

Журнал физической химии, 2021, T. 95, № 2, стр. 207-212
Кислотно-основное равновесие нового замещенного 1,2,3-триазольным фрагментом производного 6-метилурацила в водных растворах
А. А. Ахияров a, Л. М. Губайдуллина b, Л. Ф. Сайфина b, В. Э. Семенов b, Л. А. Рамазанова c, А. Н. Лобов a, И. С. Файзрахманов c, И. Е. Алехина c, С. П. Иванов a, *
a Российская академия наук, Уфимский федеральный исследовательский центр,
Уфимский институт химии
Уфа, Россия
b Российская академия наук, Казанский научный центр,
Институт органической и физической химии им. А.Е. Арбузова
г. Казань, Россия
c Башкирский государственный университет
г. Уфа, Россия
* E-mail: ivanov_sp@anrb.ru
Поступила в редакцию 09.04.2020
После доработки 09.04.2020
Принята к публикации 26.05.2020
Аннотация
Методом потенциометрического титрования определены константы и термодинамические характеристики кислотно-основного равновесия синтезированного впервые производного 6-метилурацила, несущего при С(5) урацилового кольца 1,2,3-триазольный цикл – 5-(1-пентил-4-метил-1,2,3-триазол-4-ил)-6-метилурацила (1) и его модельного соединения – 5,6-диметилурацила в водных растворах. Показано, что 1,2,3-триазольный цикл существенно не влияет на кислотно-основные свойства урацильного фрагмента в составе соединения 1. Предположены места депротонирования соединения 1 в водных щелочных растворах и растворах в ДМСО. Структура соединения 1 доказана методами ЯМР- и ИК-спектроскопии, элементного анализа.
Известно, что в щелочных водных растворах производные урацила выступают как слабые двухосновные кислоты [1]. В зависимости от природы заместителей в положениях 5 и 6 пиримидинового кольца, диссоциация может происходить как от азота N(1), так и N(3) [2].
Диссоциацию по первой ступени можно представить следующими равновесиями:
если отрывается протон от N(1) или при диссоциации по N(3) положению.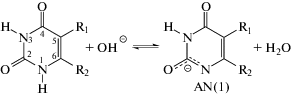
Термодинамической характеристикой равновесий (1) и (2) является константа диссоциации pKa1. Для 5- и 6-замещенных производных урацила величины pKa1 в водных растворах при 298 K составляют от 5.3 у 5-нитроурацила [3] до 9.8 у тимина [4]. Наиболее распространенными экспериментальными методами определения pKa производных урацила в растворах являются спектрофотометрия (СФ) и потенциометрическое титрование (ПТ). В последние десятилетия также эффективно применяются квантово-химические методы расчета pKa [1, 2].
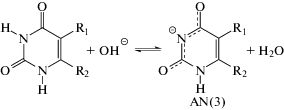
Часто в щелочных водных растворах диссоциация наблюдается одновременно как от азота N(1), так и N(3). Например, для 5-галогенурацилов методами ЯМР-спектроскопии было показано, что в щелочной водной среде одновременно существуют анионы с отрывом протона от N1 (AN(1)) и N3 (AN(3)) [5, 6]. Мольное соотношение AN(1) : AN(3) в водных щелочных растворах составляет 0.35 : 0.65 для 5-фторурацила [5] и 0.72 : 0.28 для 5-бромурацила [6]. При этом в щелочных диметилсульфоксидных растворах существует только форма AN(1) для всех исследованных 5-галогенурацилов [5, 6].
1,2,3-Триазольный цикл обладает высокой химической устойчивостью (инертность к окислению, восстановлению, гидролизу), ароматическим характером, высоким дипольным моментом, способностью выступать в качестве акцептора при образовании водородных связей [7]. В свою очередь урациловый цикл способен выступать и как донор, и как акцептор при образовании водородных связей, участвовать в π–π-контактах. Ковалентное связывание урацилового и 1,2,3-триазольного циклов в единую структуру, и в частности, в соединение 1 представляется перспективным в плане создания нового мотива, несущего нуклеотидное основание, и который может быть использован в создании новых супрамолекулярных систем, а также новых комплексообразователей. Механизм комплексообразования зависит, в большинстве случаев, от кислотно-основных свойств исходных лигандов в используемых растворителях.
В данной работе синтезирован 5-(1-пентил-4-метил-1,2,3-триазол-4-ил)-6-метилурацил (1) и изучено его кислотно-основное равновесие в воде методами УФ- и ЯМР-спектроскопии и потенциометрического титрования, исследовано влияние триазольного заместителя на рКа урацилового фрагмента с использованием модельных соединений – 6-MeU и 5,6-diMeU.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Эксперименты ЯМР (1H, 13С, δ, м.д., J, Гц) соединения (1) проводились на фурье-спектрометре AVANCE-500 (Bruker) с рабочей частотой 500.13 МГц (1Н) и 125.77 МГц (13С) при температуре 30°С, внешний стандарт тетраметилсилан. ИК-спектры соединений (νmax, см–1) записаны в таблетке в KBr на фурье-спектрометре Vector 22 (Bruker) при стандартных условиях в диапазоне 4000–400 см–1 при разрешении 4 см–1. Элементный анализ проводили на C, H, N-анализаторе EA3000 (EuroVector).
Производное урацила 1:
где R = H – 6-метилурацил (6-MeU)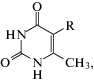
R = CH3 – 5,6-диметилурацил (5,6-diMeU)
R = 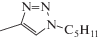 – 5-(1-пентил-4-метил-1,2,3-триазол-4-ил)-6-метилурацил (1),
– 5-(1-пентил-4-метил-1,2,3-триазол-4-ил)-6-метилурацил (1),
синтезировали в условиях реакции диполярного 1,3-циклоприсоединения Хьюсгена–Мельдаля–Шарплесса (CuAAC) производного урацила 2 с диазидом 3 (схема 1). В условиях этой реакции специфично образуются 1,4-дизамещенные 1,2,3-триазолы [8–10]. Поскольку производное урацила 2 и целевой продукт 1 имеют весьма ограниченную растворимость, реакцию проводили в ДМСО.
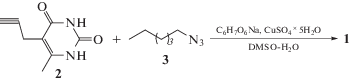
(5-(1-Пентил-4-метил-1,2,3-триазол-4-ил)-6-метил-1,2,3,4-тетрагидропиримидин-2,4-дион (1)) К раствору 1 г (6.5 ммоль) 5-пропаргил-6-метилурацила (2) [11] в 30.0 мл ДМСО добавляли раствор 0.90 г (3.6 ммоль) н-пентилазида (3) [12], 0.24 г (1.20 ммоль) аскорбата натрия и 0.90 г (8.0 ммоль) пентагидрата сульфата меди в 3.0 мл воды. Реакционную смесь перемешивали при комнатной температуре 16 ч. В раствор добавляли 30.0 мл воды, выпавший осадок отфильтровывали, промывали водой, сушили на воздухе. Выход составил 1.41 г (78%). Порошок кремового цвета, т. пл. 223°С. ИК-спектр, ν, см–1: 3044, 2952, 2862, 1703, 1679, 1652, 1454, 1416, 1324, 1219, 1049, 848, 798, 758, 536. Спектр ЯМР 1Н (ДМСО-d6), δ, м.д. (J, Гц): 10.05, 10.81 оба с (по 1Н, N$_{{{\text{ур}}}}^{1}$H, N$_{{{\text{ур}}}}^{3}$H), 7.77 с (1Н, C$_{{{\text{тр}}}}^{{5'}}$H), 4.26 т (2Н, 2N$_{{{\text{тр}}}}^{1}$CH2, 3JНН 7.0), 3.61 с (2H, 2С$_{{{\text{тр}}}}^{4}$CH2C$_{{{\text{ур}}}}^{5}$), 2.13 с (3Н, C$_{{{\text{ур}}}}^{6}$СH3), 1.80–1.72 м (2Н, СН2), 1.37–1.04 м (4Н, 2СН2), 0.84 т (3Н, СН3, 3JНН 7.1). Спектр ЯМР 13C (ДМСО-d6), δ, м.д.: 164.3, 150.8, 149.6, 145.7, 122.2, 106.8, 49.2, 29.2, 27.9, 21.4, 20.3, 16.2, 13.7. Спектр ЯМР 15N (ДМСО-d6), δ, м.д.: 137.06, 155.30, 250.34, 359.82. Найдено, %: C 56.26; H 6.93; N 25.36. C13H19N5O2. Вычислено, %: C 56.30; H 6.91; N 25.25.
В работе использовались коммерчески доступные соединения без дополнительной очистки.
Величины рKа1 определяли по стандартной методике [13] методом потенциометрического титрования (ПТ) в одногорлом термостатируемом реакторе объемом 25 мл с обратным холодильником при четырех температурах: 20, 25, 35 и 45°С. Температуру поддерживали с точностью ±0.1°С термостатом LOIP LT-205. Титрование проводили на рН-метре рН-150МИ с использованием комбинированного стеклянного электрода ЭСК-10307. Калибровку электрода проводили с помощью стандартных буферных растворов. Для поддержания постоянной ионной силы в титруемом растворе использовали 0.1 М раствор KNO3. В качестве растворителя применяли свежеперегнанную бидистиллированную воду. Концентрацию свежеприготовленного раствора KOH устанавливали титрованием раствором 0.01 М HCl c индикатором фенолфталеином.
Также были определены величины рКа1 спектрофотометрическим методом (СФ) по методике [13]. УФ-спектры регистрировали на спектрофотометре Shimadzu UV-1800 в диапазоне длин волн 200–350 нм. Раствором сравнения служила вода. Использовали кварцевые кюветы с толщиной поглощающего слоя 1 см. Для поддержания постоянного значения рН использовали буферную систему в диапазоне 5.8–9.2 (KH2PO4–Na2B4O7) и 9.2–11.0 (Na2B4O7–NaOH) в соответствующих соотношениях. Концентрация полученных растворов составляет 5.0 × 10–5 моль/л.
В связи с малой растворимостью 1 в воде, для потенциометрического титрования готовили его растворы с концентрацией 0.01 моль/л следующим образом: навеску соединения 1 массой 0.1454 г помещали в мерную колбу объемом 500 мл, добавляли 25 мл ацетонитрила, после полного растворения добавляли навеску КNO3 массой 5.0539 г и доводили до метки дистиллированной водой. Доля ацетонитрила в полученном растворе составляла 5%. Растворы 6-MeU и 5,6-diMeU готовили аналогично растворением навесок соответствующих соединений в воде без добавления ацетонитрила.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
С целью изучения влияния рН на кислотно-основное равновесие 1, 6-MeU и 5,6-diMeU в растворах записывали УФ-спектры их водных растворов при различных pH (рис. 1).
Рис. 1.
УФ-спектры 6-метилурацила (а), 5,6-диметилурацила (б) и 5-(1-пентил-4-метил-1,2,3-триазол-4-ил)-6-метилурацила (в) в водных растворах при различных рН.
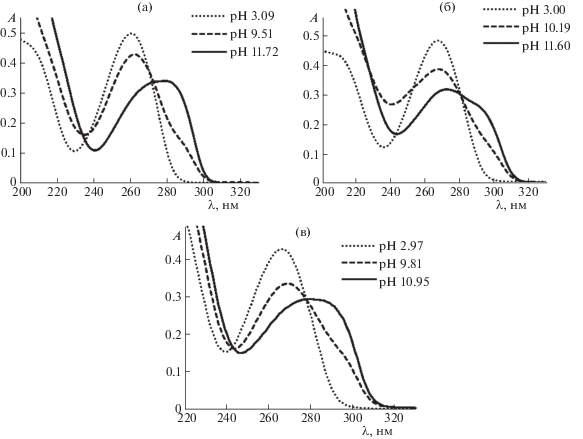
Как видно из рис. 1 в УФ-спектрах всех трех соединений в кислой и нейтральной среде наблюдаются максимумы поглощения в близких областях и примерно одинаковой интенсивности: 260 нм (ε = 9980 л моль–1 см–1) у 6-MeU, 268 нм (ε = 9702 л моль–1 см–1) у 5,6-diMeU и 266 нм (ε = = 8575 л моль–1 см–1) у 1, обусловленный поглощением сопряженной группы –С6=С5–С4=O урацильного фрагмента. При увеличении рН более 9.0 наблюдается батохромное смещение максимумов данных полос поглощения примерно на 17–20 нм. Аналогичные изменения в УФ-спектрах при увеличении pH водных растворов наблюдаются у урацила [14], тимина [15] и большинства других 5- и (или) 6-замещенных производных урацила [16]. Наличие триазольного кольца не влияет на спектр поглощения, о чем свидетельствуют спектры модельных соединений 6-MeU (рис. 1а) и 5,6-diMeU (рис. 1б) в водных растворах при различных рН. Такие изменения, по мнению [17], обусловлены смещением кислотно-основного равновесия в сторону образования анионной формы:
(3)
${{{\text{H}}}_{{\text{2}}}}{\text{U}} + {\text{O}}{{{\text{H}}}^{--}} \to {\text{H}}{{{\text{U}}}^{--}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}{\text{.}}$Методом потенциометрического титрования определены константы и термодинамические характеристики кислотно-основного равновесия (3) вещества 1, а также 6-MeU и 5,6-diMeU в качестве соединений сравнения в водных растворах (табл. 1).
Таблица 1.
Константы диссоциации и термодинамические характеристики кислотно-основного равновесия 6-MeU, 5,6-diMeU и 1 в водных растворах (0.1М KNO3)
| Соединение | Т, °С | рKa1 | ΔG298, кДж/моль |
ΔН, кДж/моль |
ΔS298, Дж/(моль K) |
|
|---|---|---|---|---|---|---|
| (ПТ) | (СФ) | |||||
| 6-MeU | 15 25 35 45 |
9.85 ± 0.04 9.65 ± 0.03 9.62 ± 0.03 9.61 ± 0.03 |
9.53 ± 0.05 | 55.0 ± 0.2 | 13.3 ± 0.4 | –140 ± 1 |
| 5,6-diMeU | 15 25 35 45 |
10.33 ± 0.04 10.26 ± 0.03 10.20 ± 0.04 10.13 ± 0.03 |
10.28 ± 0.11 | 58.5 ± 0.2 | 11.6 ± 0.3 | –158 ± 1 |
| 1 | 15 25 35 45 |
10.14 ± 0.07 10.09 ± 0.07 9.98 ± 0.05 9.93 ± 0.05 |
9.59 ± 0.10 | 57.5 ± 0.9 | 12.9 ± 0.4 | –150 ± 2 |
Значения рKа1, полученные нами, хорошо согласуются с приведенными в литературных источниках значениями (9.68 [18] и 9.45 [19] при 25°С для 6-MeU) и 9.8 для 5,6-diMeU [20].
Также значения рKа1 были получены спектрофотометрическим методом. Как видно из результатов (табл. 1), значения рKа, полученные спектрофотометрически при 25°С для 6-MeU и 5,6-diMeU, хорошо согласуются со значениями, полученными методом потенциометрического титрования. Различия, в значениях, полученных двумя методами для 1 можно объяснить наличием небольшого количества ацетонитрила в исходном растворе при потенциометрическом титровании при определении рKа.
Присутствие в молекуле 6-метилурацила 1-пентил-4-метил-1,2,3-триазол-4-илного заместителя у пятого углеродного атома приводит к увеличению рКа примерно на 0.5 единицы по сравнению с 6-метилурацилом, причем термодинамические характеристики кислотно-основного равновесия обоих веществ имеют близкие значения. Влияние 1,2,3-триазольного цикла на величину рKа1 не существенно, о чем свидетельствуют константы и термодинамические характеристики кислотно-основного равновесия 5,6-diMeU (табл. 1).
С целью изучения структурных изменений 1 в щелочных растворах были записаны ЯМР-спектры 1 и образца 1 + КОН в диметилсульфоксидных и водно-диметилсульфоксидных растворах (табл. 2). Отнесение сигналов произведено на основании корреляционных спектров.
Таблица 2.
Данные спектров ЯМР (м.д.) 5-(1-пентил-4-метил-1,2,3-триазол-4-ил)-6-метилурацила в диметилсульфоксидных и водно-диметилсульфоксидных растворах
| Атом | ДМСО-d6 | ДМСО-d6 + + D2O (1 : 1) | |||
|---|---|---|---|---|---|
| δ | δ | Δδ | δ | Δδ | |
| 1 | 1 + KOH (1 : 1) | 1 + KOH (1 : 1) | |||
| C(2) | 150.84 | 158.15 | 7.31 | 160.72 | 9.88 |
| C(4) | 164.25 | 166.48 | 2.23 | 171.13 | 6.88 |
| C(5) | 106.84 | 103.36 | –3.48 | 108.48 | 1.64 |
| C(6) | 149.55 | 161.15 | 11.6 | 161.74 | 12.19 |
| C(7) | 16.15 | 20.71 | 4.56 | 21.45 | 5.3 |
| C(8) | 20.31 | 21.24 | 0.93 | 23.15 | 2.84 |
| C(4') | 145.69 | 147.46 | 1.77 | 149.31 | 3.62 |
| C(5') | 122.2 | 121.56 | –0.64 | 125.1 | 2.9 |
| C(1'') | 49.24 | 48.99 | –0.25 | 52.28 | 3.04 |
| C(2'') | 29.24 | 29.37 | 0.13 | 31.55 | 2.31 |
| C(3'') | 27.89 | 27.97 | 0.08 | 30.22 | 2.33 |
| C(4'') | 21.39 | 21.42 | 0.03 | 23.76 | 2.37 |
| C(5'') | 13.67 | 13.69 | 0.02 | 15.94 | 2.27 |
| N(1) | 137.06 | 164.27 | 27.21 | 182.36 | 45.3 |
| N(3) | 155.3 | – | – | – | – |
| N(1') | 250.34 | 248.79 | –1.55 | 252.2 | 1.86 |
| N(2') | 359.82 | 360.29 | 0.47 | 353.79 | –6.03 |
| N(3') | – | 349.93 | – | 340.64 | – |
| HN(1) | 10.81 | – | – | – | – |
| HN(3) | 11.05 | – | – | – | – |
| CH3 | 2.13 | 1.98 | –0.15 | 2.25 | 0.12 |
| HC(8) | 3.61 | 3.59 | –0.02 | 3.83 | 0.22 |
| H(C5') | 7.77 | 7.56 | –0.21 | 7.82 | 0.05 |
| HC(1'') | 4.26 | 4.23 | –0.03 | 4.43 | 0.17 |
| HC(2'') | 1.77 | 1.75 | –0.02 | 1.93 | 0.16 |
| HC(3'') | 1.17 | 1.17 | 0 | 1.29 | 0.12 |
| HC(4'') | 1.27 | 1.27 | 0 | 1.39 | 0.12 |
| HC(5'') | 0.83 | 0.83 | 0 | 0.95 | 0.12 |
При добавлении к раствору 1 гидроксида калия в мольном соотношении 1 : 1 в диметилсульфоксидных растворах наблюдается существенное смещение сигналов углерода С(2), С(6), С(7) и N(1) (нумерацию атомов см. схему 2) в слабое поле относительно спектра исходного 1. Смещение сигналов углеродов пентильного радикала практически не наблюдается. Подобные изменения в спектрах ЯМР наблюдаются в щелочных диметилсульфоксидных растворах 5-галогенурацилов и, вероятно, обусловлены образованием анионной формы AN(1) [5, 6]. В ЯМР-спектрах щелочных водно-диметилсульфоксидных растворов 1 наблюдается похожая картина, что и в диметилсульфоксидных. При этом, более выражено смещение в слабое поле сигналов атомов С(4), С(5) и N(1), что может свидетельствовать о содержании в щелочных водно-диметилсульфоксидных растворах 1 кроме основного AN(1), некоторого количества аниона AN(3). К сожалению, из-за плохой растворимости соединения 1 в воде, нам не удалось растворить в чистом D2O достаточного количества 1 для записи ЯМР-спектров исходного соединения до добавления КОН.
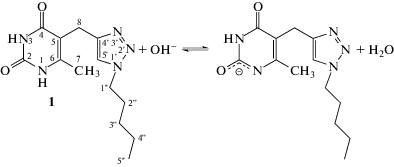
Несмотря на это, полученные методом ЯМР-спектроскопии данные свидетельствуют, видимо, о преимущественном образовании анионной формы с отрывом протона от азота N(1) пиримидинового кольца в щелочных растворах 1 в воде и диметилсульфоксиде (схема 2).
Таким образом, синтезировано новое производное урацила, несущее при С(5) урацилового цикла 1,2,3-триазольный цикл, и доказана его структура различными физико-химическими методами. Определены константы и термодинамические характеристики кислотно-основного равновесия синтезированного соединения и модельного соединения – 5,6-diMeU в водных растворах. Показано, что 1,2,3-триазольный цикл существенно не влияет на кислотно-основные свойства урацильного фрагмента в составе целевого соединения.
Исходя из полученных результатов можно предположить координацию соединения 1 с d‑металлами через азот N(1) урацильного фрагмента. При этом, наличие атомов азота в триазольном кольце, а также три ненасыщенных связи, пространственно близко расположенных относительно атома N(1) пиримидинового фрагмента, выделяют соединение 1 как перспективный лиганд при взаимодействии с ионами d- и f-металлов. Присутствие в данном соединении пентильной группы способствует увеличению его растворимости, по сравнению с другими урацилами и 1,2,3-триазолами, в менее полярных, чем вода и диметилсульфоксид, растворителях, что, безусловно, увеличит диапазон условий для синтеза комплексных соединений с металлами.
Спектры записаны с использованием оборудования ЦКП “Химия” УфИХ УФИЦ РАН и РЦКП “Агидель” УФИЦ РАН.
Работа выполнена в рамках государственного задания Министерства образования и науки (№ AAAA-A20-120012090029-0 и АААА-А18-118040390114-8).
Список литературы
Ilyina M.G., Khamitov E.M., Ivanov S.P. et al. // J. Phys. Chem. A. 2018. V. 122. № 1. P. 341. https://doi.org/10.1021/acs.jpca.7b09330
Ilyina M.G., Khamitov E.M., Ivanov S.P. et al. // Comput. Theor. Chem. 2016. V. 1078. P. 81. https://doi.org/10.1016/j.comptc.2015.12.024
Privat E.S., Sowers L.C. // Mutat. Res. 1996. V. 354. P. 151. https://doi.org/10.1016/0027-5107(96)00005-X
Wittenburg E. // Chemische Berichte. 1966. V. 99. P. 2391. https://doi.org/10.1002/cber.19660990737
Abdrakhimova G.S., Ovchinnikov M.Yu., Lobov A.N. et al. // J. Phys. Org. Chem. 2014. V. 27. P. 876. https://doi.org/10.1002/poc.3350
Abdrakhimova G.S., Ovchinnikov M.Yu., Lobov A.N. et al. // J. Mol. Struct. 2018. V. 1158. P. 51. https://doi.org/10.1016/j.molstruc.2018.01.013
Tome A.C. Product class 13: 1,2,3-triazoles, Eds. Storr R.C., Gilchrist T.L. (In: Science of Synthesis, Stuttgart-N.-Y. Thieme, 2004), p. 415.
Huisgen R. // Angew. Chem. Int. Edit. 1963. V. 2. P. 565. https://doi.org/10.1002/anie.196305651
Rostovtsev V.V., Green L.G., Fokin V.V., Sharpless K.B. // Angew. Chem. Int. Ed. 2002. V. 41. № 14. P. 2596. https://doi.org/10.1002/1521-3773(20020715)41
Tornoe C.W., Christensen C., Meldal M. // J. Org. Chem. 2002. V.67. № 9. P. 3057. https://doi.org/10.1021/jo011148j
Semenov V.E., Voloshina A.D., Kulik N.V. et al. // Russ. Chem. Bull. 2015. V. 64. № 12. P. 2885. https://doi.org/10.1007/s11172-015-1243-5
Yan F., Lartey V., Jariwala K. et al. // J. Phys. Chem. B. 2014. V. 118. № 47. P. 13609. https://doi.org/10.1021/jp506972w
Albert A., Serjeant E.P. Ionization constants of acids and bases; a laboratory manual (London, Methuen; New York, Wiley. 1962) p. 179.
Billinghurst B.E., Oladepo S.A., Loppnow G.R. // J. Phys. Chem. B. 2009. V. 113. № 20. P. 7392. https://doi.org/10.1021/jp811327w
Stimson M.M. // J. Am. Chem. Soc. 1949. V. 71. № 4. P. 1470. https://doi.org/10.1021/ja01172a093
Иванов С.П., Муринов Ю.И. // Башкирский хим. журнал. 2006. V. 13. № 1. P. 22.
Clark L.B. // J. Am. Chem. Soc. 1965. V. 87. № 1. P. 11. https://doi.org/10.1021/ja01079a003
Jonas J., Gut J. // Coll. Czech. Chem. Commun. 1962. V. 27. № 27. P. 716. https://doi.org/10.1135/cccc19620716
Blagoy Yu.P., Sheina G.G., Luzanov A.V. et al. // Int. J. Quant. Chem. 1980. V. 18. P. 913. https://doi.org/10.1002/qua.560180402
Luke T.L., Mohan H., Jacob T.A. et al. // J. Phys. Org. Chem. 2002. V. 15. P. 293–305. https://doi.org/10.1002/poc.478
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии