекул могут быть предсказаны и объяснены из анализа положений и значений минимумов и максимумов электростатического потенциала (ESP) на ван-дер-ваальсовой поверхности молекул. Как правило, связывание мономеров друг с другом происходит таким образом, что экстремумы ESP молекул максимально комплементарны. Выполненные в работе [4] расчеты показали, что молекула аммиака имеет два минимума ESP на ван-дер-ваальсовой поверхности, которые лежат на оси симметрии С3v молекулы по разные стороны от ядра атома азота. Эти минимумы отвечают за водородное связывание молекулы аммиака с акцептором электронов с образованием комплексов А- и Б-типа. А-комплекс H3N···HCl был исследован в целом ряде более ранних работ, например, [3, 11–17]; Б-комплекс детально рассмотрен в работах [3, 4].
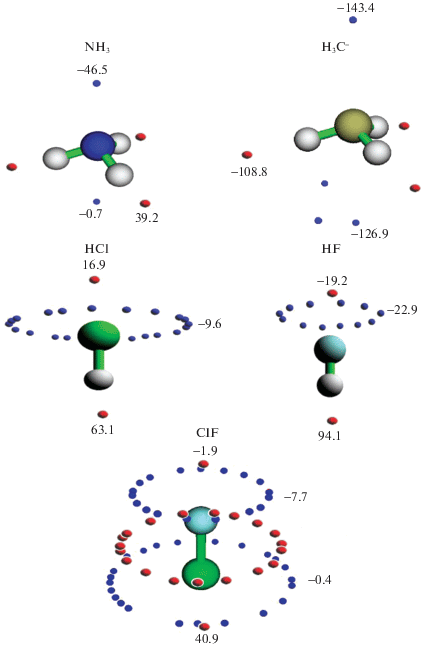
На рис. 2 приведены карты ESP для аммиака и -аниона, а также молекул HCl, HF и ClF. На рисунке видно, что ван-дер-ваальсова поверхность молекулы HСl имеет максимум ESP, лежащий на оси молекулы вблизи атома Cl. По-видимому, именно это делает возможным связывание молекулы HCl с молекулой NH3 по вариантам в и г. По аналогии с Н-связанными комплексами, вариант в отвечает ориентации мономеров “голова к хвосту” (это В-комплексы в тексте статьи), а вариант г – ориентации “хвост к хвосту” (Г-комплексы). Квантово-химические расчеты комплекса H3N···ClH В-типа были ранее проведены в теоретическом исследовании [18], Г-комплекс впервые описан в настоящей статье. Проведено сравнение нейтральных молекулярных систем с ионными В- и Г-комплексами, образованными молекулой HCl и метид-анионом (H3C–).
Рис. 2.
Положения минимумов (синие сферы) и максимумов (красные сферы) электростатического потенциала на ван-дер-ваальсовой молекулярной поверхности (изолиния 0.001 а.е.) для мономеров, участвующих в комплексообразовании. Числа показывают значения экстремумов.
МЕТОДЫ РАСЧЕТОВ
Квантово-химические расчеты молекулярных комплексов проведены по программе Gaussian 09 [19] методом MP2/aug-cc-pVTZ, который в последние годы широко используется при исследовании нековалентных взаимодействий. В исследовании [20] межмолекулярных взаимодействий различной природы было показано, что энергии связи, найденные методом MP2 [21] теории возмущений Меллера–Плессета второго порядка и методом связанных кластеров CCSD(T) [22, 23], имеют близкие значения при проведении расчетов в корреляционно-согласованных базисах Даннинга aug-cc-pVxZ (x = T, Q), дополненных диффузными функциями [24]. В настоящей статье приведены энергии связи по данным расчетов методом CCSD(T)/aug-cc-pVTZ для геометрии молекулярных комплексов, оптимизированной в MP2/aug-cc-pVTZ- расчетах.
Структуры, найденные в расчетах с полной оптимизацией геометрии, были проверены на отсутствие мнимых частот в матрице силовых констант. Энергии связи в молекулярных комплексах определены как разность между полной энергией комплекса и суммой энергий изолированных мономеров с учетом суперпозиционной ошибки базисного набора (BSSE) по Бойсу–Бернарди [25]. Анализ орбиталей натуральных связей (NBO) [26, 27] и топологический анализ электронной плотности по теории Бейдера [28, 29] проводились с использованием процедур, включенных в Gaussian 09. Карты электростатического потенциала и распределения электронной плотности, а также молекулярные графы построены на основе данных квантово-химических расчетов с помощью программы Multiwfn [30].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Геометрия и энергия связи. Рассмотренные в работе молекулярные комплексы, образованные аммиаком c молекулами HCl и ClF, показаны на рис. 3. Характерные геометрические параметры комплексов, рассчитанные методом MP2/aug-cc-pVTZ, приведены также в табл. 1. Как видно из данных таблицы, А-комплексы H3N···HCl и H3N···ClF с водородной и галогенной связью заметно прочнее комплексов, образованных при других вариантах ориентации мономеров. Расчетные значения межмолекулярного расстояния и энергии связи в этих комплексах находятся в согласии с известными из литературы данными [8, 9, 14, 18], которые также приведены в табл. 1. Во всех комплексах отмечается удлинение ковалентных связей в молекулах акцептора электронной пары и длинноволновый сдвиг полос HCl и ClF в ИК-спектре (красное смещение). Можно отметить близкие расчетные значения для удлинения ковалентной связи H−Cl и красного смещения ΔνHCl полосы HCl в ИК-спектре А-комплекса H3N···HCl и ионного комплекса В-типа H3C−···ClH, образованного метид-анионом.
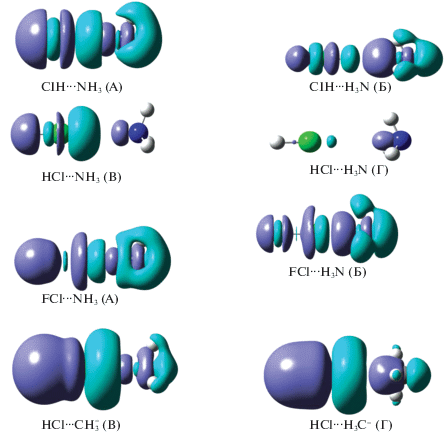
Рис. 3.
Комплексы аммиака и метанида с HCl и ClF при различной взаимной ориентации молекул. Комплексы H3N···ClF с галогенной связью определены как комплексы типа А и Б по аналогии с Н-связанными комплексами, так как ближним к азоту находится менее электрооотрицательный атом Cl. Межатомное расстояние указано в Ангстремах.

Таблица 1.
Межатомное расстояние N···Cl (R), угол Cl···NH (θ) при связывании мономеров, изменение в длине ковалентной связи H–Cl (ΔrHCl), красный сдвиг в частоте ($\Delta \nu _{{{\text{H}} - {\text{Cl}}}}^{{{\text{asym}}}}$), химический сдвиг ЯМР (δH) на атоме водорода и энергия связи (Ebind) в молекулярных комплексах, образованных аммиаком с молекулами HCl и ClF при их различной ориентации
| Молекулярный комплекс | R | θ | ΔrHCl | $\Delta \nu _{{{\text{H}} - {\text{Cl}}}}^{{{\text{asym}}}}$ | δH | Ebind | |
|---|---|---|---|---|---|---|---|
| Без учета BSSE | С учетом BSSE | ||||||
| H3N···HCl (A) | 3.066 | 111.4 | 53.3 | –748.1 | –10.13 | 9.22 (8.37) | 9.51 (8.52) |
| а) | 2.986 | 9.70 | |||||
| NH3···HCl (Б) | 3.654 | 71.0 | 7.6 | –111.6 | –1.81 | 1.59 (1.44) | 1.54 (1.45) |
| H3N···ClH (В) | 3.293 | 112.1 | 1.6 | –14.2 | 0.33 | 1.05 (1.03) | 0.90 (0.89) |
| б) | 3.244 | 0.70 | |||||
| NH3···ClH (Г) | 3.558 | 68.0 | 1.2 | –10.6 | –0.14 | 0.79 (0.80) | 0.64 (0.65) |
| H3N···ClF (A) | 2.232 | 110.1 | 76.2 | –174.8 | 497.23 | 11.82 (9.95) | 12.75 (10.50) |
| в) | 2.24 | 11.0 | |||||
| г) | 2.27 | 11.64 | |||||
| NH3···ClF (Б) | 2.825 | 72.1 | 15.6 | –74.5 | 175.41 | 2.72 (2.37) | 2.87 (2.54) |
| H3C–···ClH (В) | 2.831 | 108.4 | 51.3 | –613.5 | –2.57 | 3.42 (3.34) | 3.50 (3.48) |
| CH$_{3}^{ - }$···ClH (Г) | 3.310 | 73.0 | 17.1 | –234.0 | –0.53 | 1.23 (1.67) | 0.75 (1.32) |
Примечание. R и ΔrHCl даны в Å и мÅ соответственно, Δν дано в см–1, δH – в мд. Величины Ebind приведены в ккал/моль; значения в круглых скобках показывают Ebind, рассчитанную методом CCSD(T)/aug-cc-pVTZ при геометрии комплекса, оптимизированной в расчетах MP2/aug-cc-pVTZ. В двух нижних строках таблицы даны расчетные данные для комплекса метанида. В третьем столбце дано значение угла θ, усредненное по атомам водорода молекулы акцептора. Для комплексов с галогенной связью в четвертом и пятом стобцах приведены данные для ковалентной связи Cl–F и химический сдвиг δ на атоме хлора в шестом столбце. Для комплексов метанида во втором столбце указано межатомное расстояние C···Cl. Для А- и Б-комплексов аммиака с молекулой HCl приведены результаты расчетов [3] и [4]; в строках а), б), в) и г) – данные работ [14], [18], [8] и [9] соответственно.
Известно, что межмолекулярное расстояние часто рассматривается как один из критериев силы межмолекулярного взаимодействия [31, 32]. Из табл. 1 видно, что в ионных В- и Г-комплексах метанида межмолекулярное расстояние С···Cl на 0.3–0.5 Å меньше, чем межмолекулярное расстояние N···Cl в нейтральных В- и Г-комплексах аммиака, что находится в согласии с большей стабильностью комплексов метанида. С другой стороны, комплексы NH3···ClF (Б) и H3C−···ClH (В) с примерно одинаковым межмолекулярным расстоянием R(N···Cl) и R(C···Cl) имеют близкие значения энергии связи. Как видно из таблицы, энергии связи, рассчитанные методом CCSD(T)/aug-cc-pVTZ находятся в хорошем согласии с данными расчетов MP2/aug-cc-pVTZ.
В равновесных комплексах А, Б и Г аммиака с молекулой HCl, а также в комплексах метанида, наблюдается химический сдвиг δH водорода со смещением сигнала протона в ЯМР-спектре в сторону более слабого поля, а величина химического сдвига коррелирует с энергией связи. Следует отметить, что комплекс H3N···ClH В-типа рассчитывался ранее методом MP2/aug-cc-pVDZ в работе [18] и был описан как равновесная структура; найденные в [18] значения R(N···Cl) и энергии связи даны в табл. 1. Однако, наши расчеты методом MP2/aug-cc-pVTZ показали, что матрица силовых констант для этой структуры имеет два отрицательных собственных значения, т.е. В-комплекс H3N···ClH отвечает критической точке второго порядка.
Анализ натуральных орбиталей (NBO анализ). Данные NBO-анализа для комплексов позволяют выделить четыре основных типа межорбитальных взаимодействий, которые определяют связывание мономеров. В последнем столбце табл. 2 приведены значения энергии E(2) возмущения второго порядка для доминирующих взаимодействий по данным расчетов методом MP2/aug-cc-pVTZ. Из табл. 2 видно, что в А- и Б-комплексах аммиака с водородной и галогенной связью основной вклад в связывание вносит взаимодействие nN → σ*HCl (nN → σ*ClF) неподеленной пары азота с антисвязывающей орбиталью ковалентной связи HCl или ClF-акцептора. В более слабых комплексах H3N···ClH (В) и NH3···ClH (Г) становится заметным вклад взаимодействия σHCl → Ry*N с переносом электронного заряда на Ридберговы орбитали атома азота, энергия $E_{{n{\text{N}} \to \sigma {\text{*HCl}}}}^{{(2)}}$ в этих комплексах невелика.
Таблица 2.
Вклад p-орбитали в натуральную гибридную орбиталь (% p), заселенность орбиталей натуральных связей (заселенность NBO η), изменения зарядов (Δq) на атомах при комплексообразовании, перенесенный на молекулу HCl (ClF) электронный заряд (Q) и энергия E(2) возмущения второго порядка в комплексах аммиака при различной ориентации мономеров
| Молекулярный комплекс | n-орбиталь азота | σ*HCl и Ry*Н (Ry*Cl) орбитали | Δq | Q | $\begin{gathered} E_{{n{\text{N}} \to \sigma {\text{*HCl}}}}^{{(2)}} \\ E_{{\sigma {\text{HCl}} \to {\text{Ry*N}}}}^{{(2)}} \\ E_{{n{\text{Cl}} \to {\text{Ry*N}}}}^{{(2)}} \\ E_{{n{\text{N}} \to {\text{Ry*H(Cl)}}}}^{{(2)}} \\ \end{gathered} $ | ||||
|---|---|---|---|---|---|---|---|---|---|
| % p | η | % p | η | акцептор N | мостик H(Cl) | донор | |||
| H3N···HCl (A) | 77.2 (sp3.38) | 1.9236 | 80.0 (sp4.13)/ 19.2 (sp0.24) |
0.0716 | 10.7 | 61.5 | –132.2 | 70.7 | 43.37 |
| 0.0045 | –25.2 | 165.5 | –224.5 | 58.9 | 3.72 | ||||
| 516.0 | –98.6 | –94.1 | 192.7 | 0.32 | |||||
| 1.14 | |||||||||
| NH3···HCl (Б) | 81.2 (sp4.37) | 1.9844 | 83.2 (sp5.20)/ 8.43 (sp0.09) |
0.0124 | –8.9 | 3.9 | –18.4 | 14.5 | 3.34 |
| 0.0035 | –34.5 | 19.1 | –36.4 | 17.3 | 0.32 | ||||
| –213.8 | –170.3 | 55.9 | 114.4 | 0.0 | |||||
| 0.86 | |||||||||
| H3N···ClH (В) | 76.7 (sp3.31) | 1.9926 | 85.2 (sp6.13)/ 19.9 (sp1.25) |
0.0027 | 3.2 | 18.1 | –18.0 | –0.1 | 1.10 |
| 0.0003 | 6.7 | 17.9 | –25.4 | 7.5 | 1.65 | ||||
| 518.3 | –151.4 | 55.1 | 96.3 | 0.33 | |||||
| 0.09 | |||||||||
| NH3···ClH (Г) | 76.8 (sp3.35) | 1.9953 | 84.5 (sp5.75) | 0.0018 | –0.5 | –0.9 | 1.2 | –0.3 | 0.36 |
| 1.3 | –3.8 | 0.2 | 3.7 | 0.53 | |||||
| –222.1 | –153.6 | 77.8 | 75.8 | 0.09 | |||||
| 0.0 | |||||||||
| H3N···ClF (A) | 77.35 (sp3.42) | 1.8482 | 94.9 (sp29.72)/ 3.55 (sp0.08) |
0.1262 | 54.4 | –22.2 | –115.6 | 138.6 | 52.43 |
| 0.0237 | 2.6 | –52.1 | –80.0 | 133.2 | 4.08 | ||||
| 323.7 | –79.9 | –132.1 | 212.0 | 1.90 | |||||
| 8.69 | |||||||||
| NH3···ClF (Б) | 82.64 (sp4.83) | 1.9721 | 92.72 (sp17.89)/ 19.47 (sp1.78) |
0.0040 | –11.1 | –5.8 | –20.6 | 26.4 | 4.45 |
| –48.0 | –27.3 | –7.5 | 36.0 | 1.19 | |||||
| –165.9 | –142.5 | 35.3 | 107.2 | 0.36 | |||||
| 0.48 | |||||||||
| H3C–···ClH (В) | 83.50 (sp5.07) | 1.8618 | 90.13(sp10.00)/ 6.91 (sp0.14) |
0.0857/ 0.0188 |
103.2 | 39.2 | –144.1 | 105.0 | 19.55 |
| 158.0 | 44.8 | –202.2 | 157.3 | 5.68 | |||||
| 1382.2 | –159.0 | –150.2 | 309.1 | 3.09 | |||||
| 1.71 | |||||||||
| CH$_{3}^{ - }$···ClH (Г) | 86.86 (sp6.65) | 1.9523 | 87.10 (sp7.25)/ 30.56 (sp1.03) |
0.0254/ 0.0068 | 30.0 | 42.1 | –71.8 | 29.7 | 3.21 |
| 10.2 | 28.5 | –100.4 | 71.9 | 1.26 | |||||
| –433.1 | –40.9 | –112.2 | 153.0 | 0.52 | |||||
| 1.31 | |||||||||
Примечание. Значения Δq и Q даны в миллиэлектронах; при этом в первой, второй и третьей строках приведены данные, полученные с использованием NPA, AIM и ChelpG схем анализа заселенности, соответственно. Величина Q определена как сумма зарядов на атомах в молекуле аммиака. Значения энергии возмущения E(2) даны в ккал/моль. Две последние строки относятся к комплексам метид-аниона, где данные во втором, третьем и последнем столбцах относятся к n- и Ry*-орбиталям атома С, а данные шестого столбца показывают изменение заряда на атоме углерода.
Так, в А- и Б-комплексах вклад первого взаимодействия на порядок больше, чем вклад второго, а в В- и Г-комплексах эти вклады примерно равны. Из данных табл. 2 также видно, что с усилением электронодонорных свойств, при переходе от нейтральных комплексов аммиака к ионным комплексам метанида, вклад взаимодействия с переносом на орбиталь σ*(H–Cl) заметно возрастает и оно становится доминирующим. В Б-комплексе NH3···HCl заметный вклад в связывание вносит взаимодействие nN → Ry*H, а в А-комплексе H3N···ClF и в Г-комплексе CH$_{3}^{ - }$···ClH – взаимодействия nN → Ry*Cl и nС → Ry*Cl соответственно.
В табл. 2 даны также изменения зарядов на атомах триады N···HCl (N···ClF), найденные с использованием трех различных схем анализа заселенности: схемы NPA заселенности натуральных атомных орбиталей [27, 33], схемы AIM [28, 29] интегрирования электронной плотности по атомному бассейну и процедуры CHelpG [34] воспроизведения молекулярного электростатического потенциала. NPA- и AIM-заряды согласуются между собой лучше, чем со значениями CHelpG-зарядов. Например, последний метод всегда показывает заметное увеличение отрицательного заряда на атоме мостика, даже на водороде в А- и Б-комплексах с Н-связью.
Методы NPA и AIM предсказывают близкие величины электронного заряда Q, который переходит с аммиака на акцептор (см. табл. 2). В квантово-химическом исследовании [35] молекулярного комплекса HCH3···HOH корректное описание изменения зарядов на атомах было получено методом AIM, тогда как метод CHelpG показывал нереалистично большую величину перенесенного заряда. Для всех рассматриваемых в настоящей статье комплексов метод AIM показывает увеличение электронного заряда на молекулах HCl и ClF; при этом величина Q коррелирует с энергией E(2) и заселенностью η вакантной σ*-орбитали акцептора. Значительная величина Q на молекуле ClF в А-комплексе H3N···ClF с галогенной связью согласуется с определением этого комплекса как комплекса с переносом заряда [8].
Топология и карты электронной плотности. Любая схема присвоения заряда атомам допускает некоторую степень произвольности. По этой причине данные об изменении зарядов дополнены картами, которые показывают сдвиги электронной плотности в результате взаимодействия мономеров при образовании молекулярного комплекса. Такие карты, построенные для всех комплексов, представлены на рис. 4. На этих картах фиолетовым и зеленым цветом показаны области увеличения и понижения электронной плотности, соответственно. Как видно на рисунке, несмотря на различия в межмолекулярном расстоянии и энергии связи в комплексах, карты перераспределения электронной плотности очень сходны в основных чертах.
Рис. 4.
Сдвиг электронной плотности при образовании молекулярных комплексов различного типа. Зеленым цветом показаны области уменьшения электронной плотности как результат комплексообразования, а фиолетовым цветом – области повышенной электронной плотности относительно изолированных мономеров. Изолиния очерчивает области с изменением плотности более 0.0003 э.з./а.е.3.
Действительно, можно видеть одинаковое чередование областей увеличения и уменьшения электронной плотности при переходе от донора электронов (молекула аммиака) к акцептору. Так, на линии между атомом N и атомом мостика мы видим большую область высокой электронной плотности, которая примыкает к молекуле аммиака, и область понижения электронной плотности вблизи мостикового атома, водорода или хлора. Эта картина согласуется с представлениями об электростатическом связывании электронодефицитного атома мостика областью повышенной электронной плотности вблизи атома азота. Также во всех комплексах, как аммиака так и метанида, на концевом атоме акцептора отмечается область повышения электронной плотности. Для комплексов с различной ориентацией мономеров имеют место только количественные отличия: в Б- и Г-комплексах с электронной парой, ориентированной от акцептора, изменения электронной плотности выражены слабее, чем в А- и В-комплексах.
Анализ топологии электронной плотности в молекулярных комплексах методом AIM по теории Бейдера [29] показывает наличие межмолекулярного связевого пути, соединяющего атом азота с атомом H или Cl, и критической точки (КТ) связи (3, – 1) для контактов N···H и N···Cl. Значения электронной плотности ρ и лапласиана электронной плотности ${{\nabla }^{2}}$ρ в КТ соответствующих межмолекулярных контактов в комплексах аммиака и метанида приведены в табл. 3. Положительные значения лапласиана указывают на то, что все молекулярные комплексы являются системами с взаимодействием по типу закрытой оболочки, к числу которых относятся ван-дер-ваальсовы комплексы и комплексы с водородной связью. Значения электронной плотности ρ и плотности потенциальной энергии VBCP в КТ линейно коррелируют с энергией связи в молекулярном комплексе (см. рис. 5). В наиболее прочных комплексах H3N···ClF (А), H3N···HCl (А) и H3С–···ClH (В) значения плотности полной энергии HBCP в КТ отрицательны, что указывает на частично ковалентный характер межмолекулярной связи в этих комплексах [36].
Таблица 3.
Электронная плотность (ρ), лапласиан электронной плотности (${{{\nabla }^{{\text{2}}}}}$ρ), эллиптичность лапласиана (ε), плотность потенциальной энергии (VBCP) и плотность полной энергии (HBCP) в критической точке связи для контактов N···H и N···Cl в комплексах аммиака с различной ориентацией мономеров
| Молекулярный комплекс | Топологические параметры | ||||
|---|---|---|---|---|---|
| ρ | ${{{\nabla }^{{\text{2}}}}}$ρ | ε | VBCP | HBCP | |
| H3N···HCl (A) | 0.0506 | 0.0693 | <0.001 | −0.0514 | −0.0038 |
| NH3···HCl (Б) | 0.0128 | 0.0421 | 0.001 | −0.0098 | 0.0015 |
| H3N···ClH (В) | 0.0065 | 0.0269 | <0.001 | −0.0036 | 0.0015 |
| NH3···ClH (Г) | 0.0041 | 0.0166 | 0.010 | −0.0022 | 0.0010 |
| H3N···ClF (A) | 0.0618 | 0.1494 | <0.001 | −0.0637 | −0.0144 |
| NH3···ClF (Б) | 0.0166 | 0.0684 | <0.001 | −0.0136 | 0.0013 |
| H3C−···ClH (В) | 0.0247 | 0.0552 | <0.001 | −0.0144 | −0.0011 |
| CH3−···ClH (Г) | 0.0091 | 0.0270 | <0.001 | −0.0045 | 0.0009 |
Рис. 5.
Корреляция между величиной электронной плотности ρ (а) и плотности потенциальной энергии VBCP (б) в критической точке межмолекулярного контакта и энергией связи в комплексах аммиака и метанида с акцептором электронной пары.

Потенциальные кривые межмолекулярного взаимодействия. Понятие о поверхности потенциальной энергии (ППЭ) молекулярной системы является основой современных представлений о свойствах отдельных молекул и молекулярных комплексов, которые зависят от геометрических характеристик системы. Являясь функцией полной энергии молекулярной системы (в которую не входит кинетическая энергия ядер) от координат атомных ядер, в общем случае ППЭ является гиперповерхностью в многомерном пространстве. Однако для определения механизма химических взаимодействий достаточно иметь информацию об областях минимумов функции ППЭ и о критических точках первого порядка, характеризующих переходное состояние реакции.
Анализ ППЭ для молекулярной системы, образованной молекулами NH3 и HCl, предоставляет данные об относительной стабильности комплексов с различной ориентацией мономеров и барьере для реакции их взаимопревращения. На рис. 6 показан профиль ППЭ для переходов комплексов с Н-связью N···H–Cl в комплексы N···Cl–H при отклонении мостика NHCl от линейности (см. рис. 6а). На рис. 6б видно, что потенциальные кривые для переходов между конфигурациями А → В и Б → Г имеют сходную форму: А- и Б-комплексы стабильнее комплексов типа В и Г и максимум на обеих кривых отвечает улу α (∠NClH) примерно равному 90°, при наибольшем удалении атома Н мостика от прямой N···Cl.
Рис. 6.
Профиль поверхности потенциальной энергии рассчитан как функция угла α, который описывает переход конфигураций N···H–Cl → N···Cl–H для комплексов, образованных молекулами NH3 и HCl (как показано на схеме а, профиль рассчитан при фиксированном положении атома Cl на оси симметрии C3v). Потенциальные кривые соответствующих переходов А → В и Б → Г (схема б, полная энергия равновесных комплексов с Н-связью N···H–Cl принята за ноль; стрелка показывает граничное значение угла α для Н-связи) и структура переходного состояния для взаимопревращения Б- и Г-комплексов (схема в, межатомные расстояния указаны в Ангстремах).

Однако, величина барьера для второго перехода заметно меньше, чем для первого, 2 ккал/моль и 13 ккал/моль, соответственно; поэтому переход Б-комплекс → Г-комплекс и обратный переход должны происходить сравнительно легко. Как было сказано ранее, Б- и Г-комплексы отвечают минимумам на ППЭ, структура переходного состояния для реакции их взаимопревращения представлена на рис. 6в. Ее сравнение со структурой Б-комплекса показывает, что межатомные расстояния N···Cl и N···H в переходном состоянии реакции заметно увеличены, а отклонение мостика NHCl от линейности весьма значительно, угол α = 51.9°.
Представляет интерес вопрос, при каких значениях угла α молекулярные комплексы, образованные молекулами NH3 и HCl, могут рассматриваться как комплексы с водородной связью, а при каких углах α как ван-дер-ваальсовы комплексы? В работе [37] в качестве критерия, который позволяет идентифицировать природу межмолекулярного взаимодействия при заметных отклонениях мостика Y···H–X от линейности, было предложено использовать топологию (положение и природу) критической точки межмолекулярного контакта. Выполненные нами расчеты показали, что с увеличением угла α происходит трансформация топологии электронной плотности, характерной для Н-связи N···H–Cl, в топологию N···Cl.
На рис. 7а показана эволюция межмолекулярного связевого пути при переходе Б-комплекса NH3···HCl в Г-комплекс NH3···ClH. Для углов α < < 50° анализ топологии электронной плотности показывает существование КТ для контакта N···H, типичный признак водородного связывания N···H–Cl в комплексе. При углах α > 50° КТ контакта N···H исчезает и появляется КТ контакта N···Cl, что указывает на ван-дер-ваальсову связь; при этом значения ∠NClH ∼ 50° близки к значению этого угла в переходном состоянии реакции взаимопревращения комплексов. Отметим, что при переходе А-комплекса в В-комплекс замена межмолекулярного контакта N···H на контакт N···Cl происходит практически при том же значении угла α (α = 49°, см. рис. 6б). Углам α = 49° и 50° при трансформации А- и Б-конфигураций отвечают расчетные значения ∠NHCl равные 106.5° и 109.7°, соответственно. Эти значения хорошо согласуются с предельными углами (∼110°) для мостика Y···H–X при водородном связывании, которые известны из литературы [38].
Рис. 7.
Эволюция межмолекулярного связевого пути (схема а) и распределения электронной плотности (схема б, изолиния очерчивает изменение в плотности более 0.0003 э.з./а.е.3) при переходе N···H–Cl (Б-комплекс) → N···Cl–H (Г-комплекс) с увеличением угла α (см. рис. 6). При α = 50° межмолекулярный контакт N···H в комплексе “заменяется” контактом N···Cl.

Рисунок 7а показывает, что линия максимальной электронной плотности с КТ, соединяющая взаимодействующие молекулы, при переходе Б-комплекс → Г-комплекс меняет свое положение очень плавно. Видно, что при α = 49° связевый путь N···H заметно отклоняется к атому Cl, а при α = 51° связевый путь N···Cl, наоборот, сближается с атомом Н; при этом траектория от атома азота до КТ связи и положение самой КТ в обоих случаях практически одинаковы. При углах α ∼ 50° КТ находится примерно в “центре” треугольника HNCl на равном расстоянии от атомов H и Cl, что может свидетельствовать о близких по величине вкладах в связывание ван-дер-ваальсового взаимодействия N···Cl и водородного связывания N···H–Cl.
Это подтверждают также данные табл. 4, в которой приведены значения параметров КТ связи и энергии возмущения E(2) для углов α от 45° до 55°. Как видно из таблицы, с увеличением α значения $E_{{nN \to \sigma {\text{*HCl}}}}^{{(2)}}$ уменьшаются, тогда как сумма вкладов $E_{{nN \to {\text{Ry*Cl}}}}^{{(2)}}$ и $E_{{n{\text{Cl}} \to {\text{Ry*N}}}}^{{(2)}}$ растет (для Б-конфигурации соответствующие значения малы). Данные таблицы показывают, что с увеличением α происходит последовательное уменьшение электронной плотности ρ и плотности потенциальной энергии VBCP в КТ. Об ослаблении межмолекулярного взаимодействия говорит также уменьшение показателя ELF, который характеризует степень локализации электронов.
Таблица 4.
Изменение свойств критической точки связи при углах α, отвечающих трансформации А- и Б-конфигураций, с переходом от водородной связи к ван-дер-ваальсовой связи
| Угол α | Параметры критической точки межмолекулярного контакта | E(2) | ||||||
|---|---|---|---|---|---|---|---|---|
| ρ | ${{{\nabla }^{{\text{2}}}}}$ρ | VBCP | HBCP | ELF | ESP | nN → σ*HCl | σHCl → Ry*N | |
| H3N···HCl (A) | ||||||||
| 45 | 0.0171 | 0.0584 | –0.0108 | 0.0019 | 0.0614 | 0.0158 | 1.98 | 1.00 0.26 |
| 47 | 0.0162 | 0.0562 | –0.0103 | 0.0019 | 0.0561 | 0.0042 | 1.46 | 0.91 0.32 |
| 49 | 0.0154 | 0.0545 | –0.0099 | 0.0019 | 0.0515 | –0.0071 | 1.06 | 0.83 0.36 |
| 51 | 0.0148 | 0.0533 | –0.0096 | 0.0019 | 0.0476 | –0.0177 | 0.89 | 0.80 0.40 |
| 53 | 0.0143 | 0.0525 | –0.0093 | 0.0019 | 0.0445 | –0.0268 | 0.51 | 0.70 0.47 |
| 55 | 0.0140 | 0.0518 | –0.0091 | 0.0019 | 0.0425 | –0.0342 | 0.34 | 0.64 0.52 |
| NH3···HCl (Б) | ||||||||
| 45 | 0.0064 | 0.0194 | –0.0032 | 0.0008 | 0.0238 | 0.0432 | 0.24 | 0.08 <0.05 |
| 47 | 0.0061 | 0.0188 | –0.0030 | 0.0008 | 0.0223 | 0.0374 | 0.19 | 0.07 <0.05 |
| 49 | 0.0059 | 0.0183 | –0.0029 | 0.0008 | 0.0211 | 0.0314 | 0.14 | 0.07 <0.05 |
| 51 | 0.0057 | 0.0178 | –0.0027 | 0.0009 | 0.0201 | 0.0254 | 0.10 | <0.05 <0.05 |
| 53 | 0.0055 | 0.0174 | –0.0026 | 0.0009 | 0.0193 | 0.0195 | 0.07 | <0.05 <0.05 |
| 55 | 0.0054 | 0.0171 | –0.0025 | 0.0009 | 0.0188 | 0.0139 | 0.05 | <0.05 <0.05 |
Наглядную картину сдвига электронной плотности с увеличением α дает рис. 7б, на котором видно постепенное смещение области пониженной электронной плотности от атома H к атому Cl; при этом размер этой области и области избыточной электронной плотности, прилегающей к атому N, уменьшается при трансформации Б-конфигурации в Г-конфигурацию. При α ∼ 50° электронодефицитная область занимает среднее положение между атомами H и Cl, что согласуется с геометрией связевого пути на рис. 7а, который на значительном протяжении представлен линией, равноудаленной от атомов Н и Cl. Полученные данные можно рассматривать как свидетельство того, что при углах α близких к 50°, оба этих атома участвуют в связывании с азотом аммиака; замыкание связевого пути на атоме Cl или H определяет доминирование одного из каналов взаимодействия, N···Cl или N···H–Cl.
Полезную информацию о природе связывания в комплексах с различной ориентацией мономеров дает анализ поведения потенциальной функции межмолекулярного взаимодействия с изменением расстояния между молекулами. На рис. 8а приведены потенциальные кривые для четырех молекулярных комплексов: H3N···HF, H3N···HCl (A), NH3···ClH (Г) и H3N···ClF (А), в которых равновесные межмолекулярные расстояния и энергии связи заметно отличаются; два первых комплекса являются комплексами с классической Н-связью. Вопрос о том, как с расстоянием между мономерами изменяется энергия межмолекулярного взаимодействия в комплексах различной природы, исследовался в работе [9]. Энергии межмолекулярного взаимодействия при удалении молекул друг от друга были выражены в % от энергии связи в равновесных комплексах и при R ≥ 1 Å аппроксимированы функцией вида Rn–, где R – увеличение межмолекулярного расстояния по сравнению с равновесным расстоянием.
Рис. 8.
Потенциальные кривые взаимодействия мономеров при образовании аммиаком А-комплексов с молекулами HCl и ClF с водородной и галогенной связью, а также Г-комплекса NH3···ClH. Кривая для Н-связанного комплекса H3N···HF приведена для сравнения, R(N···Cl) – равновесное расстояние между мономерами (а). Масштабированные потенциальные кривые для тех же комплексов, R(N···Cl) показывает изменение расстояния между мономерами по сравнению с равновесным расстоянием, которому отвечает нулевое значение R(N···Cl) по оси Ox. Масштабирование выполнено относительно энергии связи в равновесном А-комплексе H3N···HCl (б).
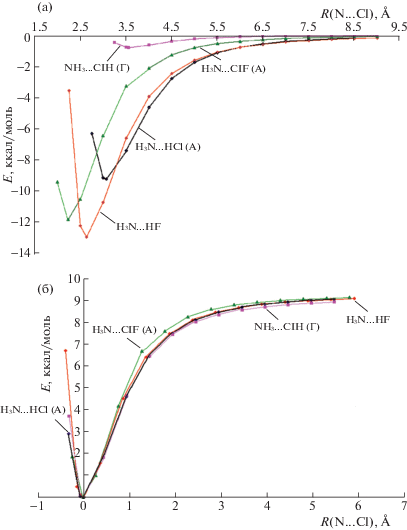
Та же задача сравнительного анализа может быть решена на качественном уровне с использованием более простого подхода масштабирования потенциальных кривых с учетом энергий связи в равновесных комплексах и последующим наложением кривых. Потенциальные кривые, построенные с поправкой на относительную стабильность молекулярных комплексов, представлены на рис. 8б. На рисунке видно, что полученные таким методом потенциальные кривые для комплексов H3N···HF, H3N···HCl (A) и NH3···ClH (Г) практически совпадают, т.е. межмолекулярное взаимодействие в этих комплексах описывается одной и той же потенциальной функцией. Это означает, что природа связывания в комплексе с классической Н-связью H3N···HCl (A) и в комплексе NH3···ClH (Г) с обратной ориентацией мономеров оказывается весьма сходной. Действительно, как видно на рис. 8а, при увеличении межмолекулярного расстояния N···Cl на 1 Å больше равновесного энергия связи в обоих комплексах уменьшается в 2 раза.
На рис. 8б также видно, что потенциальная кривая для комплекса H3N···ClF (А) с галогенной связью показывает более быстрый подъем при удалении мономеров друг от друга на расстояние, превышающее равновесное на 1–2 Å. Это может указывать на заметный вклад в связывание в этом комплексе донорно-акцепторного взаимодействия и дисперсионной энергии, которые убывают с расстоянием быстрее, чем электростатическое взаимодействие. Как показал анализ [39] компонент энергии взаимодействия, дисперсионная энергия вносит один из основных вкладов в стабилизацию комплексов с галогенной связью.
Таким образом, рассмотрены комплексы, образованные NH3 и HCl при различных вариантах взаимной ориентации молекул (неподеленной пары атома азота и ковалентной связи H–Cl): H3N···HCl (А), NH3···HCl (Б), H3N···ClH (В) и NH3···ClH (Г). С целью сравнения проведены также квантово-химические расчеты методом MP2/aug-cc-pVTZ В- и Г-комплексов метанида с HCl, а также комплексов H3N···ClF (А) и NH3···ClF (Б) с галогенной связью. Комплексы N···H–Cl с водородной связью найдены более стабильными по сравнению с комплексами типа N···Cl–H; NBO-анализ показывает доминирование взаимодействия nN → σ*HCl в комплексах первого типа и значительный вклад взаимодействия σHCl → → Ry*N в комплексах второго типа. Во всех рассмотренных комплексах отмечается переход электронного заряда на молекулу акцептора электронной пары, HCl или ClF, величина которого коррелирует с энергией возмущения E(2) и заселенностью вакантной σ*-орбитали акцептора.
А-комплекс H3N···ClF с галогенной связью и В-комплекс метанида H3С–···ClH показывают сходство в свойствах с А-комплексом H3N···HCl: большое значение электронной плотности ρ и отрицательные значения плотности потенциальной энергии VBCP в критической точке (КТ) межмолекулярного контакта. Сдвиг электронной плотности при образовании комплексов показывает во всех случаях принципиально сходную картину с увеличением электронной плотности на атомах N и C и ее уменьшением в области, прилегающей к атомам H и Cl акцептора.
Для комплексов, образованных NH3 и HCl, потенциальные кривые конфигурационных переходов А → В и Б → Г имеют сходную форму с максимумом при значении угла α (∠NClH) равном примерно 90°. Эволюция межмолекулярного связевого пути, сопровождающая трансформацию Б-комплекса NH3···HCl в Г-комплекс NH3···ClH, связана с “заменой” КТ контакта N···H на КТ контакта N···Cl при значениях α ∼ 50°, которые близки к значению этого угла в переходном состоянии реакции взаимопревращения комплексов. Геометрия связевого пути, а также сдвиги электронной плотности и расчетные значения энергий E(2) указывают на то, что при углах α близких к 50° оба атома молекулы HCl могут участвовать в связывании с азотом аммиака.
Сравнение потенциальных кривых взаимодействия мономеров позволяет сделать вывод о сходной природе межмолекулярных сил, определяющих связывание при сближении молекул NH3 и HCl с образованием А- и Г-комплексов.
Список литературы
Scheiner S. Hydrogen Bonding: A Theoretical Perspective. New York: Oxford University Press., 1997.
Isaev A.N. // Comput. Theor. Chem. 2017. V. 1117. P. 141.
Исаев А.Н. // Журн. физ. химии. 2018. Т. 92. С. 1588.
Isaev A.N. // Comput. Theor. Chem. 2018. V. 1142. P. 28.
Umeyama H. // J. Am. Chem. Soc. 1977. V. 99. P. 330.
Roeggen I., Dahl T. // J. Am. Chem. Soc. 1992. V. 114. P. 511.
Bloemink H.I., Evans C.M., Holloway J.H., Legon A.C. // Chem. Phys. Lett. 1996. V. 248. P. 260.
Karpfen A. // J. Phys. Chem. A. 2001. V. 105. P. 2064.
Nepal B., Scheiner S. // Chem. Phys. 2015. V. 456. P. 34.
Bartashevich E., Tsirelson V. // J. Comput. Chem. 2018. V. 39. P. 573.
Topp W.C., Allen L.C. // J. Am. Chem. Soc. 1974. V. 96. P. 5291.
Aldrich H.S., Cusachs L.C., Gary L.P. // Int. J. Quant. Chem., Quantum Biology Symposium. 1974. V. 1. P. 55.
Latajka Z., Scheiner S. // J. Chem. Phys. 1985. V. 82. P. 4131.
Panek J.J., Jezierska A. // J. Phys. Chem. A. 2007. V. 111. P. 650.
Scheiner S. // Int. J. Quant. Chem. 2013. V. 113. P. 1609.
Andrews L., Wang X., Mielke Z. // J. Phys. Chem. A 2001. V. 105. P. 6054.
Ma F., Li Z.-R., Xu H.-L., Li Z.-J. et al. // Chemphyschem: a European Journal of Chemical Physics and Physical Chemistry. 2009. V. 10. P. 1112.
Scheiner S. // J. Chem. Phys. 2011. V. 134. P. 164313/1.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E. et al. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009.
Quiñonero D., Estarellas C., Frontera A., Deyà P.M. // Chem. Phys. Lett. 2011. V. 508. P. 144.
Moller C., Plesset M.S. // Phys. Rev. 1934. V. 46. P. 618.
Purvis III G.D., Bartlett R.J. // J. Chem. Phys. 1982. V. 76. P. 1910.
Scuseria G.E., Schaefer III H.F. // J. Chem. Phys. 1989. V. 90. P. 3700.
Kendall R.A., Dunning T.H. Jr., Harrison R.J. // J. Chem. Phys. 1992. V. 96. P. 6796.
Boys S.F., Bernardi F. // Mol. Phys. 1970. V. 19. P. 553.
Reed A.E., Weinhold F., Curtiss L.A., Pochatko D.J. // J. Chem. Phys. 1986. V. 84. P. 5687.
Reed A.E., Curtiss L.A., Weinhold F. // Chem. Rev. 1988. V. 88. P. 899.
Bader R.F.W. // Chem. Rev. 1991. V. 91. P. 893.
Bader R.F.W. Atoms in Molecules, a Quantum Theory. Oxford: Clarendon Press. 1993.
Lu T., Chen F. // J. Comp. Chem. 2012. V. 33. P. 580.
Hydrogen Bonding – New Insights. Ed.: Grabowski S.J. New York: Springer, 2006.
Del Bene J.E., Alkorta I., Elguero J. // Chem. Phys. Lett. 2017. V. 675. P. 46.
Reed A.E., Weinstock R.B., Weinhold F.A. // J. Chem. Phys. 1985. V. 83. P. 735.
Breneman C.M., Wiberg K.B. // J. Comput. Chem. 1990. V. 11. P. 361.
Isaev A.N. // Comput. Theor. Chem. 2016. V. 1090. P. 180.
Cremer D., Kraka E. // Angew. Chem., Int. Ed. Engl. 1984. V. 23. P. 627.
Novoa J.J., Lafuente P., Mota F. // Chem. Phys. Lett. 1998. V. 290. P. 519.
Steiner T. // Angew. Chem. Int. Ed. 2002. V. 41. P. 48.
Riley K.E., Hobza P. // Phys. Chem. Chem. Phys. 2013. V. 15. P. 17742.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии