

Журнал физической химии, 2021, T. 95, № 2, стр. 238-246
Молекулярные структуры гетеролигандных макротетрациклических комплексов ионов 3d-элементов с фталоцианином и фторид-анионом в рамках теории функционала плотности
Д. В. Чачков a, *, О. В. Михайлов b, **, ***
a Казанское отделение Межведомственного суперкомпьютерного центра Российской академии наук – филиал Федерального государственного учреждения “Федеральный научный центр Научно-исследовательский институт системных исследований Российской академии наук”
420111 Казань, Россия
b Казанский национальный исследовательский технологический университет
420015 Казань, Россия
* E-mail: de2005c@gmail.com
** E-mail: olegmkhlv@gmail.com
*** E-mail: ovm@kstu.ru
Поступила в редакцию 22.04.2020
После доработки 22.04.2020
Принята к публикации 26.05.2020
Аннотация
Посредством теории функционала плотности (DFT) в варианте OPBE/TZVP осуществлен расчет молекулярных структур гетеролигандных (6666)макротетрациклических хелатов M(III) (M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu) с (NNNN)-донорноатомным лигандом – фталоцианином и F-анионом. Обнаружено, что все эти металлокомплексы обладают структурой слегка искаженной тетрагональной пирамиды, где комплексообразователь M(III) в той или иной степени приподнят над ее основанием, которое составляют донорные атомы азота. Приведены значения важнейших длин связей, валентных и невалентных углов в данных комплексах. Констатировано, что все шестичленные металлохелатные циклы в восьми из девяти этих металлохелатов идентичны друг другу, причем как по сумме входящих в них валентных углов, так и по их ассортименту; единственным исключением является комплекс Mn(III), в котором эти металлоциклы равны лишь попарно. Отмечено, что в каждом из вышеуказанных координационных соединений имеет место весьма сильно выраженная делокализация электронной плотности в пределах всего макроцикла. Рассчитаны также значения стандартной энтальпии, энтропии и энергии Гиббса образования этих соединений.
Фталоцианин или тетра[бензо]порфиразин I принадлежит к числу тех азотсодержащих макроциклических органических соединений, которые в настоящее время нашли весьма значительное приложение в самых различных отраслях науки и техники. Уже достаточно давно было обнаружено, что вещества такого типа благодаря наличию в них четырех донорных атомов азота способны выступать в качестве макроциклических лигандов и формировать чрезвычайно устойчивые (6666)макротетрациклические хелатные комплексы с ионами почти всех металлов Периодической системы элементов Д.И. Менделеева, причем с самыми различными состояниями окисления комплексообразователей (см., в частности, [1–7]). Соответственно этому общее количество публикаций, посвященных данному
макроциклическому лиганду и его производным, измеряется согласно [1–7] уже как минимум четырехзначным числом. В целом ряде этих работ так или иначе рассматривались гетеролигандные тетрагонально-пирамидальные или тетрагонально-бипирамидальные металлохелаты, содержащие различные (NNNN)-донорноатомные лиганды из категории порфиринов, порфиразинов и/или фталоцианинов, донорные атомы которых располагаются по вершинам плоского четырехугольника (как правило, квадрата, ромба или прямоугольника), и один или два монодентатных ацидолиганда (в частности, галогенид-ионы), располагающиеся в вершинах тетрагональной пирамиды (при КЧ = 5) или же бипирамиды (при КЧ = 6) [5].
Обращает на себя внимание, однако, тот факт, что в практически необъятной ныне литературе по металлмакроциклическим соединениям практически отсутствуют какие-либо сведения о таких гетеролигандных макроциклических комплексах, в состав которых наряду с различными порфиринами, порфиразинами и фталоцианинами входят также фторид-анионы. Информация о подобных координационных соединениях присутствует лишь в очень немногих публикациях, а именно в [7–11]; в их числе – комплексы двух 3d-элементов (Cr(III) [7], Fe(III), Fe(IV) [8]) и одного 4d-элемента (Nb(V) [9–11]). (Справедливости ради, однако, следует сказать, что имеется довольно большое количество публикаций, в которых были рассмотрены комплексы, содержащие фторзамещенные порфирины и фталоцианины [12, 13]). В связи с этим представляется интересным осуществить квантово-химический расчет молекулярных структур гетеролигандных металлохелатов с дважды депротонированной формой фталоцианина I и фторид-анионом общей формулы II (M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu)
с использованием популярного ныне метода, базирующегося на теории функционала плотности (DFT), результаты которого, с одной стороны, могли бы способствовать устранению вышеуказанного пробела в современной координационной химии (NNNN)-донорноатомных органических соединений вышеуказанной категории (имеющих, как уже указывалось выше, весьма важное практическое значение), с другой стороны, расширить наши представления о специфике координации “аксиальных” лигандов вообще и галогенидных в частности к комплексообразователям M(III) различных 3d-элементов. Рассмотрение всего этого и будет предметом данной статьи.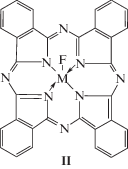
МЕТОД РАСЧЕТА
Для проведения квантово-химического расчета в данной работе, равно как и в более ранних наших публикациях [14–16], был задействован метод функционала плотности (DFT) OPBE/TZVP со стандартным расширенным базисом TZVP [17, 18] и функционалом OPBE [19, 20], который согласно данным [20–24] в случае комплексов 3d-элементов дает достаточно точное соотношение между энергиями состояний с различной спиновой мультиплетностью и в то же время надежно характеризует важнейшие параметры их молекулярных структур. Соответствие найденных стационарных точек минимумам энергии во всех случаях доказывалось расчетом вторых производных энергии по координатам атомов; при этом все равновесные структуры, соответствовавшие точкам минимума на поверхностях потенциальной энергии, имели лишь положительные значения частот. При этом для комплексов Ti(III), Cr(III) и Fe(III) рассматривались спиновые мультиплетности 2, 4 и 6, для комплексов V(III), Mn(III), Co(III) – мультиплетности 1, 3 и 5, для комплексов Sc(III) и Cu(III) – мультиплетности 1 и 3, для комплекса Ni(III) – мультиплетности 2 и 4.
Из оптимизированных при указанных мультиплетностях структур выбиралась та, которая обладала наименьшей энергией. Расчет параметров молекулярных структур при мультиплетностях, отличных от 1, всегда проводился неограниченным методом (UOPBE); при мультиплетности 1 – ограниченным методом (ROPBE). При мультиплетности 1 применялся также вариант расчета неограниченным методом, в сочетании с опцией GUESS=Mix; при этом получаемые результаты всегда были аналогичны результатам, получаемым с использованием ограниченного метода. Расчеты проводились с использованием пакета программ Gaussian09 [25], визуализация полученных при этом данных осуществлялась посредством программы ChemCraft (Version 1.8). Во всех случаях осуществлялась также проверка волновых функций полученных оптимизированных молекулярных структур соответствующих металлокомплексов на стабильность по стандартной процедуре STABLE = OPT; согласно результатам этой проверки, все эти волновые функции соответствовали указанному критерию. Расчет термодинамических параметров осуществляли с использованием методики, описанной в [26].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Согласно полученным нами расчетным данным, основным состоянием координационных соединений Sc(III) (3p63d0), Ti(III) (3d1), V(III) (3d2) и Cr(III) (3d3) типа II, как и следовало ожидать теоретически исходя из электронных конфигураций поименованных комплексообразователей, являются спиновый синглет, дублет, триплет и квартет соответственно. Для комплексов других рассматриваемых здесь M(III), где в силу специфики электронных конфигураций комплексообразователя могут быть реализованы два и большее число значений спиновых мультиплетностей основного состояния, в качестве таковых реализуются спиновый синглет [Co(III) (3d6), Cu(III) (3d8)], дублет [Ni(III) (3d7)], триплет [Mn(III) (3d4)] и квартет [Fe(III) (3d5)].
Как следует из только что приведенных данных, первые четыре из пяти последних металлохелатов однозначно принадлежат к числу низкоспиновых координационных соединений; хелат же Fe(III) следует отнести к категории соединений с промежуточными между высокоспиновыми и низкоспиновыми комплексами значениями спиновой мультиплетности. У многих из рассматриваемых здесь соединений характерно весьма значительное различие между энергией основного и энергией ближайшего к нему возбужденного состояния с иной спиновой мультиплетностью [триплетной в случае Sc(III), квартетной в случае Ti(III), синглетной – V(III), дублетной – Cr(III), квинтетной – Mn(III), дублетной – Fe(III), триплетной – Co(III), квартетной – Ni(III), триплетной – Cu(III)], которая согласно данным нашего расчета составляет 107.6, 79.9, 44.3, 92.1, 58.6, 61.4, 1.9, 202.1 и 189.6 кДж/моль. Соответствующие этому различию цифры весьма велики для того, чтобы можно было надеяться на проявление здесь спин-кроссовера; определенные ожидания на этот счет дает лишь комплекс Co(III), где различие в энергиях между основным синглетным и ближайшим возбужденным триплетным уровнями оказывается менее 2.0 кДж/моль.
Согласно данным проведенного нами NBO анализа, значения оператора квадрата собственного углового момента (S^2) в рассматриваемых нами комплексах II составляют 0.0000 (Sc), 0.7532 (Ti), 2.0261 (V), 3.7965 (Cr), 2.0561 (Mn), 3.7899 (Fe), 0.0000 (Co), 0.7563 (Ni) 0.0000 (Cu); для эффективных зарядов на атомах M в них имеют место величины +1.43 ē (Sc), +1.07 ē (Ti), +0.73 ē (V), +0.72 ē (Cr), +0.53 ē (Mn), +0.69 ē (Fe), +0.51 ē (Co), +0.68 ē (Ni) и +0.81 ē (Cu). Как можно видеть из приведенных цифр, эффективные заряды на атомах 3d-элементов, входящих в состав этих химических соединений, существенно меньше того формального заряда +3.00 ē, который имел бы место в том случае (гипотетическом, конечно), когда все имеющиеся в этих самых соединениях двухцентровые химические связи были бы чисто ионными. При этом наибольшее значение данного параметра (и соответственно, наиболее выраженная степень ионности связи 3d-элемент – донорный атом азота) имеет место в комплексе Sc(III), что с учетом наиболее низкой электроотрицательности данного химического элемента (1.3 по шкале Полинга) по сравнению с таковой для остальных рассматриваемых нами 3d-элементов M представляется вполне естественным. Аналогичная ситуация имеет место и для эффективных зарядов на других атомах в этих комплексах (см. Supplementary Materials); все это является прямым указанием на весьма высокую степень делокализации электронной плотности в их макроциклах.
Рассчитанные в рамках метода DFT OPBE/TZVP важнейшие длины связей и валентные углы для рассматриваемых нами гетеролигандных металлохелатов M(III) (M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu) с фталоцианином и фторидным ацидолигандом представлены в табл. 1. Как можно видеть из представленных в ней данных, длины связей (d) между атомами M и N в хелатном узле MN4, а именно (M1N1), (M1N2), (M1N3) и (M1N4), в ряду Sc–Co в целом уменьшаются, тогда как в ряду Co–Cu они, напротив, возрастают. Из этой динамики выпадает лишь комплекс Mn(III), для которого длины связей (M–N) оказываются более короткими, нежели в таковые в аналогичных комплексах типа II, образованных ближайшими к нему 3d-элементами – Cr и Fe (табл. 1). [Такая же динамика изменения длин связей металл – азот, но без только что указанного исключения наблюдается и для монолигандных фталоцианиновых комплексов M(II) этих же самых 3d-элементов (см. Supplementary Materials)]. При этом в шести из девяти рассматриваемых здесь комплексов (где M = Sc, Cr, Fe, Co, Ni, Cu) все длины связей длины связей (M–N) в MN4 совершенно одинаковы, в двух других (M = Ti, V) они попарно различны меж собой [d(M1N1) = d(M1N3), d(M1N2) = d(M1N4)]. А вот в комплексе Mn(III) для связей 3d-элемент–азот отмечается равенство длин лишь двух из них, две же остальные не равны друг другу; при этом, как можно видеть из табл. 1, между ними имеет место соотношение d(M1N2) < d(M1N1) = d(M1N3) < d(M1N4). Причины столь необычного момента скорее всего следует связывать с асимметричным распределением электронной плотности на связывающих и разрыхляющих МО, образованных с участием 3d-орбиталей данного элемента, но почему оно столь сильно проявляется именно в комплексе Mn(III), остается пока что непонятно. Отметим, однако, что в тех случаях, когда указанное различие между величинами d(M–N) вообще имеет место, оно оказывается не слишком значительным [1.0 пм в комплексе Ti(III), 1.8 пм – в комплексе Mn(III) и 2.4 пм – в комплексе V(III)].
Таблица 1.
Длины связей, валентные и невалентные углы в комплексах M(III) с фталоцианином и фторид-анионом типа II
| M | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu |
|---|---|---|---|---|---|---|---|---|---|
| Длины связей в хелатном узле MN4, пм | |||||||||
| (M1N1) | 215.6 | 204.0 | 202.7 | 198.8 | 195.8 | 196.1 | 192.1 | 192.6 | 194.5 |
| (M1N2) | 215.6 | 203.0 | 200.3 | 198.8 | 195.1 | 196.1 | 192.1 | 192.6 | 194.5 |
| (M1N3) | 215.6 | 204.0 | 202.7 | 198.8 | 195.8 | 196.1 | 192.1 | 192.6 | 194.5 |
| (M1N4) | 215.6 | 203.0 | 200.3 | 198.8 | 196.9 | 196.1 | 192.1 | 192.6 | 194.5 |
| Длины связей M–F, пм | |||||||||
| (M1F1) | 185.7 | 177.9 | 175.9 | 178.1 | 174.1 | 183.2 | 178.5 | 192.7 | 200.5 |
| Валентные углы в хелатном узле MN4, град | |||||||||
| (N1M1N2) | 81.6 | 86.0 | 86.9 | 88.8 | 89.4 | 88.6 | 89.7 | 89.7 | 89.5 |
| (N2M1N3) | 81.6 | 86.0 | 86.9 | 88.8 | 89.4 | 88.6 | 89.7 | 89.7 | 89.5 |
| (N3M1N4) | 81.6 | 86.0 | 86.9 | 88.8 | 88.3 | 88.6 | 89.7 | 89.7 | 89.5 |
| (N4M1N1) | 81.6 | 86.0 | 86.9 | 88.8 | 88.3 | 88.6 | 89.7 | 89.7 | 89.5 |
| (VAS) | 326.4 | 344.0 | 349.6 | 355.2 | 355.4 | 354.4 | 358.8 | 358.8 | 358.0 |
| Невалентные углы в группировке N4, град | |||||||||
| (N1N2N3) | 90.0 | 91.1 | 91.5 | 90.0 | 89.7 | 90.0 | 90.0 | 90.0 | 90.0 |
| (N2N3N4) | 90.0 | 88.7 | 88.2 | 90.0 | 90.0 | 90.0 | 90.0 | 90.0 | 90.0 |
| (N3N4N1) | 90.0 | 91.1 | 91.5 | 90.0 | 90.3 | 90.0 | 90.0 | 90.0 | 90.0 |
| (N4N1N2) | 90.0 | 88.7 | 88.2 | 90.0 | 90.0 | 90.0 | 90.0 | 90.0 | 90.0 |
| (NVAS) | 360.0 | 359.6 | 359.4 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 | 360.0 |
| Валентные углы в 6-членном хелатном цикле (N1C4N7C5N4M1), град | |||||||||
| (M1N1C4) | 124.4 | 126.1 | 126.0 | 125.4 | 126.5 | 125.6 | 126.2 | 126.0 | 125.3 |
| (N1C4N7) | 127.6 | 127.0 | 127.1 | 127.6 | 127.5 | 128.0 | 127.9 | 128.2 | 128.8 |
| (C4N7C5) | 124.3 | 123.5 | 123.2 | 123.6 | 122.3 | 122.0 | 121.5 | 121.2 | 121.2 |
| (N7C5N4) | 127.6 | 127.0 | 127.1 | 127.6 | 127.5 | 128.0 | 127.9 | 128.2 | 128.8 |
| (C5N4M1) | 124.4 | 126.1 | 126.0 | 125.4 | 126.5 | 125.6 | 126.2 | 126.0 | 125.3 |
| (N4M1N1) | 81.6 | 86.0 | 86.9 | 88.8 | 88.3 | 88.6 | 89.7 | 89.7 | 89.5 |
| (VAS6) | 709.9 | 712.8 | 715.6 | 718.4 | 718.0 | 717.8 | 718.4 | 719.3 | 718.9 |
| Валентные углы в 6-членном хелатном цикле (M1N2C2N5C3N1), град | |||||||||
| (M1N2C2) | 124.4 | 126.1 | 126.0 | 125.4 | 125.8 | 125.6 | 126.2 | 126.0 | 125.3 |
| (N2C2N5) | 127.6 | 127.0 | 127.1 | 127.6 | 127.6 | 128.0 | 127.9 | 128.2 | 128.8 |
| (C2N5C3) | 124.3 | 123.5 | 123.2 | 123.6 | 122.6 | 122.0 | 121.5 | 121.2 | 121.2 |
| (N5C3N1) | 127.6 | 127.0 | 127.1 | 127.6 | 127.9 | 128.0 | 127.9 | 128.2 | 128.8 |
| (C3N1M1) | 124.4 | 126.1 | 126.0 | 125.4 | 125.1 | 125.6 | 126.2 | 126.0 | 125.3 |
| (N1M1N2) | 81.6 | 86.0 | 86.9 | 88.8 | 89.4 | 88.6 | 89.7 | 89.7 | 89.5 |
| (VAS6) | 709.9 | 712.8 | 715.6 | 718.4 | 718.4 | 717.8 | 718.4 | 719.3 | 718.9 |
| Валентные углы в 5-членном цикле (C3N1C4C9C10), град | |||||||||
| (C3N1C4) | 109.0 | 107.8 | 108.0 | 108.8 | 107.8 | 108.0 | 107.2 | 107.7 | 108.6 |
| (N1C4C9) | 109.0 | 109.3 | 109.5 | 109.0 | 109.7 | 109.6 | 110.1 | 109.8 | 109.2 |
| (C4C9C10) | 106.5 | 106.8 | 106.5 | 106.6 | 106.4 | 106.4 | 106.3 | 106.3 | 106.5 |
| (C9C10C3) | 106.5 | 106.8 | 106.5 | 106.6 | 106.5 | 106.4 | 106.3 | 106.3 | 106.5 |
| (C10C3N1) | 109.0 | 109.3 | 109.5 | 109.0 | 109.6 | 109.6 | 110.1 | 109.8 | 109.2 |
| (VAS5) | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 | 540.0 |
| Валентные углы ∠FMN, град | |||||||||
| (F1M1N1) | 112.5 | 103.8 | 101.5 | 98.3 | 98.0 | 99.0 | 94.1 | 94.2 | 95.5 |
| (F1M1N2) | 112.5 | 107.1 | 105.6 | 98.3 | 97.0 | 99.0 | 94.1 | 94.2 | 95.5 |
| (F1M1N3) | 112.5 | 103.8 | 101.5 | 98.3 | 98.0 | 99.0 | 94.1 | 94.2 | 95.5 |
| (F1M1N4) | 112.5 | 107.1 | 105.6 | 98.3 | 99.2 | 99.0 | 94.1 | 94.2 | 95.5 |
Длина связи 3d-элемент – фтор в комплексах типа II при переходе от Sc к Cu изменяется, однако, по более сложному (можно сказать, “зигзагообразному”) закону: в ряду Sc–V она уменьшается, при переходе к Cr – несколько увеличивается, к Mn – вновь уменьшается, к Fe – вновь увеличивается, к Co – опять уменьшается, и далее до Cu – опять возрастает, достигая своего максимума в комплексе Cu(III) (табл. 1). Длины остальных связей, в частности углерод–азот и углерод–углерод также зависят от природы комплексообразователя M(III), но влияние природы иона 3d-элемента на них сказывается уже в меньшей степени. В целом же эта зависимость проявляется тем сильнее, чем ближе эти связи к металлохелатному узлу MN4, что, впрочем, также вполне ожидаемо (см. Supplementary Materials).
Данные нашего расчета свидетельствуют о том, что для всех перечисленных выше комплексов типа II независимо от природы входящего в них 3d-элемента имеет место тетрагонально-пирамидальная координация донорных центров фталоцианина относительно M(III), причем степень отклонения хелатного узла MN4 от компланарности в некоторых случаях весьма значительна. При этом в ряду Sc–Ni имеет место рост суммы четырех валентных углов (N1M1N2), (N2M1N3), (N3M1N4) и (N4M1N1) (VAS) от 322.4° (Sc) до 358.8° (Co, Ni) (табл. 1) и соответственно – снижение степени “пирамидальности” хелатного узла (или, что в данном случае то же самое, его некомпланарности); при переходе же от Ni к Cu эта сумма немного (на 0.8°) снижается. Составляющие же хелатный узел валентные углы в каждом из этих металлохелатов, как правило, равны между собой (но всегда меньше 90.0°); единственным исключением здесь оказывается комплекс Mn(III), в котором имеет место попарное равенство этих углов [причем, что любопытно – углов (N1M1N2) и (N2M1N3), (N3M1N4) и (N4M1N1), т.е. углов, смежных друг с другом]. В намного меньшей степени проявляется некомпланарность группировки, включающей четыре донорных атома азота, ибо в семи комплексах типа II из девяти рассматриваемых в данной статье сумма четырех невалентных углов (NVAS), этими атомами образованных, равна точно 360.0°, а в тех случаях, когда это не так, а именно в комплексах Ti(III) и V(III), эта сумма отличается от только что указанной всего на 0.6°; при этом в большинстве случаев эти невалентные углы равны 90.0°. Интересно в связи с этим отметить, что вышеуказанная сумма невалентных углов равна 360.0° и в комплексе Mn(III), в котором три угла из четырех не равны между собой.
Входящие в состав каждого из таких координационных соединений 6-членные металлохелатные циклы ни в одном из них не являются строго компланарными [отклонение суммы внутренних углов в них (VAS6) от суммы внутренних углов в плоском шестиугольнике (720°) варьируется от 0.7° в комплексе Ni(III) до 10.1° в комплексе Sc(III)] (табл. 1). В большинстве случаев в соответствии с теоретическими ожиданиями исходя из структурной формулы II все четыре металлохелатных цикла в конкретно взятом комплексе идентичны друг другу как по сумме внутренних валентных углов, так и по их наборам. Исключением опять-таки оказывается комплекс Mn(III), где циклы обнаруживают лишь попарную идентичность (см. Supplementary Materials). Справедливости ради надо отметить, что различие в значениях VAS6 для этих циклов весьма незначительно (718.0° и 718.4° соответственно). В отличие от металлохелатных циклов, 6-членные нехелатные циклы, содержащие лишь атомы углерода, являются идеально плоскими [значения VAS6 в каждом из них в точности совпадают с суммой внутренних углов в плоском шестиугольнике (720°) и все они совершенно идентичны друг другу по наборам внутренних углов. Аналогичная идентичность наблюдается и для 5-членных нехелатных циклов, содержащих один атом азота и четыре атома углерода; все они являются строго компланарными [суммы валентных углов в каждом из них (VAS5) в точности совпадают с суммой внутренних углов в плоском пятиугольнике (540°)] и совершенно идентичными друг другу по наборам внутренних углов (см. Supplementary Materials).
Как уже упоминалось выше, металлохелатный узел MN4 ни в одном из этих металлокомплексов не является строго плоским; в связи с этим можно ожидать, что валентные углы, образованные атомами фтора, комплексообразователем M и любым из донорных атомов азота, не будут равны 90°. Представленные в табл. 1 данные на этот счет полностью согласуются с указанным ожиданием. При этом в шести из девяти изучаемых нами комплексов все эти четыре угла равны между собой, еще в двух – комплексах Ti(III) и V(III) углы (FMN) равны лишь попарно, а в комплексе Mn(III) меж собой равны лишь два угла из четырех (табл. 1). Различие в величинах этих углов в этих комплексах оказывается более 2°.
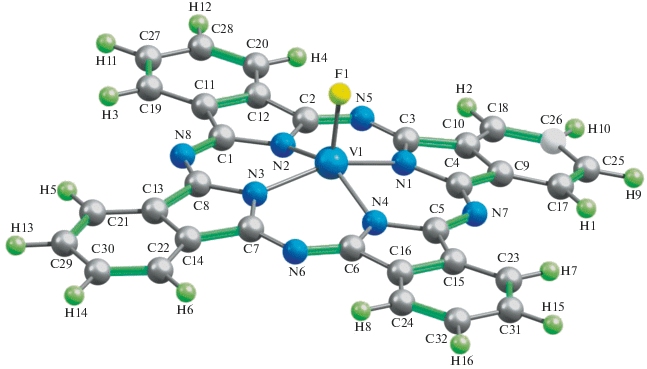
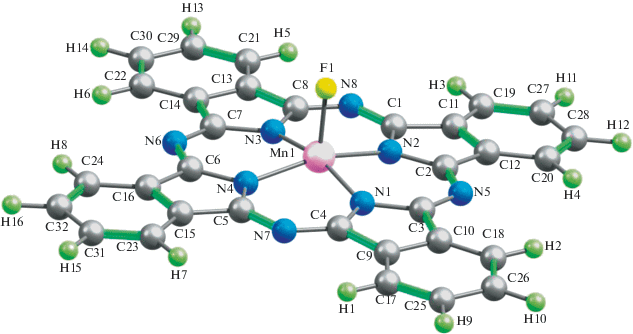
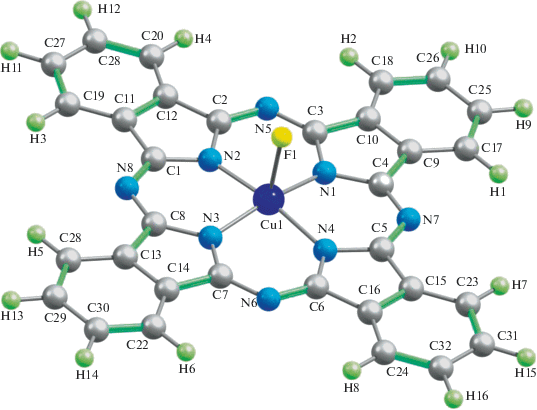
Несмотря на указанные выше различия в количественных параметрах молекулярных структур рассматриваемых металлокомплексов, внешне они мало отличаются друг от друга. Примеры этих структур представлены на рис. 1–3; на них отчетливо видна их довольно-таки заметно выраженная “пирамидальность” металлохелатного узла MN4. Расчет электрических моментов диполя рассматриваемых соединений (μ) по методу OPBE/TZVP дает для комплекса Sc(III) значение 2.56 ед. Дебая, для комплекса Ti(III) – 1.71, V(III) – 2.01, Cr(III) – 2.63, Mn(III) – 2.16, Fe(III) – 2.85, Co(III) – 2.39, Ni(III) – 3.19 и Cu(III) – 4.77 ед. Дебая. Как можно видеть из приведенных цифр, они сами по себе довольно-таки значительны, что, однако, вполне объяснимо, если учесть факт отсутствия центра симметрии в любом из этих металлокомплексов. В принципе можно было бы ожидать определенной взаимосвязи между значениями валентных углов (FMN) и величинами μ, однако таковая, как нетрудно заметить, для таких структурных параметров в комплексах типа II не обнаруживается. Не обнаруживается какой-либо взаимосвязи также между “традиционными” параметрами ионов M(III) (ионными радиусами, энергиями ионизации, типичными значениями координационных чисел комплексообразователя M(III) и т.п.) и представленными в табл. 1 структурными параметрами рассматриваемых нами металлохелатов; судя по всему, трактовку этих данных следует искать именно в специфике волновых функций, описывающих основное (а возможно, и возбужденные) состояния соответствующих M(III) и/или комплексов типа II, ими образуемых.
Значения ключевых термодинамических параметров рассматриваемых нами металлохелатов типа II, а именно стандартных энтальпии образования $\Delta H_{{\operatorname{f} ,298}}^{0}$, стандартной энтропии образования $S_{\operatorname{f} }^{0}$, 298 и стандартной энергии Гиббса образования $\Delta G_{{\operatorname{f} ,298}}^{0}$ представлены в Supplementary Materials, Table 2 . Как можно видеть из этой таблицы, величина $\Delta H_{{\operatorname{f} ,298}}^{0}$ отрицательна для пяти из девяти комплексов, а именно для хелатов Sc(III), Ti(III), V(III), Cr(III) и Mn(III), для остальных же она положительна; величина же $\Delta G_{{\operatorname{f} ,298}}^{0}$ отрицательна лишь для первых двух из них. Данное обстоятельство свидетельствует о невозможности образования большинства изучаемых нами металлохелатов из простых веществ тех химических элементов, которые входят в их состав. Тем не менее согласно данным проведенного нами расчета методом DFT OPBE/TZVP оптимизированные молекулярные структуры каждого из рассматриваемых здесь комплексов устойчивы и отвечающие им химические соединения в принципе способны к самостоятельному существованию. Это дает нам возможность надеяться, что описанные в данной статье координационные соединения будут обнаружены также и в эксперименте.
ЗАКЛЮЧЕНИЕ
Как следует из всего вышеизложенного, все рассмотренные нами в данной статье макроциклические металлохелаты M(III) (M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu) типа II в целом являются некомпланарными, что прежде всего связано с отклонением (подчас весьма значительным) от компланарности хелатного узла MN4. Неплоскими в этих комплексах оказываются также все имеющиеся в них шестичленные металлохелатные циклы; пятичленные же циклы, напротив, отклонений от компланарности не обнаруживают. При этом степени некомпланарности как самих входящих в состав хелатов II шестичленных металлохелатных циклов, так и макроцикла в целом для различных комплексообразователей M(III) 3d-элементов довольно-таки существенно различаются между собой, что свидетельствует о достаточно хорошо выраженной зависимости этого параметра от природы входящего в состав комплекса атома M. Как правило, в этих комплексах длины связей между донорными атомами азота и атомом M или совершенно одинаковы, или по крайней мере попарно равны между собой; на этом фоне выделяется комплекс Mn(III), в котором меж собой равны лишь две такие связи из четырех. В тех случаях, где электронная конфигурация M(III) допускает возможность реализации высоко- и низкоспиновых комплексов [Mn(III), Fe(III), Co(III), Ni(III) и Cu(III)], для комплексов типа II всегда реализуется низкоспиновое основное состояние. Во всех этих комплексах имеет место весьма сильно выраженная делокализация электронной плотности в пределах всего макроцикла, что находит свое отражение в довольно низких значениях эффективных зарядов как на комплексообразователях, так и на других слагающих макроцикл атомов.
Все квантово-химические расчеты были осуществлены в Казанском отделении Межведомственного суперкомпьютерного центра Российской академии наук – филиал Федерального государственного учреждения “Федеральный научный центр Научно-исследовательский институт системных исследований Российской академии наук” (http://www.jscc.ru), которому авторы выражают свою искреннюю признательность за оказанную техническую поддержку.
Вклад Чачкова Д.В. финансировался в рамках государственного задания Федерального научного центра “Научно-исследовательского института системных исследований РАН”.
Авторы подтверждают отсутствие конфликта интересов с какими-либо физическими или юридическими лицами.
Список литературы
Kasuda K., Tsutsui M. // Coord. Chem. Revs. 1980. V. 32. № 1. P. 67.
Thomas A.L. Phthalocyanines. Research & Applications, CRC Press, 1990.
Sliva W., Mianovska B. // Transit. Met. Chem. 2000. V. 25. № 5. P. 491.
Mamardashvili G.M., Mamardashvili N.Z., Koifman O.I. // Russ. Chem. Revs. 2008. V. 77. № 1. P. 59.
Ломова Т.Н. Аксиально координированные металлопорфирины в науке и практике. М.: URSS-КРАСАНД, 2018. 700 с.
Khelevina O.G., Malyasova A.S. // J. Porphyrins & Phthalocyanines, 2019. V. 23. № 11. P. 1251.
Kikuko Okada, Atsumi Sumida, Rie Inagaki, Masahiko Inamo // Inorg. Chim. Acta. 2012. V. 392. № 1. P. 473.
Colomban C., Kudric E.V., Afanasiev P., Sorokin A.B. // J. Am. Chem. Soc. 2014. V. 136. № 32. P. 11321. https://doi.org/10.1021/ja505437h
Buchler J.W., Rohbock K. // Inorg. Nucl. Chem. Lett. 1972. V. 8. № 12. P. 1073.
Guilard R., Richard P., El Borai M., Laviron E. // J. Chem. Soc., Chem Commun., 1980. № 11. P. 516. https://doi.org/10.1039/C39800000516
Lecomte C., Protas J., Richard P. et al. // J. Chem. Soc. Dalton Trans. 1982. № 2. P. 247. https://doi.org/10.1039/DT9820000247
Stuzhin P.A. // Fluorine in Heterocyclic Chemistry, Ed. V.G. Nenajdenko, V. 1, 5-Membered Heterocycles and Macrocycles, Springer, Heidelberg, 2014. P. 621.
Lebedeva (Yablokova) I.A., Ivanova S.S., Zhabanov Y.A. et al. // J. Fluor. Chem. 2018. V. 214. № 1. P. 86.
Chachkov D.V., Mikhailov O.V. // Russ. J. Inorg. Chem. 2013. V. 58. № 2. P. 174. https://doi.org/10.1134/S0036023613020186
Chachkov D.V., Mikhailov O.V. // Russ. J. Inorg. Chem. 2014. V. 59. № 3. P. 218. https://doi.org/10.1134/S0036023614030024
Mikhailov O.V., Chachkov D.V. // Russ. J. Inorg. Chem. 2015. V. 60. № 11. P. 1354. https://doi.org/10.1134/S003602361511011X
Schaefer A., Horn H., Ahlrichs R. // J. Chem. Phys. 1992. V. 97. № 4. P. 2571. https://doi.org/10.1063/1.463096
Schaefer A., Huber C., Ahlrichs R. // Ibid. 1994. V. 100. № 8. P. 5829. https://doi.org/10.1063/1.467146
Hoe W.-M., Cohen A., Handy N.C. // Chem. Phys. Lett. 2001. V. 341. № 1. P. 319. https://doi.org/10.1016/S0009-2614(01)00581-4
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865. https://doi.org/10.1103/PhysRevLett.77.3865
Paulsen H., Duelund L., Winkler H. et al. // Inorg. Chem. 2001. V. 40. № 9. P. 2201. https://doi.org/10.1021/ic000954q
Swart M., Groenhof A.R., Ehlers A.W., Lammertsma K. // J. Phys. Chem. A. 2004. V. 108. № 25. P. 5479. https://doi.org/10.1021/jp049043i
Swart M., Ehlers A.W., Lammertsma K. // Mol. Phys. 2004. V. 102. № 23. P. 2467. https://doi.org/10.1080/0026897042000275017
Swart M. // Inorg. Chim. Acta, 2007. V. 360. № 1. P. 179. https://doi.org/10.1016/j.ica.2006.07.073
Gaussian 09, Revision A.01, Frisch M.J, Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li H., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., Jr., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas O., Foresman J.B., Ortiz J.V., Cioslowski J., and Fox D.J., Gaussian, Inc., Wallingford CT, 2009.
Ochterski J.W. Thermochemistry in Gaussian. Gaussian, Inc., Wallingford CT, 2000.
Дополнительные материалы
- скачать ESM.docx
- Дополнительные материалы
Инструменты
Журнал физической химии