

Журнал физической химии, 2020, T. 94, № 7, стр. 1038-1053
Квантово-химическое исследование ионно-парных состояний молекул галогенов
С. В. Алексеева a, В. А. Алексеев b, c, *
a Санкт-Петербургский государственный лесотехнический университет имени С.М. Кирова
194021 Санкт-Петербург, Россия
b Университет ИТМО
197101 Санкт-Петербург, Россия
c Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Россия
* E-mail: vadim-alekseev@mail.ru
Поступила в редакцию 09.07.2019
После доработки 03.12.2019
Принята к публикации 10.12.2019
Аннотация
Ионно-парные состояния молекул IBr, ICl и BrCl, коррелирующие к пределам диссоциации X+(3P2,1,0, 1D2) + Y–(1S0), исследованы ССП (самосогласованного поля) методом полного активного пространства орбиталей с учетом динамических электронных корреляций и спин-орбитального взаимодействия. В согласии с экспериментом, расчетные значения равновесной энергии состояний X+Y–(3P2), коррелирующих к нижнему состоянию иона X+(3P2), группируются в интервале ΔTe ∼ 100 см–1, при этом ошибка их относительного расположения также порядка 100 см–1. Расхождение с экспериментом по межъядерному расстоянию для большинства состояний не превышает величины $R_{e}^{{\exp }}$ – $R_{e}^{{{\text{calc}}}}$ = 0.02 Å. С использованием результатов выполненных ранее неэмпирических расчетов для молекулы I2 проведен сравнительный анализ структуры ионно-парных состояний гомоядерных и гетероядерных молекул галогенов. Обсуждаются вопросы, касающиеся состояний X–Y+ с “инвертированной” локализацией заряда, связанных с X+Y–-состояниями двухэлектронным переходом, и потенциальной возможности создания структур с двумя стабильными зарядовыми состояниями X+-D-Y– и X–-D-Y+, где X, Y – центры сродства к электрону (атомы, молекулы, кластеры), D – диэлектрические прослойки, которые могут представлять интерес для микроэлектроники.
ВВЕДЕНИЕ
Двухатомные молекулы, содержащие атом с положительным сродством к электрону, имеют ионно-парные (ИП) состояния, коррелирующие к пределу диссоциации X+ + Y–. Как правило, энергия предела X+ + Y– значительно превышает энергию диссоциации нижних ридберговских состояний атомов и, как следствие этого, кулоновские потенциалы пересекаются с потенциалами многочисленных ридберговских состояний. В результате конфигурационного взаимодействия, кулоновский потенциал электронного состояния данной симметрии фрагментарно входит в состав нескольких электронных состояний той же симметрии, коррелирующих к разным пределам диссоциации. Во многих случаях кулоновский потенциал пересекается также с потенциалами отталкивательных валентных состояний, которые коррелируют с высоколежащими возбужденными валентными состояниями атомов.
Галогены относятся к немногочисленной группе молекул, которые имеют невозмущенные ИП-состояния. Благодаря большому сродству к электрону атома галогена, область пересечения ИП-состояний и нижних ридберговских состояний располагается ∼1 эВ ниже ионно-парного предела диссоциации. Для сравнения, типичное значение энергии диссоциации ИП-состояния галогена составляет ∼4 эВ. Существенно также, что атом галогена не имеет высоколежащих возбужденных валентных состояний и, как следствие этого, молекула галогена не имеет отталкивательных состояний, потенциалы которых могли бы пересекать потенциалы ИП-состояний вблизи их минимума.
В 1970–1980-е годы ИП-состояния галогенов были предметом многочисленных экспериментальных исследований, что во многом было обусловлено возможностью создания газофазного лазера на смесях галогеносодержащих молекул с инертными газами, генерирующего на переходе из нижнего ИП-состояния D' в валентное состояние A'. Генерация на переходе D' → A' была продемонстрирована для всех гомо- и гетероядерных молекул (см. [1, 2]). Однако эти лазеры не получили широкого распространения, так как их характеристики уступали лазерам на аналогичном переходе с переносом заряда в молекулах галогенидов инертных газов (переход RgX(B2Σ+ → → X2Σ+)). Исключением является лазер на переходе D' → A' молекулы F2. Благодаря рекордно короткой длине волны генерации (157 нм), этот лазер применяется в фотолитографическом процессе при производстве интегральных схем.
Гетероядерная молекула галогена XY имеет девять ИП-состояний, коррелирующих к четырем пределам X+(3PJ = 2,1,0, 1D2) + Y–(1S0):
Ионно-парные состояния галогенов изучены с разной степенью подробности. К настоящему времени известны спектроскопические параметры всех 18 ИП-состояний молекулы I2, коррелирующих к пределам I+(3PJ = 2,1,0, 1D2), а также большинства соответствующих состояний молекул Br2 и Cl2 (спектроскопические параметры и ссылки на литературу приводятся в [3]). Ионно-парные состояния F2 остаются почти неизученными вследствие технических сложностей, связанных с нахождением ионно-ковалентных переходов в области вакуумного ультрафиолета.
Среди гетероядерных галогенов наиболее подробно изучены молекулы ICl, IBr (см. разделы 2.2 и 2.3) и ClF ([4] и ссылки в этой работе). За исключением h 0–, известны спектроскопические параметры всех ИП-состояний этих молекул, коррелирующих к X+(3PJ = 2,1,0). Имеются также данные по колебательной структуре состояний f ' 0+ и g 1 молекулы ICl, коррелирующих к верхнему пределу I+(1D2). Остальные гетероатомные молекулы изучены хуже. Например, в случае молекулы BrF спектроскопические параметры известны только для состояния D' 2 [5].
Благодаря доминантной роли электростатического взаимодействия, ИП-состояния данной молекулы ИП имеют весьма похожие потенциальные кривые. При этом для ИП-состояний гетероядерной молекулы, коррелирующих к одному пределу диссоциации, характерно значительно меньшее различие равновесных электронных энергий Te по сравнению со случаем гомоядерной молекулы. В качестве примера на рис. 1a и рис. 1б сравниваются состояния, коррелирующие к нижнему ионно-парному пределу молекул I2 и IBr.
Рис. 1.
Потенциалы ионно-парных состояний (а): E $0_{g}^{ + }$, β 1g, D' 2g, D $0_{{\text{u}}}^{ + }$, γ 1u и (6) δ 2u молекулы I2, рассчитанные с использованием экспериментальных спектроскопических параметров (см. [3] и ссылки в этой работе). Потенциалы ионно-парных состояний (б): E 0+, β 1 и D' 2 молекулы IBr, рассчитанные с использованием экспериментальных спектроскопических параметров. Для сравнения показаны потенциальные кривые, полученные при усреднении энергий четных и нечетных состояний I2 (a) согласно формуле E(Ωgu) = (E(Ωg) + E(Ωu))/2: $0_{{{\text{gu}}}}^{ + }$, 1gu и 2gu. Потенциалы триплетных ls-состояний I2 (в) [6]. Асимптотическая энергия принята равной нулю. Потенциалы триплетных ls-состояний IBr (г). Асимптотическая энергия принята равной нулю.

Как видно из рис. 1б, ИП-состояния IBr группируются в узком энергетическом интервале ΔTe ∼ 100 см–1. Это характерно и для других гетероядерных галогенов, для ИП-состояний которых имеются экспериментальные данные, включая ICl (см. ниже) и IF ([7] и ссылки в этой работе) также группируются в пределах ΔTe ∼ 100 см–1; в случае молекулы СlF различие значений Te несколько больше: ΔTe ∼ 250 см–1 [4].
Для ИП-состояний йода характерно значительно большее различие значений Te (рис. 1a). Состояния D' 2g и δ 2u являются соответственно нижним и верхним ИП-состояниями в рассматриваемой группе. Это справедливо и для соответствующих состояний Br2 и Cl2, причем для всех гомоядерных молекул величина зазора Te(δ 2u) – Te (D' 2g) приблизительно одинакова и равна ≈1500 см–1.
В работах [6, 8] представлены результаты квантово-химического исследования ИП-состояний молекул I2 и Br2. Расчеты выполнены ССП-методом (метод самосогласованного поля) полного активного пространства орбиталей с учетом (методом теории возмущений) динамических электронных корреляций и спин-орбитального взаимодействия. Активное пространство было ограниченно валентными орбиталями атомов без включения возбужденных ридберговских состояний. Результаты [6, 8] для I2 и Br2 весьма хорошо согласуются с экспериментальными данными. В частности, расчет воспроизводит такое “тонкое” различие в структуре ИП-состояний I2 и Br2 как относительное расположение состояний E $0_{g}^{ + }$ и D $0_{{\text{u}}}^{ + }$, коррелирующих к нижнему пределу X+(3P2) (X = I, Br). Как видно из рис. 1а, состояние I2(E $0_{{\text{g}}}^{ + }$) располагается выше I2(D $0_{{\text{u}}}^{ + }$) (экспериментальное значение ΔTe = Te(E $0_{{\text{g}}}^{ + }$) – Te(D $0_{{\text{u}}}^{ + }$) = = 383 см–1), тогда как для соответствующих состояний Br2 и Сl2 справедливо обратное (ΔTe = = ‒150 и –667 см–1, соответственно (см. таблицу 1 в [3]). Отметим, что “неправильное” расположение E $0_{{\text{g}}}^{ + }$ и D $0_{{\text{u}}}^{ + }$ состояний молекулы йода обусловлено спин-орбитальным взаимодействием с вышележащей ионно-парной конфигурацией $^{1}\Sigma _{{\text{u}}}^{ + }$ (коррелирует к I+(1D2) + I– (1S0)).
Целью настоящей работы являлось выполнение аналогичных расчетов для ИП-состояний гетероядерных молекул IBr, ICl и BrCl. Квантово-химические исследования структуры этих молекул проводились ранее: IBr [9], ICl [10], и BrCl [11] (и ссылки в этих работах). Однако исследования касались прежде всего валентных состояний, поэтому расчеты проводились или без включения ИП-конфигураций или с использованием неполного набора ИП-конфигураций, необходимых для описания всех ИП-состояний, коррелирующих к пределам X+(3PJ=2,1,0, 1D2) + Y–(1S0).
В данной статье представлены результаты расчетов потенциалов ИП-состояний IBr, ICl и BrCl. В связи с тем, что различия в структуре ИП-состояний этих молекул не имеют качественного характера, в статье подробно обсуждаются только результаты для молекулы IBr. При этом для всех трех молекул проводится детальное сравнение расчетных спектроскопических параметров с имеющимися экспериментальными данными. На примере молекул IBr и I2 представлен сравнительный анализ конфигурационного состава ИП-состояний гомо- и гетероядерных молекул.
Ионно-парные состояния галогенов являются уникальным объектом для получения знаний о свойствах этого типа связи в молекулах, что объясняет актуальность их исследования. Фактически аналогичными невозмущенными ИП-состояниями обладают только эксимерные молекулы галогенидов инертных газов RgX. Однако группа ИП-состояний этих молекул RgX сравнительно малочисленна и состоит из трех состояний, коррелирующих к Rg+ (2P3/2,1/2)+ X–(1S0). В отличие от RgX, гетероядерные галогены имеют два типа пределов диссоциации X+ + Y– и X– + Y+. Экспериментальные сведения, касающиеся состояний X–Y+ с “инвертированной” локализацией заряда к настоящему времени отсутствуют. Исследования этих состояний могут представлять не только научный, но и прикладной интерес, включая, в частности, вопрос о возможности создания структур с двумя стабильными зарядовыми состояниями X+-D-Y– и X–-D-Y+, где X, Y – центры сродства к электрону (атомы, молекулы, кластеры), D – диэлектрические прослойки, которые могут найти применение в устройствах микроэлектроники.
МЕТОДИКА РАСЧЕТА
Методика расчета аналогична использованной ранее для I2, Br2 [6, 8] и молекул галогенидов инертных газов [12]. Расчеты выполнены с использованием пакета программ MOLCAS [13]. Энергии электронных состояний с учетом статической составляющей корреляционной энергии электронов рассчитываются ССП-методом полного активного пространства орбиталей CASSCF (complete active space self consistent field). Динамические электронные корреляции учитываются методом теории возмущений CASPT2 (complete active space with second order perturbation theory correction) [14]. Энергии и волновые функции, рассчитанные методом СASSCF/CASPT2, далее используются программой RASSI (restricted active space state interaction) [15] для расчета энергий с учетом спин-орбитального взаимодействия, а также расчета дипольных моментов и скоростей излучательных переходов.
Расчеты проводились в симметрии ${{C}_{{2{v}}}}$. Активное пространство включало шесть орбиталей для 10 активных электронов. Как показали результаты расчетов для I2, Br2 [6, 8] и галогенидов инертных газов [12], энергии ИП-состояний, полученные методом СASSCF/CASPT2/RASSI для активного пространства, включающего только валентные р-орбитали, весьма хорошо согласуются с экспериментом.
Представленные в работе результаты получены с использованием базисных наборов
I.ano-rcc.Roos.22s19p13d5f3g.10s9p8d5f3g
Br.ano-rcc.Roos.20s17p11d4f2g.9s8p6d4f2g
Cl.ano-rcc.Roos.17s12p5d4f2g.8s7p5d4f2g
(ano – atomic natural orbital; rcc – relativistic correlation consistent). Точность расчетов методом СASSCF/CASPT2/RASSI с базисами этого типа обсуждается в [16].
Для иллюстрации точности расчетов с выбранными базисами в табл. 1 представлены энергии ионов Br+, I+ и Cl+, а также сродство к электрону и энергия спин-возбужденного состояния 2P1/2 нейтральных атомов. Результаты получены для пяти (четырех – для ионов) активных электронов в активном пространстве, ограниченном валентными р-орбиталями.
Таблица 1.
Сродство к электрону, потенциал ионизации и энергии нижних состояний атомов и ионов галогенов (см–1)
| Параметр | I | Br | Cl | |||
|---|---|---|---|---|---|---|
| Расчет | Эксперимент [17] | Расчет | Эксперимент | Расчет | Эксперимент | |
| EA | 25 022 | 24 670 | 26 293 | 27 131 | 27 571 | 29 139 |
| IP | 83 957 | 84 295.1 | 94 085 | 95 284.8 | 103 691 | 104 591.01 |
| X(2P1/2 – 2P3/2) | 6863 | 7603.0 | 3460 | 3685.2 | 902 | 882.3515 |
| X+(3P1 – 3P2) | 6423 | 7086.9 | 2896 | 3136.4 | 652 | 696 |
| X+(3P0 – 3P2) | 6231 | 6447.9 | 3638 | 3837.5 | 941 | 996.47 |
| X+(1D2 – 3P2) | 13 231 | 13 727.2 | 12 018 | 12 089.1 | 11 488 | 11 653.58 |
| X+(1S0 – 3P2) | 28 837 | 29 501.3 | 28 495 | 27 867.1 | 29 078 | 27 878.02 |
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Валентные состояния молекулы IBr
На рис. 2 представлены потенциальные кривые валентных и ИП-состояний молекулы IBr рассчитанные без учета и с учетом спин-орбитального взаимодействия (ls- и ωω-состояния соответственно). Группа валентных состояний молекулы IBr включает в себя 23 состояния, коррелирующих к четырем пределам диссоциации I(2PJ) + Br (2PJ'), J, J' = 3/2, 1/2. Потенциалы валентных состояний показаны в нижней части рис. 2. Для иллюстрации точности расчетов валентных состояний, в табл. 2 представлены основные спектроскопические параметры основного электронного состояния X0+ и двух возбужденных валентных состояний A 1u и A' 2u. Для всех трех состояний различие расчетного и экспериментального значений энергии диссоциации составляет величину ≈500 cм–1. Результаты аналогичной точности были получены для энергии диссоциации валентных состояний молекул I2 [6] и Br2 [8]. Отметим хорошее согласие расчетных значений равновесного межъядерного расстояния. Для гомоядерных молекул согласие несколько хуже. Детальное квантовохимическое исследование валентных состояний IBr представлено в [9].
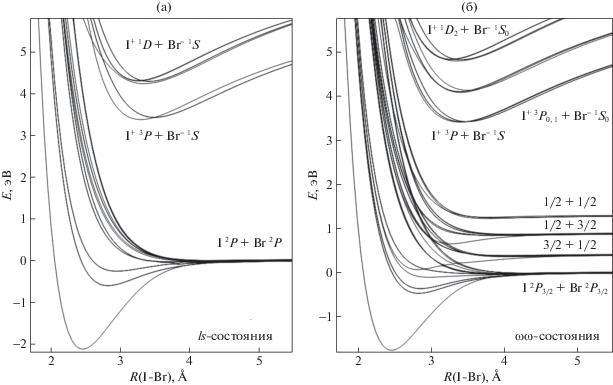
Таблица 2.
Спектроскопические параметры валентных состояний X 0+, A' 3Π2 и A 3Π1 молекулы IBr
| Состояние | Расчет/Экспериментa | ||
|---|---|---|---|
| Re, Å | De, см–1 | ωe, см–1 | |
| X0+ | 2.470/2.469 | 14 204/14 660 | 267.0/268.7 |
| A' 3Π2 | 2.839/2.842 | 3783/3357 | 148.5/147.6 |
| A 3Π1 | 2.865/2.858 | 2910/2424 | 136.6/135.2 |
a [9] и ссылки в этой работе
Ионно-парные состояния молекулы IBr
На рис. 3a представлены потенциалы ионно-парных ls- и ωω-состояний IBr. Спектроскопические параметры ls-состояний приведены в табл. 3. Для характеризации относительного расположения потенциалов состояний, коррелирующих к общему пределу диссоциации, в табл. 3 приводятся разности равновесных электронных энергий ΔTe и равновесных межъядерных расстояний ΔRe.
Рис. 3.
Расчетные потенциальные кривые ионно-парных состояний молекулы IBr (a) и ICl(б). ls-состояния показаны линиями с символами и ωω-состояния линиями без символов. Величины энергии в атомных единицах смещены на 715 и 572 а.u. для IBr и ICl соответственно. Энергия в единицах эВ отсчитывается от нижнего предела диссоциации (I 2P3/2 + Br 2P3/2 для IBr и I 2P3/2 + Сl 2P3/2 для ICl).
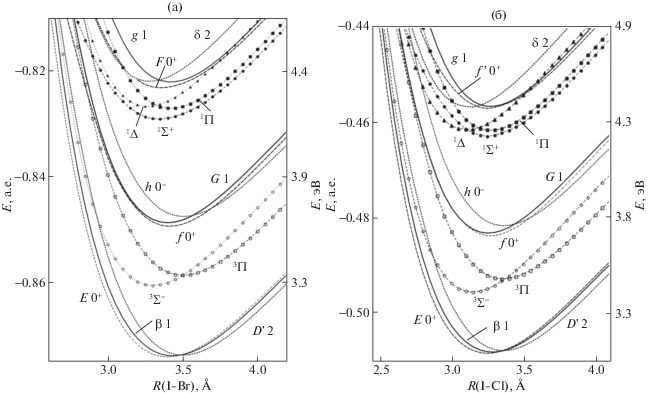
Таблица 3.
Расчетные спектроскопические параметры ионно-парных ls-состояний IBr, ICl и BrCl
| Параметры | Re, Å | ωe, см–1 | ωexe, см–1 | ΔRe, Å | ΔTe, см–1 | |
|---|---|---|---|---|---|---|
| IBr | ||||||
| 3P | 3Σ– | 3.304 | 135.0 | 0.356 | 3Π – 3Σ– = 0.198 1Π – 1Σ+ = 0.083 1Π – 1Δ = 0.155 |
3Π – 3Σ– = 394 1Π – 1Σ+ = 419 1Π – 1Δ = –73 |
| 3Π | 3.502 | 126.2 | 0.374 | |||
| 1D2 | 1Π | 3.423 | 130.6 | 0.319 | ||
| 1Σ+ | 3.340 | 115.4 | 0.204 | |||
| 1Δ | 3.268 | 135.7 | 0.348 | |||
| ICl | ||||||
| 3P | 3Σ– | 3.149 | 194.1 | 0.729 | 3Π – 3Σ– = 0.211 1Π – 1Σ+ = 0.030 1Π – 1Δ = 0.172 |
3Π – 3Σ– = 571 1Π – 1Σ+ = 250 Π – 1Δ = –102 |
| 3Π | 3.360 | 178.9 | 0.781 | |||
| 1D2 | 1Π | 3.273 | 187.0 | 0.660 | ||
| 1Σ+ | 3.243 | 192.9 | 0.650 | |||
| 1Δ | 3.101 | 193.8 | 0.667 | |||
| BrCl | ||||||
| 3P | 3Σ– | 3.013 | 210.1 | 0.796 | 3Π – 3Σ– = 0.162 1Π – 1Σ+ = 0.057 1Π – 1Δ = 0.119 |
3Π – 3Σ– = 214 1Π – 1Σ+ = 507 Π – 1Δ = –462 |
| 3Π | 3.175 | 199.5 | 0.715 | |||
| 1D2 | 1Π | 3.098 | 206.7 | 0.706 | ||
| 1Σ+ | 3.041 | 183.4 | 0.429 | |||
| 1Δ | 2.979 | 210.3 | 0.749 | |||
| I2u-состояния | ||||||
| 3P | 3Σ– | 3.75 | 3Π – $^{3}{{\Sigma }^{ - }}$ = 0.35 1Π – 1Σ+ = 0.45 1Π – 1Δ = 0.30 |
3Π – 3Σ– = 998 1Π – 1Σ+ = 4422 1Π – 1Δ = 916 |
||
| 3Π | 3.40 | |||||
| 1D2 | 1Π | 3.70 | ||||
| 1Σ+ | 3.25 | |||||
| 1Δ | 3.40 | |||||
| I2g-состояния | ||||||
| 3P | 3Σ– | 3.50 | 3Π – $^{3}{{\Sigma }^{ - }}$ = 0.00 1Π – 1Σ+ = 0.20 1Π – 1Δ = –0.05 |
3Π – 3Σ– = – 1575 1Π – 1Σ+ (иррегулярность) 1Π – 1Δ = –2011 |
||
| 3Π | 3.50 | |||||
| 1D2 | 1Π | 3.45 | ||||
| 1Σ+ | 3.65 | |||||
| 1Δ | 3.50 | |||||
Состояния 3Σ– и 3Π отвечают соответственно параллельной и перпендикулярной ориентациям дважды заполненной р-орбитали Br+ относительно молекулярной оси. Как следует из рис. 3 и данных в табл. 3, потенциал 3Π располагается несколько выше 3Σ– и имеет большее межъядерное расстояние. Такое расположение состояний 3Π и 3Σ– характерно и для других гетероядерных молекул галогенов, включая ClF [4], ICl и BrCl (см. табл. 3).
Вследствие спин-орбитального (СО) взаимодействия триплетные состояния E 0+, f 0+, β 1, G 1 являются смесью характеров 3Σ– и 3Π, состояния D' 2 и h 0– являются 3Π-состояниями. Триплетные ИП-состояния 3Σ– и 3Π также связаны СО взаимодействием с вышележащими синглетными ИП-состояниями. При этом матричный элемент синглет-триплетного СО-взаимодействия в 21/2 раза меньше триплет-триплетного СО-взаимодействия [18]. Вследствие большой величины матричного элемента СО взаимодействия в ионе йода, синглетные ωω-состояния в йодсодержащих галогенах заметно смещены вверх по энергии относительно соответствующих ls-cостояний (рис. 3). В свою очередь, триплетные ωω-состояния смещены вниз. Сдвиг составляет величину ∼1000 см–1, а приобретаемая в результате синглет-триплетного СО-взаимодействия примесь противоположной мультиплетности имеет вес ∼7%. Отметим, что это относится не только к состояниям E 0+, f 0+, β 1, и G 1, имеющим смешанный 3Σ– ∼ 3Π характер, но и к 3Π-состоянию D' 2, так как конфигурация 3Π связана СО взаимодействием с 1Δ. Таким образом, единственным “чистым” триплетным состоянием является h 0–. Вследствие запретов налагаемых правилами отбора для дипольных переходов, это состояние гетероядерных галогенов до сих пор не наблюдалось экспериментально (см. [19, 20] о методике исследования состояний $0_{{{\text{g/u}}}}^{ + }$ молекулы йода).
Расчетные и экспериментальные спектроскопические параметры ИП-состояний IBr сравниваются в табл. 4. Как можно видеть, величина ΔR = $R_{e}^{{{\text{exp}}}}$ – $R_{e}^{{{\text{calc}}}}$ не превышает +0.02 Å, различие расчетных и экспериментальных значений ωe не превышает 2%. Исключением является состояние G 1, для которого ΔR = –0.036 Å и ($\omega _{e}^{{{\text{exp}}}}$ – ‒ $\omega _{e}^{{{\text{calc}}}}$)/$\omega _{e}^{{{\text{exp}}}}$ ≈ 3%.
Таблица 4.
Расчетные и экспериментальные спектроскопические параметры ионно-парных состояний IBr, коррелирующих к пределам диссоциации I+(3P2,1,0, 1D2) + Br–(1S0)
| Te, см–1 (эксперимент) |
Расчет/Эксперимент | |||||
|---|---|---|---|---|---|---|
| Te – Te(E 0+) | Re, Å | ωe, см–1 | ωexe, см–1 | |||
| 3P2 | D' 2 | 39 456.6 [21] | 73/–31 | 3.502/3.4806 | 125.5/123.06 | 0.38/0.2813 |
| β 1 | 39 507.8 [22] | 47/20 | 3.435/3.4344 | 122.9/122.09 | 0.34/0.2546 | |
| E 0+ | 39 487.8 [23]* | 0/0 | 3.401/3.4067 | 120.8/119.43 | 0.20/0.2055 | |
| 3P1 | h 0– | 5828/ | 3.507/ | 125.9/ | 0.37/ | |
| G 1 | 45 996.0 [24] | 5568/6508 | 3.402/3.3636 | 132.4/128.5 | 0.46/0.3188 | |
| 3P0 | f 0+ | 45 382.6 [23] | 5424/5894 | 3.415/3.3937 | 131.1/128.8 | 0.42/0.363 |
| 1D2 | f ' 0+ | 11 169/ | 3.357/ | 118.6/ | 0.20/ | |
| g 1 | 11 414/ | 3.422/ | 130.8/ | 0.33/ | ||
| δ 2 | ∼11 490**/ | 3.280/ | 134.6/ | 0.34/ | ||
Равновесная электронная энергия Te ИП-состояния относительно минимума основного электронного состояния X0+ определяется из соотношения
Для характеристики точности расчета ИП-состояний относительно друг друга удобно сравнивать расчетные и экспериментальные энергии относительно одного из ионно-парных состояний. В качестве такого состояния мы выбрали состояние E 0+. Расчетные и экспериментальные значения энергетических зазоров Te(IP) – Te(E 0+) сравниваются в табл. 4.
Как следует из представленных данных, три нижних ИП-состояния располагаются в пределах ∼100 см–1, при этом состояние E 0+ является нижним по энергии. Согласно экспериментальным данным, нижним является состояние D' 2, однако расхождение экспериментальных и расчетных значений Te (D' 2) – Te(E 0+) составляет величину ∼100 см–1 (табл. 4). Близость энергий трех нижних ИП-состояний характерна и для IBr и BrCl (см. ниже).
Для ИП-состояний, коррелирующих к вышележащим пределам диссоциации, точность величины Te(IPS) – Te(E 0+) во многом определяется точностью расчета энергетических зазоров между состоянием I+(3P2) и соответствующими вышележащими состояниями иона. Сравнение показывает, что расхождение между расчетными и экспериментальными энергиями ИП-состояний коррелирует с величиной расхождения для соответствующих состояний иона I+. Так экспериментальное и расчетное значения относительной энергии состояния IBr (G 1) различаются на 940 см–1 (табл. 3). Для сравнения, экспериментальные и расчетные значения энергетического зазора между состояниями I+(3P1) и I+(3P2) различаются на 664 см–1 (табл. 1). В свою очередь, для состояния IBr(f 0+) соответствующие величины различаются на 470 см–1, а экспериментальные и расчетные значения энергетического зазора между состояниями I+(3P0) и I+(3P2) на 217 см–1.
Ионно-парные состояния молекулы ICl
Результаты расчетов ИП-состояний ICl представлены на рис. 3б. Как можно видеть из сравнения с рис. 3а, относительное расположение потенциалов триплетных ωω-состояний молекул ICl и IBr практически одинаково. Интересно, что при этом зазор между ls-состояниями 3Σ– и 3Π молекулы ICl приблизительно на 30% больше чем в молекуле IBr: 571 и 394 см–1 соответственно (табл. 3).
Расчетные и экспериментальные спектроскопические параметры ИП-состояний ICl сравниваются в табл. 5. Как можно видеть, для большинства состояний величина ΔR = $R_{e}^{{{\text{exp}}}}$ – $R_{e}^{{{\text{calc}}}}$ не превышает ±0.02 Å. Исключением является состояние G 1, для которого ΔR = –0.027 Å. Относительная точность расчета ωe составляет величину ($\omega _{e}^{{{\text{exp}}}}$ – $\omega _{e}^{{{\text{calc}}}}$)/$\omega _{e}^{{{\text{exp}}}}$ ∼ 3%. Здесь исключением является состояние f ' 0+, для которого ($\omega _{e}^{{{\text{exp}}}}$ – ‒ $\omega _{e}^{{{\text{calc}}}}$)/$\omega _{e}^{{{\text{exp}}}}$ ≈ 7%. Это состояние также характеризуется весьма малым значением ангармонизма колебаний, ωexe = 0.195 см–1, что почти в два с половиной раза меньше расчетного значения. Причина расхождений расчетных и экспериментальных значений спектроскопических параметров состояния f ' 0+ остается не ясной.
Таблица 5.
Расчетные и экспериментальные спектроскопические параметры ионно-парных состояний ICl, коррелирующих к пределам диссоциации I+ (3P2,1,0, 1D2) + Cl–(1S0)
| Te, см–1 (эксперимент) |
Расчет/Эксперимент | |||||
|---|---|---|---|---|---|---|
| Te – Te(E 0+) | Re, Å | ωe, см–1 | ωexe, см–1 | |||
| 3P2 | D' 2 | 39 061.83 [25] | 122/3 | 3.359/3.3502 | 177.1/173.63 | 0.770/0.5572 |
| β 1 | 39 103.6 [25] | 60/44 | 3.287/3.2930 | 170.6/170.31 | 0.462/0.4706 | |
| E 0+ | 39 059.48 [26]* | 0/0 | 3.251/3.2553 | 169.9/165.68 | 0.445/0.288 | |
| 3P1 | h 0– | 5857/ | 3.364/ | 178.4/ | 0.800/ | |
| G 1 | 45 552.8 [27] | 5515/6493 | 3.257/3.2294 | 185.8/184.85 | 0.631/0.6737 | |
| 3P0 | f 0+ | 44 924.44 [26] | 5398/5865 | 3.273/3.2582 | 190.2/184.16 | 1.118 /0.7439 |
| 1D2 | f ' 0+ | 51 199.96 [28] | 11232/12140 | 3.248/ | 172.0/160.72 | 0.483/0.195 |
| g 1 | 51 615.6 [29] | 11330/12556 | 3.273/ | 187.5/184.03 | 0.669/0.596 | |
| δ 2 | ∼11330**/ | 3.130/ | 192.3/ | 0.64/ | ||
Аналогично IBr, экспериментальные значения равновесных энергий Te трех нижних ИП-состояний ICl различаются не более чем на 100 см–1. В целом, расчеты воспроизводят этот результат. Однако для состояний ICl(g 1) и ICl(f ' 0+), также коррелирующих к общему пределу диссоциации, расхождение значительно: экспериментальное и расчетное значения величины Te(g 1) – Te(f ' 0+) равны 416 и 98 см–1 соответственно.
Как отмечалось выше, точность расчета относительной энергий ИП-состояний (относительно E 0+), коррелирующих к вышележащим пределам диссоциации, во многом определяется точностью расчета энергетических зазоров между состоянием I+(3P2) и вышележащими состояниями иона йода. Как следует из данных в табл. 4 и 5, экспериментальное значение Te(f 0+) – Te(E 0+) для IBr и ICl различаются не более чем на 50 см–1, что составляет менее 1% величины Te(f 0+) – Te(E 0+). Интересно, что столь же хорошо согласуются и расчетные значения Te(f 0+) – Te(E 0+) (см. табл. 4 и 5); это справедливо и для экспериментальных и расчетных значений Te(G 1) – Te(E 0+) для IBr и ICl. Отметим также, что расчетные значения относительных энергий состояний, коррелирующих к I+(1D2), также оказались весьма близки. Например Te(f ' 0+) – Te(E 0+) = 11169 и 11232 см–1 для IBr и ICl соответственно (см. табл. 4 и 5). Необходимые для сравнения экспериментальные данные отсутствуют.
Ионно-парные состояния молекулы BrCl
Результаты расчетов для молекулы BrCl представлены на рис. 4. Отличие от IBr и ICl (рис. 3a, 4б) носит количественный характер и обусловлено главным образом различием в величине СО-взаимодействия в ионах I+ и Br+. Матричный элемент СО-взаимодействия в ионе Br+ приблизительно в два раза меньше. Этим, в частности, объясняется сравнительно малое смещение ωω-состояний, коррелирующих к Br+(1D2), относительно ls-состояний, коррелирующих к этому пределу. В молекулах галогенов, состоящих из легких атомов (Cl2, F2, ClF), обсуждаемое смещение становится пренебрежимо малым, вследствие малой величины СО-взаимодействия (при этом зазор между ls-состояниями X+(1D) и X+(3P) приблизительно одинаков для всех галогенов).
Рис. 4.
Расчетные потенциальные кривые ионно-парных состояний молекулы BrCl. ls-состояния показаны линиями с символами и ωω-состояния линиями без символов. Величина энергии в атомных единицах смещена на 65 а.u. Энергия в единицах эВ отсчитывается от нижнего предела диссоциации Br 2P3/2 + Cl 2P3/2.

В литературе имеются неполные экспериментальные данные только для трех нижних ИП-состояний. Расчетные и экспериментальные спектроскопические параметры ИП-состояний BrCl сравниваются в табл. 6.
Таблица 6.
Расчетные и экспериментальные спектроскопические параметры ионно-парных состояний BrCl, коррелирующих к пределам диссоциации Br+(3P2,1,0, 1D2) + Cl–(1S0)
| Te, cм–1 | Расчет/Эксперимент | |||||
|---|---|---|---|---|---|---|
| Te – Te(E 0+) | Re, Å | ωe, см–1 | ωexe, см–1 | |||
| 3P2 | D' 2 | 17/ | 3.176/3.113 [30] | 199.4/197.88 | 0.729/0.71 | |
| β 1 | 48830 ± 3 [31] | 50/–24 | 3.103/3.082 | 186.3/ | 0.435 | |
| E 0+ | 48854.29 ± 0.05 [31]* | 0/0 | 3.075/3.042 | 191.1/196.4 | 0.514/1.7 | |
| 3P1 | h 0– | 2584/ | 3.175 | 199.3 | 0.710 | |
| G 1 | 2496/ | 3.101 | 216.7 | 1.029 | ||
| 3P0 | f 0+ | 3186/ | 3.119 | 211.4 | 0.943 | |
| 1D2 | f ' 0+ | 10 185/ | 3.044 | 184.7 | 0.401 | |
| g 1 | 10 670/ | 3.097 | 206.9 | 0.713 | ||
| δ 2** | ∼11 070/ | 2.981 | 210.6 | 0.768 | ||
Наряду с BrF, молекула BrCl остается одной из наименее изученных. Эффективным методом исследования ИП-состояний галогенов является метод двойного оптического резонанса с использованием валентного состояния A 1 в качестве промежуточного. Состояние A 1 может быть оптически заселено из основного состояния молекулы. В свою очередь, A 1 связано разрешенными переходами с ИП-состояниями D' 2, β 1 и E 0+, а также с вышележащими ИП-состояниями симметрии Ω = 0+, 1, 2. В частности, эта методика использовалась для исследования ИП-состояний ClF [32].
СРАВНЕНИЕ СТРУКТУРЫ ИОННО-ПАРНЫХ СОСТОЯНИЙ ГОМОЯДЕРНЫХ И ГЕТЕРОЯДЕРНЫХ МОЛЕКУЛ ГАЛОГЕНОВ
Как отмечалось во введении, существенным отличием структуры ИП-состояний гомоядерных молекул от гетероядерных является сравнительно большой разброс значений равновесной энергии Те (см. рис. 11a, 2 б). Расчеты достаточно точно воспроизводят указанное различие. Интересно, что это достигается в результате весьма тонкого баланса факторов, влияющих на относительное расположение состояний. Для иллюстрации на рис. 1в и рис. 1г сравниваются потенциальные кривые триплетных ls-состояний 3Π- и 3Σ−-молекул I2 и IBr. Как можно видеть, в смысле относительного расположения, потенциалы IBr весьма похожи на потенциалы нечетных ls-состояний I2. Величины Re(3Π) – Re(3Σ−) и Te(3Π) – Te(3Σ−) для состояний IBr и нечетных состояний I2 различаются на ≈0.15 Å и ≈600 см–1 соответственно (табл. 3). При этом для трех нижних нечетных ωω-состояний I2, которые “получаются” из ls-состояний $^{3}\Sigma _{{\text{u}}}^{ - }$ и 3Πu при диагонализации матрицы гамильтониана спин-орбитального взаимодействия, различие равновесных энергий составляет ∼800 см–1, тогда как для состояний E 0+, β 1 и D' 2 IBr указанное различие не превышает 100 см–1, при этом величина спин-орбитального взаимодействия в рассматриваемых молекулах одинакова.
Представляет интерес более подробно рассмотреть МО конфигурации ИП-состояний гомо- и гетероядерных молекул галогенов. На рис. 5a–5в представлены зависимости весовых коэффициентов МО-конфигураций от межъядерного расстояния для ионно-парных состояний молекулы IBr; дополнительно в табл. 7 приведены численные значения весовых коэффициентов для трех значений межъядерного расстояния $R_{e}^{{\Sigma \Pi }}$ и $R_{e}^{{\Sigma \Pi }}$ ± 1, где $R_{e}^{{\Sigma \Pi }}$ = 3.4 Å, что приблизительно соответствует среднему значению равновесных межъядерных расстояний состояний 3Σ– и 3Π, $R_{e}^{{\Sigma \Pi }}$ ≈ (Re(3Σ–) + Re(3Π))/2. Как следует из этих данных, при R = $R_{e}^{{\Sigma \Pi }}$, состояния 3Π и 1Π являются смесью конфигураций 2431 (∼80%) и 1432 (∼20%). В свою очередь для состояний 3Σ– и 1Δ доминантной конфигурацией вблизи $R_{e}^{{\Sigma \Pi }}$ является 2422 (>95%). Наконец, синглетное состояние 1Σ+ является смесью трех конфигураций 2440 (58%), 1441 (21%) и 2422 (21%).
Рис. 5.
МО-конфигурации σkπlπ*mσ*n ионно-парных ls-состояний молекулы IBr (a–в). МО-конфигурации $\sigma _{{\text{g}}}^{k}\pi _{{\text{u}}}^{l}\pi _{{\text{g}}}^{m}\sigma _{{\text{u}}}^{n}$ ионно-парных ls-состояний молекулы I2 (г–е). Линии без символов соответствуют g-состояниям, линии с символами − u-состояниям.
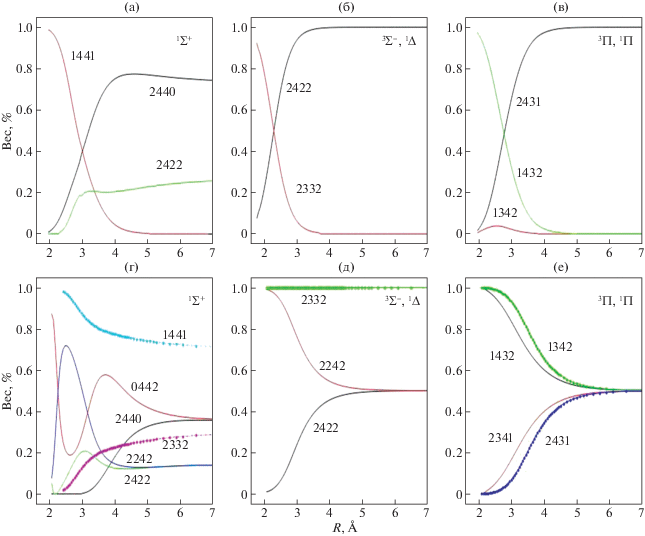
Таблица 7.
МО-конфигурации ИП-состояний молекул IBr и I2 и их весовые коэффициенты (%)
| Cостояние иона I+ | IBr | I2g-состояния | I2u-состояния | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| σkπlπ*mσ*n | вес. (%) | $\sigma _{{\text{g}}}^{k}\pi _{{\text{u}}}^{l}\pi _{{\text{g}}}^{m}\sigma _{{\text{u}}}^{n}$ | вес. (%) | $\sigma _{{\text{g}}}^{k}\pi _{{\text{u}}}^{l}\pi _{{\text{g}}}^{m}\sigma _{{\text{u}}}^{n}$ | вес. (%) | ||||||||
| 3P | 3Σ− | 2422 2332 |
46 54 |
97 3 |
100 0 |
2242 2422 |
95 5 |
63 37 |
52 48 |
2332 | 100 | 100 | 100 |
| 3Π | 2431 1432 2341 |
10 88 2 |
78 21 1 |
98 2 0 |
1432 2341 |
97 3 |
74 26 |
56 44 |
2431 1342 |
1 99 |
18 82 |
42 58 |
|
| 1D2 | 1Σ+ | 2440 2422 1441 2332 |
9 2 87 2 |
58 21 21 0 |
77 21 2 0 |
2440 2422 2242 0442 |
0 10 71 19 |
7 16 20 57 |
28 12 12 46 |
1441 2332 |
96 4 |
80 20 |
76 24 |
| 1Π | 2431 1432 2341 |
17 79 3 |
85 14 1 |
99 1 0 |
1432 2341 |
93 7 |
67 33 |
54 46 |
2431 1342 |
2 98 |
24 76 |
44 56 |
|
| 1Δ | 2422 2332 |
58 42 |
98 2 |
100 0 |
2422 2242 |
8 92 |
40 60 |
48 52 |
2332 | 100 | 100 | 100 | |
| R (Å)* | 2.4 | 3.4 | 4.4 | 2.6 | 3.6 | 4.6 | 2.6 | 3.6 | 4.6 | ||||
* Веса конфигураций приводятся для трех значений межъядерного расстояния $R_{e}^{{\Sigma \Pi }}$ – 1, $R_{e}^{{\Sigma \Pi }}$ и $R_{e}^{{\Sigma \Pi }}$ + 1, где $R_{e}^{{\Sigma \Pi }}$ ≈ (Re(3Σ–) + + Re(3Π))/2 для ИП состояний IBr и $R_{e}^{{\Sigma \Pi }}$ ≈ (Re(${}^{3}\Sigma _{{\text{g}}}^{ - }$) + Re(3Πg))/2 ≈ (Re(${}^{3}\Sigma _{{\text{u}}}^{ - }$) + Re(3Πu))/2 для ИП состояний I2.
Для сравнения на рис. 5 (см. также табл. 7) представлены зависимости весовых коэффициентов МО-конфигураций ИП-состояний I2. При больших межъядерных расстояниях, при которых можно пренебречь обменным взаимодействием, конфигурации $\sigma _{{\text{g}}}^{2}\sigma _{{\text{u}}}^{0}$ и $\sigma _{{\text{g}}}^{0}\sigma _{{\text{u}}}^{2}$ энергетически эквивалентны и имеют одинаковый вес; это также относится к парам $\sigma _{{\text{g}}}^{1}\sigma _{{\text{u}}}^{2}{\text{/}}\sigma _{{\text{g}}}^{2}\sigma _{{\text{u}}}^{1}$, $\pi _{{\text{u}}}^{4}\pi _{{\text{g}}}^{2}{\text{/}}\pi _{{\text{u}}}^{2}\pi _{{\text{g}}}^{4}$ и $\pi _{{\text{u}}}^{4}\pi _{{\text{g}}}^{3}{\text{/}}\pi _{{\text{u}}}^{3}\pi _{{\text{g}}}^{4}$. Например 3Πg является смесью 1432(50%) + 2341(50%) (рис. 5). Конфигурации 1441 и 2332 являются “уникальными”. Как следует из рис. 5г $^{1}\Sigma _{{\text{u}}}^{ + }$ является смесью конфигураций 1441 и 2332, причем в области больших межъядерных расстояний веса этих конфигураций составляют 76 и 24% соответственно. Состояния ${}^{3}\Sigma _{{\text{u}}}^{ - }$ и 1Δu имеют конфигурацию 2332 во всей области межъядерного расстояния.
В отличие от орбиталей σg и σu молекулы I2, орбитали σ и σ* молекулы IBr не являются энергетически эквивалентными при больших межъядерных расстояниях. Это также относится и к орбиталям π и π*. Разрыхляющие орбитали σ* и π* преимущественно локализованы на атоме I, тогда как связывающие орбитали σ и π локализованы на атоме Br. В соответствии с этим, ИП-состояния IBr при больших межъядерных расстояниях имеют МО конфигурации σ2π4π*mσ*n (m + n = = 4) с полностью заполненными связывающими σ- и π-орбиталями. Так состояниям 3Π и 1Π отвечает конфигурация π*3σ*1, состояниям 3Σ– и 1Δ конфигурация π*2σ*2, а 1Σ+ является смесью π*4σ*0 и π*2σ*2 (рис. 4).
Наряду с ИП-состояниями с полностью заполненными связывающими орбиталями, молекула IBr имеет также ИП-состояния с полностью заполненными (при больших межъядерных расстояниях) разрыхляющими σ*- и π*-орбиталями. Эти состояния имеют конфигурации σnπmπ*4σ*2 (m + n = 4) и коррелируют с распадом на I– + Br+. Энергетический зазор между асимптотами I+(3P2) + Br–(1S0) и I–(1S0) + Br+(3P2) можно оценить с использованием известных значений потенциалов ионизации и сродства к электрону атомов I и Br. Расчеты показывают, что величина зазора составляет 1.66 эВ. В силу стечения обстоятельств, эта величина близка к величине зазора между состояниями 1D2 и 3P2 иона I+ (1.702 эВ). Аналогичный расчет для BrCl и ICl дает 1.40 и 3.01 эВ соответственно.
Выполненные в ходе настоящей работы пробные расчеты показали, что X+Y–- и X–Y+-состояния располагаются приблизительно параллельно и имеют близкие равновесные межъядерные расстояния и энергии диссоциации. В частности, состояния X– (1S0) + Y+ (3P2) молекул IBr и BrCl оказались вблизи состояний X+(1D2) + Y–(1S0), так как зазор между этими асимптотами близок к зазору между X+ (3P2) и X+ (1D2).
Отметим, что пробные расчеты проводились без включения ридберговских конфигураций. Взаимодействие между ионно-парным и ридберговским состояниями связано с перемещением электрона между орбиталями ионно-парного и ридберговского состояний и, как следствие этого, пропорционально интегралу перекрывания орбиталей. Нижние ридберговские состояния коррелируют с возбужденными состояниями атома X* и, соответственно, при больших и средних (порядка Re) расстояниях ридберговские орбитали локализованы на атоме X. В свою очередь, в случае состояний X+Y– заряд локализован на атоме Y. Как следствие этого, взаимодействие конфигураций X*Y и X+Y– сравнительно слабое. Однако в случае X–Y+-состояний, перемещение электрона происходит между орбиталями локализованными на атоме X. Можно ожидать, что взаимодействие конфигураций X*Y и X–Y+ существенно сильнее. Для ответа на этот вопрос требуются более детальные расчеты с включением ридберговских конфигураций.
С точки зрения общих положений квантовой механики, расщепление между потенциальными кривыми состояний Ωg и Ωu, сходящимися к одному пределу диссоциации, является результатом проницаемости потенциального барьера, разделяющего энергетически эквивалентные конфигурации X+X– и X–X+. В отличие от туннельного перехода X+X ↔ XX+ между эквивалентными конфигурациями иона гомоядерной молекулы, переход X+X– ↔ X–X+ предполагают туннелирование пары электронов [33]. В рамках представлений о туннелировании, состояния противоположной четности с одинаковым значением Ω располагаются симметрично относительно пары гипотетических состояний X+X– и X–X+, в которых “разрешена” локализация электронов аналогично случаю X+Y– и X–Y+ состояний гетероядерных галогенов. Как можно видеть из рис. 1б, относительное расположение таких “усредненных” ионно-парных состояний йода замечательно похоже на расположение потенциалов состояний E 0+, β 1 и D' 2 молекулы IBr. Усреднение энергий соответствующих ИП-состояний молекулы Br2 также дает группу из трех близколежащих состояний (ΔTe < 100 см–1), относительное расположение которых весьма схоже с расположением состояний E 0+, β 1 и D' 2 молекулы BrCl (рис. 4).
В зависимости от МО-конфигурации туннельный переход X+X– ↔ X–X+ предполагает туннелирование двух π-электронов, одного π-электрона и одного σ-электрона, или двух σ-электронов. Переходам π−π отвечают конфигурации $\sigma _{{\text{g}}}^{2}\pi _{{\text{u}}}^{m}\pi _{{\text{g}}}^{n}\sigma _{{\text{u}}}^{2}$ (m + n = 6) состояний 3Σ− и 1Δ, переходам σ−π-конфигурации $\sigma _{{\text{g}}}^{{1(2)}}\pi _{{\text{u}}}^{m}\pi _{{\text{g}}}^{n}\sigma _{{\text{u}}}^{{2(1)}}$ (m + n = 7) состояний 1,3Π, и переходам σ−σ-конфигурации $\sigma _{{\text{g}}}^{m}\pi _{{\text{u}}}^{4}\pi _{{\text{g}}}^{4}\sigma _{{\text{u}}}^{n}$ (m + n = 2) состояний 1Σ+. Отметим, что электроны являются энергетически эквивалентными только в случае π−π-туннелирования в конфигурациях $\sigma _{{\text{g}}}^{2}\pi _{{\text{u}}}^{2}\pi _{{\text{g}}}^{4}\sigma _{{\text{u}}}^{2}$ и $\sigma _{{\text{g}}}^{2}\pi _{{\text{u}}}^{4}\pi _{{\text{g}}}^{2}\sigma _{{\text{u}}}^{2}$ и σ–σ-туннелирования в конфигурациях $\sigma _{{\text{g}}}^{2}\pi _{{\text{u}}}^{4}\pi _{{\text{g}}}^{4}\sigma _{{\text{u}}}^{0}$ и $\sigma _{{\text{g}}}^{0}\pi _{{\text{u}}}^{4}\pi _{{\text{g}}}^{4}\sigma _{{\text{u}}}^{2}$.
Как следует из рис. 1в, расщепление между состояниями 3Σ−, МО конфигурации которых отвечают случаю π−π-туннелирования, существенно меньше чем между состояниями 3Π, отвечающими случаю σ−π-туннелирования: |Te($^{3}\Sigma _{{\text{u}}}^{ - }$) – Te($^{3}\Sigma _{{\text{g}}}^{ - }$)| ≈ ≈ 0.1 эВ и |Te(3Πu) – Te(3Πg)| ≈ 0.22, соответственно. С этой точки зрения, тот факт, что среди ИП-состояний I2, коррелирующих к I+(3P2) (рис. 1a), расщепление между состояниями Ω = 2, объясняется тем, что эти состояния имеют “чистый” 3Π характер, тогда как Ω = 0+, 1 имеют смешанный 3Π ∼ $^{3}{{\Sigma }^{ - }}$ характер. Сравнение потенциалов других ИП состояний I2 показывает [6], что для синглетных и триплетных пар состояний с одинаковыми МО-конфигурациями величина расщепления приблизительно одинакова. Так 1Πu/g и 1Δ2g/u имеют такие же МО-конфигурации как соответственно 3Πu/g и $^{3}\Sigma _{{{\text{g/u}}}}^{ - }$ (см. рис. 5 и табл. 7), поэтому |Te(1Πu) – Te(1Πg)| ≈ |Te(3Πu) – Te(3Πg)| > Te(1Δu) – ‒ Te(1Δg) ≈ Te($^{3}\Sigma _{{\text{u}}}^{ - }$) – Te($^{3}\Sigma _{{\text{g}}}^{ - }$). Расщепление достигает максимальной величины для 1Σ+-состояний, Te($^{1}\Sigma _{{\text{g}}}^{ + }$) – Te($^{1}\Sigma _{{\text{u}}}^{ + }$) ≈ 0.7 эВ (см. рис. 1 в [6]). МО-конфигурации этих состояний отвечают случаю σ−σ-туннелирования.
В контексте обсуждаемого вопроса отметим, что механизм некоторых процессов двухэлектронной перезарядки А2+ + B → А + B2+ также предполагает коррелированное туннелирование двух электронов. В частности, это относится к процессам Rg2+ + Rg → Rg + Rg2+, которые сравнительно хорошо изучены экспериментально ([34] и ссылки в этой работе). Относительное расположение потенциалов димера ${\text{Rg}}_{2}^{{2 + }}$ исключает возможность двухэлектронной перезарядки как последовательности двух одноэлектронных процессов; одноэлектронная перезарядка Rg2+ + Rg → → Rg+ + Rg+ наблюдается при энергиях столкновения >10 эВ, при которых энергетически возможно пересечение потенциалов Rg2+ + Rg и Rg+ + Rg+. Насколько нам известно, единственным изученным процессом двухэлектронной перезарядки для анион-катионных столкновений является процесс H+ + H– → H– + H+ ([35] и ссылки в этой работе). Однако сечение этого процесса мало – основным каналом является взаимная нейтрализация ионов.
Представляет интерес вопрос об оптических переходах между ИП-состояниями. В случае гомоядерной молекулы ИП-состояния Ωg и Ωu, сходящиеся к одному пределу диссоциации, связаны разрешенным оптическим переходом. Вследствие малой величины энергетического зазора между Ωg и Ωu, рассматриваемым переходам отвечает далекая ИК-область и их скорость мала по сравнению со скоростью ионно-ковалентного перехода с ${{{\text{X}}}^{ + }}{\text{X}}_{{{\text{g/u}}}}^{ - }$ → XXu/g в видимой области спектра. С другой стороны, при лазерном возбуждении ИП-состояний большой дипольный момент является благоприятным фактором для возникновения эффекта усиленного спонтанного излучения (УСИ) , а также ряда других нелинейных оптических эффектов [36]. К настоящему времени эффект УСИ наблюдался на нескольких переходах между ИП-состояниями I2 ([37] и ссылки в этой работе) и Br2 [38]. Эффект УСИ наблюдался также на переходе EF(${}^{1}\Sigma _{{\text{g}}}^{ + }$) → B($^{1}\Sigma _{{\text{u}}}^{ + }$) молекулы H2 [39]. Следует, однако, отметить, что в отличие от состояния B($^{1}\Sigma _{{\text{u}}}^{ + }$), распределение двухэлектронной плотности в состоянии EF(${}^{1}\Sigma _{{\text{g}}}^{ + }$) не является характерным для связи ионно-парного типа [40].
Аналогично ионно-ковалентным переходам, переход X+Х– (Ωg ↔ Ωu) физически отвечает переносу заряда вдоль оси молекулы, причем величина дипольного момента линейно возрастает с ростом межъядерного расстояния [41]. Такое поведение функции дипольного момента является следствием наличия центра симметрии. Для иллюстрации на рис. 6а представлены потенциалы ИП-состояний $^{1}\Sigma _{{\text{u}}}^{ + }$ и ${}^{1}\Sigma _{{\text{g}}}^{ + }$, а также основного валентного состояния ${}^{1}\Sigma _{{\text{g}}}^{ + }$ молекулы H2, рассчитанные для активного пространства, ограниченного только 1s-орбиталями атома H; рис. 6б приводится функция дипольного момента перехода между ИП-состояниями $^{1}\Sigma _{{\text{u}}}^{ + }$ и ${}^{1}\Sigma _{{\text{g}}}^{ + }$. Аналогично ведет себя функция дипольного момента перехода между состояниями $^{2}\Sigma _{{\text{u}}}^{ + }$ и ${}^{1}\Sigma _{{\text{g}}}^{ + }$ иона H2+.
Рис. 6.
Потенциальные кривые (a) молекулы водорода (сплошные линии) и линейного трехатомного комплекса HeH2 при фиксированном расстоянии R(HeH) = 1.5 A (штриховые линии), рассчитанные для активного пространства электронов, состоящего только из 1s-орбиталей атомов H и He. Функции дипольных моментов (б) переходов между состояниями H2 и HeH2. Расчеты выполнены с помощью программы МOLCAS методом CASSCF/CASPT2.

Переход X+Х– (Ωg ↔ Ωu) имеет интересную особенность – в отличие от одноэлектронных переходов Ωg ↔ Ωu между состояниями иона гомоядерной молекулы, коррелирующими к общему пределу диссоциации, дипольный момент этого перехода обусловлен перемещением двух электронов вдоль оси молекулы. В случае гетероядерной молекулы переходу X+Х– (Ωg ↔ Ωu) отвечает переход Х+Y–(Ω) ↔ Х–Y+(Ω), двухэлектронный характер которого очевиден. Однако дипольный момент перехода Х+Y–(Ω) ↔ Х–Y+(Ω) уменьшается при больших межъядерных расстояниях, что объясняется уменьшением интеграла перекрывания орбиталей локализованных на разных атомах. В качестве иллюстрации на рис. 1 представлены результаты модельных расчетов для линейного комплекса HeH2. Эта система не имеет центра инверсии. Наличие атома He приводит к расщеплению асимптотически вырожденных термов ионно-парных состояний H2. Как можно видеть, ионно-парные состояния, коррелирующие к H– + (HHe)+ и H+ + (HHe)–, различаются по энергии при больших межъядерных расстояниях, а дипольный момент перехода, связывающий эти состояния, быстро уменьшается в области R > 2 Å. Отметим, что функция дипольного момента имеет иррегулярность в области R ∼ 4 Å, что совпадает с положением иррегулярности на потенциальных кривых ИП-состояний HeH2. Этот эффект вероятно связан с “подмешиванием” ридберговских конфигураций (в области энергий ∼10 эВ ИП-состояния H2 пересекаются c ридберговскими состояниями, коррелирующими к 2s- и 2p-состояниями атома водорода). В данной работе этот эффект подробно не анализировался и функция дипольного момента перехода между ИП-состояниями HeH2 в области R > 3.5 Å не приводится.
Насколько нам известно, к настоящему времени отсутствуют экспериментальные данные, касающиеся обнаружения ИП-состояний Х–Y+ с инверсной локализацией заряда. Резонансы X+Y– + $h{v}$ → → X–Y+ могут быть обнаружены методом ЛИФ по ослаблению люминесценции X+Y– → XY + $h{v}$ или появлению новых спектральных переходов (предполагается, что для заселения X+Y– используется методика двойного оптического резонанса). Как отмечалось выше, есть основания полагать, что состояния Х–Y+ подвержены сильному взаимодействию с ридберговскими состояниями. В матрицах инертных газов ридберговские и ионно-парные состояния молекул галогенов смещаются по энергии в противоположных направлениях, что связано с различным характером взаимодействия с матрицей (обменное отталкивание и сольватация диполя, соответственно) [17]. Смещение может достигать нескольких эВ. Благодаря этому эффекту возможна стабилизация предиссоциированных (в газовой фазе) состояний Х–Y+.
ЗАКЛЮЧЕНИЕ
В данной работе представлены результаты неэмпирических расчетов ИП-состояний молекул IBr, ICl и BrCl. Полученные результаты достаточно хорошо согласуются с имеющимися экспериментальными данными. В частности, расхождение по межъядерному расстоянию для большинства состояний не превышает величины $R_{e}^{{{\text{exp}}}}$ – $R_{e}^{{{\text{calc}}}}$ = 0.02 Å. В согласие с экспериментом, расчетные значения Te состояний первого яруса группируются в интервале ∼100 см–1 и, хотя последовательность расположения состояний отличается от экспериментальной (согласно расчетам нижним является состояние E 0+, согласно эксперименту – D' 2), ошибка расчета их относительного расположения не превышает 100 см–1. Отсутствие экспериментальных данных для вышележащих ИП-состояний, коррелирующих к X+ (1D2), не позволяет провести сравнение с результатами расчета.
Вероятно наибольший интерес для дальнейшего исследования представляет вопрос о ионно-парных состояниях X–Y+ с “инвертированным” расположением заряда. К настоящему времени отсутствуют какие-либо экспериментальные данные, касающиеся этих состояний. Насколько нам известно, систематических исследований этих состояний с использованием методов квантовой химии также не проводилось. Выполненные нами пробные расчеты показали, что потенциалы X–Y+-состояний располагаются параллельно потенциалам X+Y–-состояний и при этом заметное конфигурационное взаимодействие между этими группами состояний отсутствует. Однако есть основания полагать, что в сравнении с состояниями X+Y–, состояния X–Y+ значительно сильнее взаимодействуют с ридберговскими состояниями. Пробные расчеты проводились без включения ридберговских конфигураций. В связи с этим результаты выполненных расчетов для этой группы состояний в статье не приводятся.
В заключение отметим, что структуры типа X‑D-Y, где X, Y – центры сродства к электрону (атомы, молекулы, кластеры), D – прослойка из молекул инертной матрицы, имеющие два стабильных зарядовых состояния X+-D-Y– и X+-D-Y–, могут представлять технологический интерес для микроэлектроники, включая, в частности, для создания так называемых клеточных автоматов (cellular automata) – устройств с двумя устойчивыми расположениями дискретных зарядов, разделенных потенциальным барьером. В случае атомов галогенов в качестве центров сродства к электрону, такие структуры будут неустойчивы вследствие излучательного распада, но могут представлять интерес как модельная система для исследования свойств таких структур, так как излучение является удобным монитором. Структуры, в которых ИП-состояние является асимптотически нижним, потенциально могут быть созданы из кластеров металлов. Известно, что по мере увеличения размера кластера, сродство к электрону и потенциал ионизации приближаются к величине работы выхода электрона из металла (W): EA(Mn) = W + аe2/(rsn1/3) и IP(Mn) = W – ‒ be2/(rsn1/3), где rs– радиус Вигнера–Зейтса, n – число атомов в кластере; параметры a и b приблизительно одинаковы для всех металлов и равны a = 3/8 и b = 5/8 [42]. Для модельной структуры, состоящей из двух кластеров разных металлов M1n1 и M2n2, разделенных диэлектрическим промежутком, энергетический зазор между ковалентной и ИП асимптотами равен IP(M1n1) – ‒ EA(M2n2). Например, согласно экспериментальным данным [42], потенциал ионизации кластера атомов калия (WK = 2.22 эВ) при nK ∼ 60 и сродство к электрону кластера меди при nCu ∼ 40 приблизительно одинаковы и равны ∼2.8 эВ. В соответствии с этим, состояния K60 + Cu40 и (K60)+ + (Cu40)– имеют одинаковые асимптотические энергии. По мере увеличения размеров кластеров, энергетический зазор между ионно-парными состояниями приближается к разности работ выхода электронов из металлов. Например, WNa – WK = 0.13 эВ и, соответственно, переход между ИП-состояниями кластерной пары Nan1 + Kn2 при n1, n2 > 100 лежит в ИК-области спектра.
Расчеты выполнены в Ресурсном центре “Вычислительный Центр СПбГУ”.
В недавно опубликованной работе [43] состояние f '0+ молекулы ICl исследовалось методом двойного оптического резонанса. Анализ вращательной структуры позволил определить со спектроскопической точностью равновесное межъядерное состояние в этом состоянии. Согдасно [43], Te = 51200.42 см−1, Re = 3.2262 Å, ωe = = 160.599 см−1, ωexe = 0.1760 см−1 (см. табл. 5 для сравнения с результатами неэмперических расчетов).Мы благодарим Dr. Shoma Hoshino за репринт работы [43].
Список литературы
McCusker M.V. // Excimer Lasers, Topics in Applied Physics edited by Ch. K. Rhodes: Springer, Berlin 1979. V. 30. P. 67.
Diegelmann M., Hohla K., Rebentrost F., Kompa K.L. // J. Chem. Phys. 1982. V. 76. P. 1233.
Алексеев В.А. // Опт. и спектр. 2005. Т. 99. № 5. С. 719.
Alekseyev A.B., Liebermann H.-P., Buenker R.J., Kokh D.B. // J. Chem. Phys. 2000. V. 112. P. 2274.
Narayani R.I., Tellinghuisen J. // J. Molec. Spectrosc. 1990. V. 141. P. 79.
Алексеев В.А. // Опт. и спектр. 2014. Т. 116. № 3. С. 355.
Hoy A.R., Jordan K.J., Lipson R.H. // J. Phys. Chem. 1991. V. 95. P. 611.
Овчинникова Н.Е., Алексеев В.А. // Опт. и спектр. 2016. Т. 120. № 2. С. 200.
Patchkovskii S. // Phys. Chem. Chem. Phys. 2006. V. 8. P. 926.
Kalemos A., Prosmiti R. // J. Chem. Phys. 2014. V. 141. P. 104312.
Wang M., Wang B., Chen Z. // J. Molec. Struct.: THEOCHEM. 2007. V. 806. P. 187.
Alekseev V.A. // J. Phys. B: At. Mol. Opt. Phys. 2014. V. 47. P. 105101.
Aquilante F., DeVico L., Ferré N. et al. // J. Comput. Chem. 2010. V. 31. P. 224.
Finley J., Malmqvist P.-Å., Roos B.O., Serrano-Andrés L. // Chem. Phys. Lett. 1998. V. 288. P. 299.
Malmqvist P.-Å., Roos B.O., Schimmelpfennig B. // Ibid. 2002. V. 357. P. 230.
Roos B.O., Lindh R., Malmqvist P.-Å. et al. // J. Phys. Chem. A. 2004. V. 108. P. 2851.
Linstrom P.J., Mallard W.G. Eds. NIST Chemistry WebBook, NIST Standard Reference Database Number 69, National Institute of Standards and Technology, Gaithersburg MD, 20899.
Lefebvre-Brion H., Field R.W. Perturbations in the Spectra of Diatomic Molecules N.Y.: Academic Press, 1986.
Motohiro S., Nakajima S., Ishiwata T. // J. Chem. Phys. 2002. V. 117. P. 187.
Motohiro S., Nakajima Sh., Aoyama K. et al. // Ibid. 2002. V. 117. P. 9777.
Radzykewycz D.T., Littlejohn C.D., Carter M.B. et al. // J. Molec. Spectrosc. 1994. V. 166. P. 287.
Clevenger J.O., Ray Q.P., Tellinghuisen J., Zheng X., Heaven M.C. // Can. J. Phys. 1994. V. 72. P. 1294.
Brand J.C.D., Hoy A.R., Risbud A.C. // J. Mol. Spectrosc. 1985. V. 113. P. 47.
Brand J.C.D., Dhatt D.R., Hoy A.R., Tse D.C.P. // J. Molec. Spectrosc. 1986. V. 119. P. 398.
Bussieres D., Hoy A.R. // Can. J. Phys. 1984. V. 62. P. 1941.
Brand J.C.D., Hoy A.R., Jaywant S.M. // J. Mol. Spectrosc. 1984. V. 106. P. 388.
Hudson J.B., Sauls L.J., Tellinghuisen P.C., Tellinghuisen J. // J. Mol. Spectrosc. 1991. V. 148. P. 50.
Donovan R.J., Lawley K.P., Ridley T., Wilson P.J. // Chem. Phys. Lett., 1993. V. 207. P. 129.
Donovan R.J., Ridley T., Lawley K.P., Wilson P.J. // Ibid. 1993. V. 205. P. 129.
Chakraborty D.K., Tellinghuisen P.C., Tellinghuisen J. // Chem. Phys. Lett. 1987. V. 141. P. 36.
Brown S.W., Dowd C.J., Tellinghuisen J. // Molec. Spectrosc. 1988. V. 132. P. 178.
Alekseev V.A., Setser D.W., Tellinghuisen J. // J. Molec. Spectrosc. 1999. V. 195. P. 162.
Brand J.C.D., Hoy A.R. // Appl. Spectrosc. Rev. 1987. V. 23. P. 285.
Hadjar O., Ascenzi D., Bassi D. et al. // Chem. Phys. Lett. 2004. V. 400. P. 476.
Bräuning H., Helm H., Briggs J.S., Salzborn E. // Ibid. 2007. V. 99. P. 173202.
Алексеев В.А. // Опт. спектроск. 2002. Т. 93. № 3. С. 366.
Hoshino S., Araki M., Nakano Y. et al. // J. Chem. Phys. 2016. V. 144. P. 034302.
Hoshino S., Araki M., Ishiwata T., Tsukiyama K. // Phys. Chem. Chem. Phys. 2016. V. 18. P. 19464.
Luk T.S., Egger H., Muller W., Pummer H., Rhodes C.K. // J. Chem. Phys. 1985. V. 82. P. 4479.
Wang J., Kim K.S., Baerends E.J. // Ibid. 2011. V. 135. P. 074111.
Lawley K.P. // Chem. Phys. 1988. V. 127. P. 363.
De Heer W.A. // Rev. Mod. Phys. 1993. V. 65. P. 611.
Hoshino S., Muto Y., Nishimichi D. et al. // J. Molec. Struc. 2020. V. 1209. P. 127913.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии