

Журнал физической химии, 2021, T. 95, № 4, стр. 549-557
Кинетические закономерности медленного переноса протонов от β-замещенных порфиразинов к органическим основаниям
a Ивановский государственный химико-технологический университет
Иваново, Россия
* E-mail: poa@isuct.ru
Поступила в редакцию 06.02.2020
После доработки 29.07.2020
Принята к публикации 06.10.2020
Аннотация
Рассмотрены закономерности межмолекулярного переноса протонов от β-замещенных порфиразинов к диметилсульфоксиду, циклическим и ациклическим азотсодержащим основаниям в инертных растворителях. Установлены необычно низкие скорости процесса. Показано влияние кислотных свойств порфиразинового макроцикла, протоноакцепторной способности основания, а также диэлектрической проницаемости среды на кинетические параметры кислотно-основного взаимодействия. Обсуждены вопросы строения и устойчивости комплексов с переносом протонов порфиразинов.
Изучение физико-химических свойств порфиразиновых макроциклов (Н2РА) является предметом все более пристального внимания исследователей, поскольку наличие четких представлений о межмолекулярных взаимодействиях, протекающих с участием Н2РА, создает хорошую базу для использования результатов эксперимента в практических целях. В настоящее время порфиразины, благодаря разнообразным возможностям модификации их структуры, нашли применение в качестве жидкокристаллических, каталитических и сенсорных материалов, фотосенсибилизаторов синглетного кислорода, гасителей флуоресценции, нелинейной оптике и др. [1]. Расширить спектр полезных свойств этого класса соединений возможно при всестороннем изучении кислотно-основных взаимодействий, в которых Н2РА проявляют ряд особенностей, не свойственных порфиринам [2].
Кислотные свойства β-замещенных порфиразинов
Под действием сильных оснований (гидроокись тетрабутиламмония) в среде диметилсульфоксида Н2РА подвергаются двухстадийной кислотной ионизации по внутрициклическим NH-связям, приводящей к образованию депротонированных дианионных форм [3]. При этом величины рKа1 и рKа2 достаточно сильно зависят от сольватационных эффектов. Сравнение кислотных свойств порфиразинов в газовой фазе наиболее достоверно отражает взаимосвязь между протонодонорной способностью макроцикла и его строением, поскольку исключает влияние свойств среды [4].
Порфиразин (рKа1 = 12.36), благодаря наличию четырех мезо-атомов азота, обладает выраженными кислотными свойствами по NH‑связям в отличие от порфина (рKа1 = 22.35). Введение в β-положения порфиразинового макроцикла электронодонорных заместителей приводит к увеличению рKа1, а электроноакцепторных – к ее уменьшению. Для тетрабромпорфиразина (H2PaBr4) и тетрахлорпорфиразина (H2PaCl4) величины pKа1 равны 8.45 и 9.09 соответственно. Менее сильное влияние на кислотность молекулы оказывает фенильное замещение в порфиразине. Для октафенилпорфиразина (Н2Ра(С6Н5)8) величина pKa1 = 10.36. Дальнейший рост протонодонорной способности следует ожидать при введении в фенильные кольца Н2Ра(С6Н5)8 электроноакцепторных заместителей. Однако термодинамические данные кислотной ионизации для окта(п-бромфенил)порфиразина (Н2Ра(С6Н4Br)8), окта(п-нитрофенил)порфиразина (Н2Ра(С6Н4NO2)8), окта(м‑трифторметилфенил)порфиразина (Н2Ра(С6Н4CF3)8), гекса(м-трифторметилфенил)бензопор-фиразина (Н2Ра(С6Н4CF3)6)(С4Н4)) и тетра(м-трифторметилфенил)дибензо-порфиразина (Н2Ра(С6Н4CF3)4)(С4Н4)2) отсутствуют.
Сведения о протонодонорной активности этих порфиразинов были получены при изучении их реакционной способности в процессах кислотно-основного взаимодействия с органическими основаниями, в качестве которых были взяты пиридин (Py), 2-метилпиридин (МеPy), морфолин (Mor), бензиламин (BzNH2), пиперидин (Pip), н-бутиламин (BuNH2), трет-бутиламин (ButNH2), диэтиламин (Et2NH), триэтиламин (Et3N), три-н-бутиламин (Bu3N) и диметилсульфоксид (DMSO).
Спектральная картина кислотно-основного взаимодействия с участием β-замещенных порфиразинов
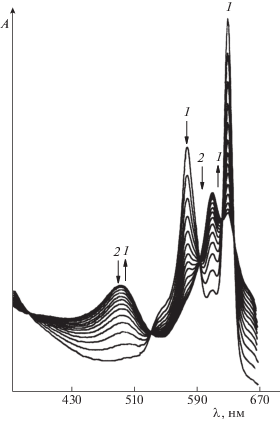
Взаимодействие порфиразинов с азотсодержащими основаниями и DMSO в инертном растворителе наблюдается только в условиях значительного избытка основания [5–15]. При этом спектральные изменения, сопровождающие реакцию, не зависят от его природы. Так, электронный спектр поглощения (ЭСП) H2PaBr4 в среде нейтрального бензола имеет в видимой области расщепленную Q-полосу с λI и λII при 643 и 577 нм соответственно (D2h-симметрия π-хромофора молекулы). При введении в бензол добавок трет-бутиламина с течением времени регистрируется уменьшение интенсивности Qх- и Qу-составляющих Q-полосы и одновременный рост интенсивности полосы поглощения с λ = 607 нм, характерной для D4h-симметрии порфиразинового макроцикла (рис. 1, изменение 1).
Рис. 1.
Изменение ЭСП H2PaBr4 в присутствии трет-бутиламина в течение 10 мин при 313 К и ${{С}_{{{\text{B}}{{{\text{u}}}^{t}}{\text{N}}{{{\text{H}}}_{{\text{2}}}}}}}$ = 1.20 моль/л в бензоле [9].
В системе бензол–DMSO спектральная картина не претерпевает существенных изменений. В ЭСП H2Pa(C6H4CF3)8 в присутствии морфолина наблюдается уменьшение интенсивности Qх- и Qу-полос поглощения с λI = 659 и λII = 594 нм и одновременный рост полосы поглощения с λ = = 632 нм (рис. 2, изменение 1). Аналогичная картина сохраняется для H2PaCl4 [9], H2Pa(C6H4Br)8 [8, 14] и H2Pa(C6H4NO2)8 [8, 14]. Электронный спектр поглощения порфиразинов с различными заместителями в пиррольных кольцах как в бензоле, так и в системе бензол–DMSO также имеет в видимой области расщепленную Q-полосу с λI = 690, λII = 636, λIII = 585 нм для H2Pa(C6H4CF3)6(С4Н4) и λI = 731, λII = 565 нм для H2Pa(C6H4CF3)4(С4Н4)2. В присутствии азотсодержащих оснований интенсивность расщепленной Q-полосы уменьшается на фоне роста полос поглощения с λI = 664, λII = 619 и λI = 711, λII = 590 нм для H2Pa(C6H4CF3)6(С4Н4) и H2Pa(C6H4CF3)4(С4Н4)2 соответственно (рис. 3, 4; изменение 1).
Рис. 2.
Изменение ЭСП Н2Ра(С6Н4CF3)8 в присутствии морфолина в течение 30 мин при 323 К и СMor = = 10.90 моль/л в системе бензол–5% DMSO [13].
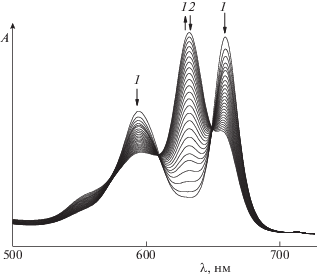
Рис. 3.
Изменение ЭСП Н2Ра(С6Н4CF3)6(С4Н4) в присутствии н-бутиламина в течение 90 мин при 338 К и ${{С}_{{{\text{BuN}}{{{\text{H}}}_{{\text{2}}}}}}}$ = 2.53 моль/л в бензоле [11].
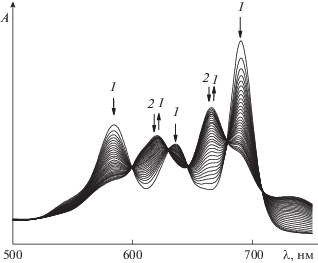
Рис. 4.
Изменение ЭСП Н2Ра(С6Н4CF3)6(С4Н4)2 в присутствии пиперидина в течение 210 мин при 328 К и СPip = 2.54 моль/л в системе бензол–5% DMSO [12].
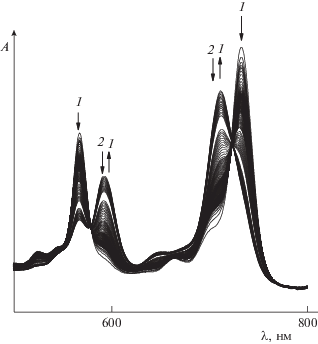
Происхождение Q-полосы и ее составляющих связано с π → π*-электронными переходами между высшими заполненными и низшими вакантными молекулярными орбиталями [16], которые в ходе кислотно-основного взаимодействия изменяются по энергии. Это приводит к изменению симметрии порфиразинового макроцикла от D2h до D4h [17]. Наблюдаемые спектральные изменения (рис. 1–4), указывающие на повышение симметрии молекулы от D2h до D4h, как и при образовании металлокомплексов [18, 19], свидетельствуют о том, что исследованные порфиразины в присутствии органических оснований проявляют свойства двухосновных NH-кислот и образуют комплексы с переносом протонов – Н2РА·2В.
Строение и устойчивость комплексов с переносом протонов β-замещенных порфиразинов
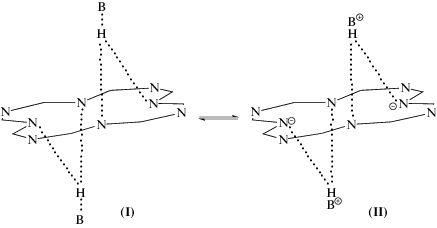
В комплексах Н2РА·2В протоны NH-групп, связанные с молекулами оснований и двумя внутрициклическими атомами азота располагаются над и под плоскостью макроцикла [20, 21], что обеспечивает соблюдение D4h-симметрии π-хромофора молекулы (рис. 1–4). При этом в малополярном бензоле (хлорбензоле) перенос протонов от NH-кислоты к основанию, приводящий к образованию разделенных растворителем ионных пар с последующей их диссоциацией представляется маловероятным [22]. Кислотно-основное взаимодействие, скорее всего, ограничивается либо стадией образования Н-комплекса (Н-ассоциата I), либо ионного комплекса (ион-ионного ассоциата), представляющего собой Н-связанную ионную пару (II). По мере увеличения кислотных свойств макроцикла, а также протоноакцепторной способности основания и диэлектрической проницаемости среды следует ожидать смещение кислотно-основного равновесия (1) в сторону образования более полярной структуры:
Комплексы Н2РА·2В в бензоле и системе бензол–DMSO подвергаются достаточно быстрому распаду с течением времени. В ЭСП регистрируется уменьшение интенсивности Q‑полосы с λ = 607 нм для H2PaBr4, λ = 632 нм для H2Pa(C6H4CF3)8, λI = 664 и λII = 619 нм для H2Pa(C6H4CF3)6(С4Н4), а также с λI = 711 и λII = = 590 нм для H2Pa(C6H4CF3)4(С4Н4)2 (рис. 1–4, изменение 2). В случае H2PaBr4 низкоинтенсивная полоса поглощения с λ = 490 нм, свидетельствующая о наличии продуктов полураспада макроцикла–дипиррометенов, не претерпевает существенных изменений. В ЭСП раствора она исчезает только ~120 ч. При этом процесс деструкции сопровождается первоначальным изменением синей окраски раствора до желтой с последующим обесцвечиванием.
Аналогичная картина наблюдается для H2PaCl4 [9]. Для октафенилзамещенных порфиразинов и порфиразинов с β-замещением и β, β-бензоаннелированием промежуточные продукты полураспада спектрально не регистрируются. Наблюдается изменение ярко-зеленой окраски раствора до бесцветной. Напротив, комплексы H2PaBr4 ⋅ 2DMSO [5], H2PaCl4 ⋅ 2DMSO [5] и H2Pa(C6H4NO2)8 ⋅ 2DMSO [6] в системе DMSO–хлорбензол обладают высокой кинетической устойчивостью. Их ЭСП не претерпевают изменений в течение ~38 ч при Т = 328 К. Они достаточно легко вступают в реакции образования металлокомплексов порфиразинов с ацетатами натрия, магния, цинка, меди, кобальта и никеля [5, 6].
Кинетические особенности межмолекулярного переноса протонов β-замещенных порфиразинов
В зависимости от особенностей геометрического строения взаимодействующих молекул перенос протонов от порфиразинов к основаниям в бензоле (хлорбензоле) и системе бензол–DMSO может осуществляться по различным альтернативным механизмам.
Кислотно-основное взаимодействие H2PaBr4 с DMSO, циклическими (Py, MePy, Mor, Pip) и третичными ациклическими аминами (Et3N, Bu3N) в бензоле (хлорбензоле) [7, 23] описывается уравнением
(2)
$ - d{{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{PaB}}{{{\text{r}}}_{{\text{4}}}}}}}{\text{/}}d\tau = k{{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{PaB}}{{{\text{r}}}_{{\text{4}}}}}}}{{C}_{{\text{B}}}},$Кинетические данные указывают на бимолекулярный характер лимитирующей стадии процесса, а повышение симметрии π-хромофора молекулы от D2h до D4h (рис. 1, изменение 1) свидетельствуют о том, что перенос протонов от кислоты к основанию осуществляется в две стадии в соответствие со следующей схемой:
(4)
${{{\text{H}}}_{{\text{2}}}}{\text{PaB}}{{{\text{r}}}_{4}} + {\text{В}}\;\xrightarrow{{{{k}_{{\text{1}}}}}}\;{{({\text{HPaB}}{{{\text{r}}}_{{\text{4}}}})}^{ - }}{\kern 1pt} \cdots {\kern 1pt} ({\text{Н}}{{{\text{В}}}^{ + }}),$(5)
${{({\text{HPaB}}{{{\text{r}}}_{4}})}^{ - }}{\kern 1pt} \cdots {\kern 1pt} ({\text{Н}}{{{\text{В}}}^{{\text{ + }}}}) + {\text{В}}\;\xrightarrow{{{{k}_{2}}}}\;{{({\text{PaB}}{{{\text{r}}}_{4}})}^{{2 - }}}{\kern 1pt} \cdots {\kern 1pt} {{({\text{Н}}{{{\text{В}}}^{ + }})}_{2}}.$Молекула основания вступает во взаимодействие с одним из двух внутрициклических протонов NH-групп H2PaBr4 и осуществляет его вывод из плоскости макроцикла. При этом электронный спектр поглощения образующегося промежуточного комплекса – (HPaBr4)–⋅⋅⋅(НВ+) должен иметь вид ЭСП H2PaBr4 по числу полос, но расщепление Q-полосы должно уменьшаться за счет гипсохромного смещения ее длинноволновой компоненты Qх [17]. Однако подобные спектральные изменения в условиях значительного избытка основания в ходе реакции не наблюдаются (рис. 1, изменение 1). Этот факт дает основание полагать, что образование спектрально не регистрируемого комплекса – (HPaBr4)–⋅⋅⋅(НВ+) происходит медленнее, чем (PaBr4)2–⋅⋅⋅(НВ+)2, т.е . k1 < k2. Поскольку скорость кислотно-основного взаимодействия определяли по уменьшению оптической плотности раствора наиболее интенсивной полосы поглощения Qх (λI = 643 нм), то k1 = kн. Образующийся на стадии (5) комплекс с переносом протонов – (PaBr4)2–⋅⋅⋅(НВ+)2 представляет собой Н-связанную ионную пару (II). При этом не исключается, что она может находиться в равновесии (1) с Н-комплексом [22]. Аналогичный двухстадийный процесс реализуется при взаимодействии H2PaCl4 с DMSO, Py, MePy, Mor, Pip, Et3N и Bu3N в бензоле (хлорбензоле) [7, 9, 23, 24].
Напротив, взаимодействие H2PaBr4 и H2PaCl4 с первичными и вторичными ациклическими аминами (BzNH2, BuNH2, ButNH2, Et2NH) в бензоле [7, 9, 24] описывается суммарным кинетическим уравнением третьего порядка
(6)
$ - d{{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{PaB}}{{{\text{r}}}_{{\text{4}}}}}}}{\text{/}}d\tau = k{{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{PaB}}{{{\text{r}}}_{{\text{4}}}}}}}C_{{\text{B}}}^{2},$(8)
${\text{В}} + {\text{В}}\;\overset {{{K}_{p}}} \longleftrightarrow \;{\text{В}}{\kern 1pt} \cdots {\kern 1pt} {\text{В}},$(9)
${{{\text{H}}}_{{\text{2}}}}{\text{PaB}}{{{\text{r}}}_{{\text{4}}}} + {\text{В}}{\kern 1pt} \cdots {\kern 1pt} {\text{В}}\;\xrightarrow{{{{k}_{1}}}}\;{{({\text{PaB}}{{{\text{r}}}_{4}})}^{{2 - }}}{\kern 1pt} \cdots {\kern 1pt} {{({\text{Н}}{{{\text{В}}}^{ + }})}_{2}},$Из-за отсутствия значений Kр для первичных и вторичных ациклических аминов в бензоле нельзя полностью исключить возможность протекания процесса в две стадии с k1 ≈ k2 в соответствии со схемой (4), (5) . Представляется вполне вероятным, что этот процесс является наиболее предпочтительным для аминов (ButNH2, Et2NH), имеющих в своем составе объемные алкильные заместители, которые противодействуют процессу межмолекулярной ассоциации [25].
Для H2Pa(C6H4CF3)8 [10, 13], H2Pa(C6H4NO2)8 [8, 14], H2Pa(C6H4Br)8 [8, 14], H2Pa(C6H4CF3)6(С4Н4) [11, 15] и H2Pa(C6H4CF3)4(С4Н4)2 [12] схема механизма переноса протонов, согласно (9), не реализуется вследствие пространственных помех, создаваемых объемными заместителями в β-положениях порфиразинового макроцикла с одной стороны и димерными молекулами оснований – с другой. Реакция кислотно-основного взаимодействия описывается суммарным кинетическим уравнением второго порядка (2), а перенос протонов от кислоты к основанию осуществляется двухстадийно, согласно (4), (5). Следует однако отметить, что детальный механизм переноса протонов NH-групп β-замещенных порфиразинов к основанию представляется чрезвычайно сложным и требует дальнейшего углубленного изучения.
Порфиразины в отличие от порфиринов [2] вступают в кинетически контролируемые взаимодействия с основаниями, которые характеризуются необычно низкими значениями констант скорости и достаточно высокими значениями Еа процесса (табл. 1–4), не свойственными для подавляющего большинства относительно простых жидкофазных кислотно-основных систем [22, 26]. Причина этого явления связана с действием геометрической и электронной (поляризационной) составляющей порфиразинового макроцикла. Последняя способствует увеличению полярности NH-связей Н2РА за счет электроноакцепторного влияния четырех мезо-атомов азота и заместителей, находящихся в β-положениях макроцикла. В результате этого создаются благоприятные условия для переноса протонов от кислоты к основанию. Напротив, геометрическая составляющая изменяется несимбатно электронной. Высокая конформационная жесткость ароматической π-системы 16-членного макроцикла (C8N8) и наличие объемных заместителей в β-положениях пиррольных колец порфиразина способствует экранированию атомами и π-электронами внутрициклических протонов NH-групп. Это противодействует благоприятному контакту реакционных центров молекул–партнеров и вносит основной вклад в кинетику взаимодействия порфиразинов с основаниями.
Таблица 1.
Кинетические параметры кислотно-основного взаимодействия галогензамещенных порфиразинов с первичными и вторичными ациклическими аминами в бензоле [7, 9, 23]
| Порфиразин (Н2РА) | Основание (В) | $k_{{\text{н}}}^{{298}}$ × 104, c–1 | k298 × 102, л2/(моль2 c) |
Еа, кДж/моль |
|---|---|---|---|---|
| H2PaBr4 | Бензиламин | 2.05 | 0.78 | 29 |
| н-Бутиламин | 16.10 | 6.60 | 11 | |
| трет-Бутиламин | 0.20 | 0.07 | 18 | |
| Диэтиламин | 8.80 | 4.00 | 15 | |
| H2PaCl4 | Бензиламин | 1.40 | 0.65 | 31 |
| н-Бутиламин | 8.40 | 3.80 | 23 | |
| трет-Бутиламин | 0.16 | 0.09 | 28 | |
| Диэтиламин | 3.60 | 1.10 | 30 |
Таблица 2.
Кинетические параметры кислотно-основного взаимодействия галогензамещенных порфиразинов с циклическими и третичными ациклическими аминами в бензоле [7–11, 13, 23] и диметилсульфоксидом в хлорбензоле [5]
| Порфиразин (Н2РА) | Основание (В) | $k_{{\text{н}}}^{{298}}$ × 104, c–1 | k298 × 104, л/(моль c) |
Еа, кДж/моль |
|---|---|---|---|---|
| H2PaBr4 | Пиридин | 2.00 | 0.23 | 69 |
| 2-Метилпиридин | 0.15 | 0.02 | 87 | |
| Морфолин | 9.00 | 72.00 | 26 | |
| Пиперидин | 21 | 2830 | 20 | |
| Триэтиламин | 3.22 | 0.24 | 46 | |
| Три-н-Бутиламин | 0.09 | 0.09 | 26 | |
| Диметилсульфоксид | 1.63 | 0.35 | 24 | |
| H2PaCl4 | Пиридин | 2.55 | 0.36 | 40 |
| 2-Метилпиридин | 0.35 | 0.04 | 49 | |
| Морфолин | 6.20 | 80.00 | 28 | |
| Пиперидин | 17 | 2280 | 23 | |
| Триэтиламин | 5.90 | 1.10 | 30 | |
| Три-н-Бутиламин | 0.24 | 0.23 | 24 | |
| Диметилсульфоксид | 1.33 | 0.53 | 26 |
Таблица 3.
Кинетические параметры кислотно-основного взаимодействия β-замещенных порфиразинов с азотсодержащими основаниями в бензоле [8, 10, 11]
| Порфиразин (Н2РА) | Основание (В) | $k_{{\text{н}}}^{{298}}$ × 105, c–1 | k298 × 106, л/(моль c) |
Еа, кДж/моль |
|---|---|---|---|---|
| Н2Ра(С6Н4CF3)8 | Морфолин | 0.12 | 0.11 | 55 |
| Бензиламин | 0.09 | 0.23 | 56 | |
| Пиперидин | 1.50 | 6.15 | 32 | |
| н-Бутиламин | 0.20 | 4.20 | 32 | |
| трет-Бутиламин | 0.05 | 0.43 | 50 | |
| Н2Ра(С6Н4NO2)8 | Морфолин | 0.04 | 0.04 | 94 |
| Бензиламин | 0.18 | 0.045 | 85 | |
| Пиперидин | 0.24 | 1.20 | 78 | |
| н-Бутиламин | 0.18 | 1.80 | 86 | |
| трет-Бутиламин | 0.20 | 0.80 | 94 | |
| Н2Ра(С6Н4CF3)6(С4Н4) | Пиперидин | 0.17 | 0.55 | 64 |
| н-Бутиламин | 0.30 | 0.60 | 61 | |
| Н2Ра(С6Н4Br)8 | н-Бутиламин | 0.11 | 0.16 | 92 |
Таблица 4.
Кинетические параметры кислотно-основного взаимодействия β-замещенных порфиразинов с азотсодержащими основаниями в системе бензол–диметилсульфоксид [12–15]
| Порфиразин (Н2РА) | Основание (В) | DMSO,% | $k_{{\text{н}}}^{{298}}$ × 105, c–1 | k298 × 106, л/(моль c) |
Еа, кДж/ моль |
|---|---|---|---|---|---|
| Н2Ра(С6Н4CF3)8 | Морфолин | 0.5 | 5.60 | 25.50 | 32 |
| Пиперидин | 0.5 | 43 | 177 | 22 | |
| н-Бутиламин | 0.5 | 67 | 252 | 23 | |
| Н2Ра(С6Н4CF3)6(С4Н4) | Морфолин | 0.5 | 0.70 | 2.70 | 45 |
| Пиперидин | 0.5 | 4.00 | 16.00 | 28 | |
| н-Бутиламин | 0.5 | 8.00 | 30.00 | 21 | |
| Н2Ра(С6Н4CF3)6(С4Н4)2 | Морфолин | 0.5 | р-я не идет | ||
| Пиперидин | 0.5 | 0.06 | 0.50 | 74 | |
| н-Бутиламин | 0.5 | 0.07 | 0.60 | 58 | |
| Н2Ра(С6Н4NO2)8 | н-Бутиламин | 0.5 | 1.18 | 110 | 41 |
| 1.25 | 848 | 54000 | 21 | ||
| Н2Ра(С6Н4Br)8 | н-Бутиламин | 2.5 | 0.18 | 0.63 | 82 |
| 7.5 | 0.70 | 3.16 | 80 | ||
| 20 | 6.36 | 87.20 | 60 | ||
| 30 | 120 | 31600 | 28 | ||
| 40 | 370 | 99000 | 29 | ||
| Морфолин | 50 | 0.20 | 6.00 | 24 | |
| Бензиламин | 50 | 0.25 | 15.30 | 35 | |
| Диэтиламин | 50 | 5.10 | 185 | 19 | |
| Триэтиламин | 50 | 0.37 | 18.90 | 32 |
Достаточно сильное влияние на кинетические параметры переноса протонов оказывает пространственное строение основания и его протоноакцепторная способность. Так, с увеличением ${\text{р}}K_{{\text{a}}}^{{298}}$-оснований скорость переноса протонов H2PaBr4 возрастает, а Еа процесса значительно уменьшается. Среди циклических оснований максимальной реакционной способностью обладает пиперидин (${\text{р}}K_{{\text{a}}}^{{298}}$ = 11.23 [27]), который является сильным акцептором протона и имеет стерически доступный атом азота в составе молекулы [28]. Введение в пиперидиновый цикл гетероатома кислорода не влияет на пространственное строение амина, однако приводит к понижению ${\text{р}}K_{{\text{a}}}^{{298}}$ на ~2.5 единицы [29]. В результате этого при переходе от пиперидина к морфолину (${\text{р}}K_{{\text{a}}}^{{298}}$ = 8.50 [27]), величина k298 уменьшается в ~39 раз на фоне незначительного роста Еа-процесса (табл. 2). Уменьшение ${\text{р}}K_{{\text{a}}}^{{298}}$-оснований на ~6 единиц в ряду Pip → Mor → Py приводит к дальнейшему ингибированию переноса протона. Минимальная скорость наблюдаются при взаимодействии H2PaBr4 с 2-метилпиридином (${\text{р}}K_{{\text{a}}}^{{298}}$ = = 6.00 [27]) вследствие более сильного, чем в пиридине (${\text{р}}K_{{\text{a}}}^{{298}}$ = 5.23 [27]) пространственного экранирования неподеленной электронной пары азота метильной группой.
Аналогичная картина наблюдается при замене Et3N (${\text{р}}K_{{\text{a}}}^{{298}}$ = 10.75 [27]) на близкий по основности Bu3N (${\text{р}}K_{{\text{a}}}^{{298}}$ = 10.97 [27]) (табл. 2), а также BuNH2 (${\text{р}}K_{{\text{a}}}^{{298}}$ = 10.60 [27]) на Et2NH (${\text{р}}K_{{\text{a}}}^{{298}}$ = 10.84 [27]) (табл. 1). Необычно низкая реакционная способность три-н-бутиламина, как и 2-метилпиридина, связана с сильным экранированием алкильными группами атома азота, в результате чего контакт кислотного и основного центров взаимодействующих молекул оказывается затруднен. Наряду с увеличением числа и длины алкильных заместителей оптимальной пространственной ориентации кислотно-основных центров противодействует разветвление углеводородной цепи в амине. Так, скорости переноса протонов NH-групп от H2PaBr4 к BuNH2 и ButNH2 (${\text{р}}K_{{\text{a}}}^{{298}}$ = 10.68 [27]) различаются в ~94 раза (табл. 1). Диметилсульфоксид, несмотря на его менее выраженную протоноакцепторную способность по сравнению с азотсодержащими основаниями [29], по реакционной способности, судя по величинам k298 (табл. 2), близок к пиридину. Этот факт не является неожиданным, если принять во внимание, что атом кислорода в DMSO пространственно более доступен для протона [30], чем атом азота в Py, который экранирован шестицентровой π-связью. В результате этого взаимодействие H2PaBr4 с DMSO и Py характеризуются близкими значениями k298, но сопровождается увеличением Еа процесса на 14 кДж/моль (табл. 2).
Замена атомов брома на хлор в порфиразине не оказывает значительного влияния на кинетические параметры переноса протонов (табл. 1, 2). Как известно, влияние атомов галогена на кислотные NH-центры передается с полуизолированных Сβ = Сβ-связей по индуктивному (‒I) эффекту и за счет эффекта n, π-сопряжения с макроциклом (+M-эффект). –I-Эффект при переходе от брома к хлору увеличивается, что способствует росту полярности NH-связей. Действие +M-эффекта, возрастая в том же порядке, напротив способствует уменьшению подвижности протонов NH-групп. В результате электронные эффекты атомов галогена нивелируют кислотность H2PaBr4 и H2PaCl4 и не проявляются в процессе кислотно-основного взаимодействия.
При переходе от H2PaBr4 и H2PaCl4 к β-фенилзамещенным порфиразинам перенос протонов существенно затрудняется. Так, Н2Ра(С6Н4CF3)8 [10] и Н2Ра(С6Н4NO2)8 [8] оказываются неактивными в реакции с основаниями, обладающими слабовыраженной протоноакцепторной способностью (Py, MePy), а также с основаниями, имеющими сильно пространственно экранированный атом азота в амине (Et2NH, Et3N, Bu3N). Несмотря на структурную близость, Н2Ра(С6Н4CF3)8 в отличие от Н2Ра(С6Н4NO2)8 более легко вступает в кислотно-основное взаимодействие с Mor, BzNH2, Pip и BuNH2 в бензоле (табл. 3). Окта(п-бромфенил)порфиразин в реакции с основаниями менее активен, чем Н2Ра(С6Н4CF3)8 и Н2Ра(С6Н4NO2)8. Из всех изученных оснований он вступает во взаимодействие только с н-бутиламином (табл. 3). При этом скорость переноса протонов NH-групп от Н2Ра(С6Н4Br)8 и Н2Ра(С6Н4CF3)8 к BuNH2, судя по величинам k298, различается в ~26 раз, а в случае с Н2Ра(С6Н4NO2)8 в ~10 раз (табл. 3). По реакционной способности Н2Ра(С6Н4CF3)6)(С4Н4) занимает промежуточное положение между Н2Ра(С6Н4NO2)8 и Н2Ра(С6Н4Br)8. При переходе от Н2Ра(С6Н4NO2)8 к Н2Ра(С6Н4CF3)6(С4Н4) и от Н2Ра(С6Н4CF3)6(С4Н4) к Н2Ра(С6Н4Br)8 скорость кислотно-основного взаимодействия с BuNH2 в бензоле уменьшается в 3 и ~4 раза соответственно (табл. 3). Среди всех изученных порфиразинов минимальной реакционной способностью обладает Н2Ра(С6Н4CF3)4)(С4Н4)2. В отличие от Н2Ра(С6Н4Br)8 он не вступает в реакцию переноса протонов к н-бутиламину и более сильному основанию – пиперидину в бензоле [12]. Следовательно, наряду с уменьшением протоноакцепторной способности азотсодержащего основания и/или с увеличением пространственного экранирования атома азота в амине перенос протонов NH-групп от H2PA в бензоле существенно затрудняется с увеличением пространственного экранирования реакционного центра в макроцикле и/или с уменьшением кислотных свойств порфиразинов в ряду: H2PaCl4 ≈ H2PaBr4 → Н2Ра(С6Н4CF3)8 → → Н2Ра(С6Н4NO2)8 → Н2Ра(С6Н4CF3)6(С4Н4) → → Н2Ра(С6Н4Br)8 → Н2Ра(С6Н4CF3)4(С4Н4)2.
Кроме протонодонорной и протоноакцепторной способности взаимодействующих молекул перенос протонов от кислоты к основанию сильно зависит от полярности среды, которая определяется его диэлектрической проницаемостью (ε). Среда с более высоким значением ε способствует более быстрому образованию продукта кислотно-основного взаимодействия, увеличивая благодаря этому его концентрацию, а значит и скорость реакции [25]. При переходе от бензола к системе бензол – 5% DMSO скорость переноса протонов от Н2Ра(С6Н4CF3)8 к морфолину, пиперидину и н‑бутиламину, судя по величинам k298, значительно возрастает на фоне уменьшения Еа-процесса (табл. 3, 4). При концентрации DMSO в бензоле в количестве более 0.5% реакция между Н2Ра(С6Н4CF3)8 и Mor (BuNH2, Pip) проходит практически мгновенно.
Аналогичное изменение величин k298 и Еа от полярности среды наблюдается при взаимодействии Н2Ра(С6Н4NO2)8 и Н2Ра(С6Н4Br)8 с н-бутиламином (табл. 4). При концентрации DMSO в бензоле в количестве 2.5 и 50% для Н2Ра(С6Н4NO2)8 [14] и Н2Ра(С6Н4Br)8 [14] соответственно константа скорости реакции резко возрастает причем так, что ее значение уже невозможно измерить обычными кинетическими методами. Напротив, в системе бензол – 50% DMSO реакция Н2Ра(С6Н4Br)8 с Mor (${\text{р}}K_{{\text{a}}}^{{298}}$ = 8.50 [27]) и BzNH2 (${\text{р}}K_{{\text{a}}}^{{298}}$ = 9.34 [27]) характеризуется достаточно низкими значениями констант скорости вследствие их пониженной протоноакцепторной способности по сравнению с BuNH2 (${\text{р}}K_{{\text{a}}}^{{298}}$ = 10.60 [27]). Как и следовало ожидать, увеличение ${\text{р}}K_{{\text{a}}}^{{298}}$ оснований на ~2 единицы в ряду Mor → BzNH2 → → Et2NH приводит к росту величин k298 в ~30 раз (табл. 4). При этом замена Et2NH (${\text{р}}K_{{\text{a}}}^{{298}}$ = 10.84 [27]) на близкий по основности Et3N (${\text{р}}K_{{\text{a}}}^{{298}}$ = = 10.75 [27]) существенно затрудняет кислотно-основное взаимодействие (табл. 4) вследствие менее благоприятной стерической доступности неподеленной электронной пары атома азота в амине. В системе бензол–5% DMSO перенос протонов Н2Ра(С6Н4CF3)8 к основаниям, обладающим слабовыраженной протоноакцепторной способностью (Py, MePy) или имеющим в своем составе пространственно экранированный атом азота (Et2NH, Et3N) не происходит [27].
Бензоаннелирование в порфиразиновом макроцикле также затрудняет перенос протона вследствие уменьшения кислотных свойств молекулы. В ряду Н2Ра(С6Н4CF3)8 → Н2Ра(С6Н4CF3)6(С4Н4) → → Н2Ра(С6Н4CF3)4(С4Н4)2 скорость переноса протонов к Pip и BuNH2 в системе бензол–5% DMSO уменьшается в 354 и 420 раз соответственно, а Еа процесса возрастает (табл. 4).
Следовательно, если молекулы-партнеры обладают слабовыраженными протонодонорными и/или протоноакцепторными свойствами и имеют при этом пространственно-экранированный реакционный центр, то увеличение диэлектрической проницаемости среды не играет ключевой роли в процессе преноса протонов от β-замещенных порфиразинов к органическим основаниям.
Список литературы
Novakova V., Donzello P.A., Ercolani C. et al. // Coord. Chem. Rev. 2018. V. 361. № 4. P. 1.
Березин Д.Б. Макроциклический эффект и структурная химия порфиринов. М.: Красанд, 2010. 424 с.
Успихи химии порфиринов / Под ред. О.А. Голубчикова. НИИ Химии СПбГУ, 2001. Т. 3. 359 с.
Stuzhin P.A. // J. Porphyrins Phthalocyanines. 2003. V. 7. № 12. P. 813.
Петров О.А., Хелевина О.Г., Чижова Н.В. // Координац. химия. 1997. Т. 23. № 2. С. 143.
Петров О.А., Чижова Н.В., Карасева Н.А. // Там же. 1999. Т. 25. № 6. С. 415.
Петров О.А., Чижова Н.В. // Там же. 1999. Т. 25. № 5. С. 393.
Петров О.А. // Журн. физ. химии. 2002. Т. 76. № 9. С. 1577.
Петров О.А. // Координац. химия. 2003. Т. 29. № 2. С. 144.
Петров О.А., Кузмина Е.Л., Хелевина О.Г., Майзлиш В.Е. // Журн. физ. химии. 2011. Т. 85. № 9. С. 1696.
Петров О.А., Кузмина Е.Л. // Там же. 2012. Т. 86. № 12. С. 1958.
Петров О.А. // Там же. 2015. Т. 89. № 2. С. 214.
Петров О.А., Осипова Г.В., Аганичева К.А. // Там же. 2020. Т. 94. № 1. С. 40.
Петров О.А., Чижова Н.В., Осипова Г.В. // Журн. общ. химии. 2009. T. 79. Вып. 4. С. 676.
Петров О.А. // Журн. физ. химии. 2017. Т. 91. № 11. С. 1845.
Toyota K., Hasegawa J., Nakatsuji H. // Chem. Phys. Lett. 1996. V. 250. № 5–6. P. 437.
Stuzhin P., Khelevina O., Berezin B. // Phthalocyanines: Properties and Applications. N.Y.: VCH Publ. Inc., 1996. V. 4. P. 23.
Stuzhin P.A., Ivanova S.S., Koifman O.I. et al. // Inorg. Chem. Com. 2014. V. 49. № 9. P. 72.
Vagin S.I., Hanack M. // Eur. J. Org. Chem. 2002. № 16. P. 2859.
Kokareva E.A., Petrov O.A., Khelevina O.G. // Macroheterocycles. 2009. V. 2. № 2. P. 157.
Петров О.А., Аганичева К.А., Гамов Г.А., Киселев А.Н. // Журн. физ. химии. 2020. Т. 94. № 9. С. 1379.
Молекулярные взаимодействия / Под ред. Г. Ратайчака, У. Орвилл-Томаса. М.: Мир, 1984. 599 с.
Петров О.А., Березин Б.Д. // Журн. физ. химии. 1999. Т. 73. № 5. С. 830.
Петров О.А., Хелевина О.Г., Чижова Н.В., Березин Б.Д. // Координац. химия. 1994. Т. 20. № 11. С. 876.
Райхардт К. Растворители и эффекты среды в органической химии. М.: Мир, 1991. 764 с.
Базилевский М.В., Венер М.В. // Успехи химии. 2003. Т. 72. № 1. С. 3.
CHC Handbook of Chemistry and Physics / Ed. by William M. Haynes. 2013. 2668 p.
Anet F.A.L., Yavari I. // J. Amer. Chem. Soc. 1977. V. 99. P. 2794.
Blackburne I.D., Katritzky A.R., Takeuchi. Y. // Accounts. Chem. Res. 1975. V. 8. № 9. P. 300.
Получение и свойства органических соединений серы / Под ред. Л. И. Беленького. М.: Химия, 1999. 557 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии