

Журнал физической химии, 2021, T. 95, № 8, стр. 1225-1231
Механизм и термодинамические характеристики дегидратации катиона ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$
А. А. Дегтярев a, *, Т. П. Дьячкова a, Д. П. Ростова a, А. В. Рухов a
a Тамбовский государственный технический университет
392000 Тамбов, Россия
* E-mail: ad.dycost@gmail.com
Поступила в редакцию 08.10.2020
После доработки 24.11.2020
Принята к публикации 03.12.2020
Аннотация
Представлены результаты оценки адекватности различных функционалов DFT для описания реакций изомеризации и дегидратации катиона ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$, выбран наиболее корректный метод. Проведено исследование механизма реакции образования катиона ${\text{HSO}}_{3}^{ + }$ в вакууме и при неявном учете растворителя по модели COSMO, в качестве растворителя выбрана безводная серная кислота, а также олеум с 5 и 15%-ным (мас. %) содержанием SO3. Оценены термодинамические характеристики реакции образования катиона ${\text{HSO}}_{3}^{ + }$. Показано, что в интервале температур от 25 до 100°С протекание этого процесса как в вакууме, так и в растворителе, маловероятно.
Катион ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$ – продукт автопротолиза серной кислоты [1] – имеет важное значение и как самостоятельный сульфирующий агент, и как промежуточная частица при образовании более сильного сульфирующего агента – частицы ${\text{HSO}}_{3}^{ + }$ [2]. Катион ${\text{HSO}}_{3}^{ + }$ может получаться из ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$ путем отщепления молекулы воды. Цель настоящего исследования – установление механизма реакции дегидратации ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$, а также влияния растворителя на энергетический барьер этой реакции.
Ранее в [3] методами Хартри–Фока, DFT, теории возмущений и экспериментально изучалась газофазная дегидратация катиона ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$. Экспериментальное значение энергии превращения ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$ → H2O + ${\text{HSO}}_{3}^{ + }$ составило 225 ± ± 18 кДж/моль, теоретическое – 148 кДж/моль (MP2/6-311+G(d,p)). Различие между реальными и вычисленными значениями объяснялось малым количеством поляризационных функций при расчете катиона ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$. В настоящей работе проведено сравнение различных функционалов DFT и метода MP2 с целью выбора наиболее корректно описывающего превращения в данной системе. В качестве эталонного метода для оценки адекватности использован метод связанных кластеров.
МЕТОДОЛОГИЧЕСКАЯ ЧАСТЬ
Катион ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$, образующийся при автопротолизе, имеет структуру с равнозначными связями S–O–H вида [1, 4]:
Согласно [3], при изомеризации возникает катион вида:
При отщеплении от него молекулы воды образуется катион ${\text{HSO}}_{3}^{ + }$.
Таким образом, в качестве объектов моделирования выбраны:
• исходный катион (${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$);
• его изомер (H2O–SO2OH+);
• переходное состояние, соединяющее их (TS);
• отдельные частицы ${\text{HSO}}_{3}^{ + }$ и H2O.
В качестве функционалов применены наиболее часто используемые из каждой группы: PBE [5] (GGA функционалы), M-06L [6] (meta-GGA- функционалы), B3LYP5 [7], PBE0 [8] (глобальные гибридные GGA-функционалы), M06, M06-2X [9] (глобальные гибридные meta-GGA-функционалы), LC-BLYP [10], ωB97x [11] (пространственно-разделенные гибридные функционалы) и B2T-PLYP [12] (дважды гибридные функционалы).
Для учета сил дисперсионного взаимодействия выбрана коррекция Гримме по методам D3BJ [131] (PBE, B3LYP5, PBE0, ωB97x), D3Zero [14] (M-06L, M06, M06-2X) и D2 [15] (LC-BLYP).
Во всех случаях использованы базисные наборы семейства Даннинга с добавлением диффузионных функций aug-cc-pVXZ [16, 17]. Для методов DFT применены наборы до трехэкспоненциальных включительно (X = D, T), для методов теории возмущений и связанных кластеров – до пятиэкспоненциальных (X = D, T, Q, 5).
При использовании метода связанных кластеров выбрана его реализация DLPNO-CCSD(T) [18]. Также использовано приближение к полному базисному набору по методу F-12 [19] (DLPNO-CCSD(T)-F12D) в сочетании с базисом cc-pVDZ-F12 [20].
Учет влияния растворителя осуществлен по континуальной модели COSMO [21]. В качестве растворителя рассмотрена безводная серная кислота в оценочных расчетах и, дополнительно, 5 и 15 (мас.)%-ные растворы SO3 в Н2SO4 при исследовании механизма образования ${\text{HSO}}_{3}^{ + }$. Диэлектрическая проницаемость растворителя составляла: ε = 101 (0% SO3), 259 (5% SO3), 359 (15% SO3) [22]. Эффективный радиус растворителя принят равным 1.3 Å для всех концентраций [23].
На уровне теории CCSD(T) проведены только одноточечные расчеты, геометрии для которых получены методом MP2/aug-cc-pV5Z.
Все расчеты осуществлены в программном комплексе ORCA [24], визуализация расчетов – в программном комплексе wxMacMolPlt [25].
Для оценки точности метода использованы два параметра: значение энергии изомеризации и длина связи S1–O5 в изомере H2O–SO2OH+. В качестве эталона сравнения использованы значение энергии, полученное на уровне теории DLPNO-CCSD(T)/aug-cc-pV5Z, и геометрия, полученная на уровне теории MP2/aug-cc-pV5Z. В качестве критерия адекватности метода принято выполнение условий “химической точности” (погрешность менее 1 ккал/моль ≈ 4.19 кДж/моль) и наименьшая погрешность расчета длины связи S1–O5.
Связь S1–O5 выбрана в качестве критерия оценки по причине сильной зависимости ее длины от метода расчета, как будет показано ниже.
Оценка энергии проведена для стадии изомеризации катиона ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$:
(1)
$\begin{gathered} {{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + } \to {{{\text{H}}}_{{\text{2}}}}{\text{O}}{\kern 1pt} - {\kern 1pt} {\text{S}}{{{\text{O}}}_{{\text{2}}}}{\text{O}}{{{\text{H}}}^{{\text{ + }}}}, \\ {{{\Delta }}{{E}^{ \ne }} = {{E}_{{{\text{TS}}}}} - {{E}_{{{{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{{\text{4}}}^{{\text{ + }}}}}}}, \\ {{{\Delta }}{{E}_{{\text{1}}}} = {{E}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}} - {\text{S}}{{{\text{O}}}_{{\text{2}}}}{\text{O}}{{{\text{H}}}^{{\text{ + }}}}}}} - {{E}_{{{{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{{\text{4}}}^{{\text{ + }}}}}}}, \\ \end{gathered} $(2)
$\begin{gathered} {{{\text{H}}}_{{\text{2}}}}{\text{O}}{\kern 1pt} - {\kern 1pt} {\text{S}}{{{\text{O}}}_{{\text{2}}}}{\text{O}}{{{\text{H}}}^{{\text{ + }}}} \to {{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{HSO}}_{3}^{ + }, \\ \Delta {{E}_{{\text{2}}}} = {{E}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}} + {{E}_{{{\text{HSO}}_{{\text{3}}}^{{\text{ + }}}}}} - {{E}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}} - {\text{S}}{{{\text{O}}}_{{\text{2}}}}{\text{O}}{{{\text{H}}}^{ + }}}}}, \\ \end{gathered} $(3)
$\begin{gathered} {{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + } \to {{{\text{H}}}_{{\text{2}}}}{\text{O}} + {\text{HSO}}_{3}^{ + }, \\ {{{\Delta }}{{E}_{{\text{3}}}} = \Delta {{E}_{{\text{1}}}} + \Delta {{E}_{{\text{2}}}} = {{E}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}} + {{E}_{{{\text{HSO}}_{{\text{3}}}^{{\text{ + }}}}}} - {{E}_{{{{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{{\text{4}}}^{{\text{ + }}}}}}}. \\ \end{gathered} $Дополнительно оценены профили энтальпии и свободной энергии Гиббса для реакций:
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Геометрические параметры исходного катиона и его изомера приведены на рис. 1.
Рис. 1.
Геометрия изомеров ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$, рассчитанная методом MP2/aug-cc-pV5Z в вакууме (a) и в растворителе (б).
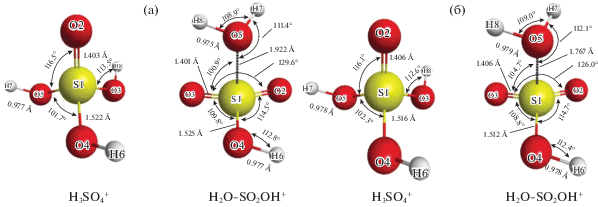
Рассчитанное расстояние S1–O5 (1.922 Å в вакууме) существенно больше длины ковалентных одинарных связей S–O (1.665 Å для пиросерных кислот [26], 1.54 Å для серной кислоты [27]). Можно предположить, что данная связь образована за счет электростатического взаимодействия частичных зарядов на атомах серы и кислорода. Значение ее длины свидетельствует о том, что эта связь слабее, чем ковалентная, следовательно, можно ожидать ее относительно легкого разрыва с образованием катиона ${\text{HSO}}_{3}^{ + }$ и молекулы воды.
При учете растворителя геометрия исходного катиона сохраняется (рис. 1), для изомера наиболее существенное изменение наблюдается в длине связи S1–O5 (уменьшение на 0.145 Å).
Результаты оценки геометрических и энергетических параметров, полученные постхартри-фоковскими методами, для вакуума приведены в табл. 1.
Таблица 1.
Оценка погрешности методов связанных кластеров и теории возмущений, вакуум
| Базис | ΔE1, кДж/моль | Eerr, кДж/моль | r(S1–O5), Å | rerr, Å |
|---|---|---|---|---|
| MP2 | ||||
| aug-cc-pVDZ | 27.15 | 41.94 | 2.073 | 0.151 |
| aug-cc-pVTZ | 51.41 | 17.67 | 1.967 | 0.046 |
| aug-cc-pVQZ | 55.43 | 13.66 | 1.936 | 0.014 |
| aug-cc-pV5Z | 57.63 | 11.45 | 1.922 | – |
| DPLNO-CCSD(T) | ||||
| aug-cc-pVDZ | 40.15 | 28.93 | – | – |
| aug-cc-pVTZ | 62.58 | 6.51 | – | – |
| aug-cc-pVQZ | 67.4 | 1.68 | – | – |
| aug-cc-pV5Z | 69.08 | – | – | – |
| cc-pVDZ-F12 | 63.95 | 5.14 | – | – |
Из табл. 1 видно, что при использовании метода теории возмущений второго порядка даже применение широких базисов не позволяет получить оценку энергии процесса с приемлемой точностью. Применение методов связанных кластеров позволяет приблизиться к точной оценке энергии на базисах не ниже QZ. Использование малых базисов приводит к завышенному значению длины связи S1–O5 и заниженному значению энергии изомеризации.
Результаты оценки геометрических и энергетических параметров, полученные методами теории функционала плотности, для вакуума приведены в табл. 2.
Таблица 2.
Оценка погрешности методов теории функционала плотности, вакуум
| Функционал | aug-cc-pVDZ | aug-cc-pVTZ | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔE1 | Eerr | r(S1–O5) | rerr | ΔE1 | Eerr | r(S1–O5) | rerr | |
| кДж/моль | Å | кДж/моль | Å | |||||
| PBE | 25.51 | 43.57 | 2.094 | 0.173 | 36.67 | 32.42 | 2.042 | 0.120 |
| M06-L | 30.34 | 38.75 | 2.090 | 0.168 | 42.68 | 26.4 | 2.008 | 0.086 |
| B3LYP5 | 39.8 | 29.29 | 2.066 | 0.144 | 51.05 | 18.04 | 2.001 | 0.079 |
| PBE0 | 52.85 | 16.23 | 2.001 | 0.079 | 63.69 | 5.4 | 1.935 | 0.014 |
| M06 | 49.29 | 19.79 | 2.008 | 0.086 | 57.95 | 11.13 | 1.939 | 0.018 |
| M06-2X | 61.9 | 7.18 | 1.954 | 0.032 | 71.4 | –2.32 | 1.908 | –0.014 |
| LC-BLYP | 54.66 | 14.42 | 1.969 | 0.048 | 63.45 | 5.63 | 1.921 | –0.0004 |
| ωB97x | 56.58 | 12.5 | 1.981 | 0.059 | 66.15 | 2.93 | 1.928 | 0.006 |
| B2T-PLYP | 41.15 | 27.93 | 2.050 | 0.129 | 57.18 | 11.91 | 1.970 | 0.048 |
Из табл. 2 видна общая тенденция уменьшения ошибок при переходе к более широкому базису. В целом, можно выделить лучшие результаты для пространственно-разделенных функционалов: ωB97x попадает в условие “химической точности”, LC-BLYP близок к нему. Функционал M06-2X демонстрирует изменение знака ошибки оценки энергии, что может указывать на случайное совпадение полученного значения с эталонным. Из остальных функционалов погрешность, близкую к допустимой, демонстрирует только PBE0.
Для сравнения приведены значения энергии изомеризации ΔE1, рассчитанные в работе [3] различными методами, кДж/моль:
которые дополнительно подтверждает низкую точность моделирования данной реакции с использованием глобальных гибридных GGA- функционалов и метода теории возмущений.Результаты оценки геометрических и энергетических параметров, полученные постхартри-фоковскими методами, для растворителя приведены в табл. 3.
Таблица 3.
Оценка погрешности методов связанных кластеров и теории возмущений (COSMO, 100% H2SO4)
| Базис | ΔE1, кДж/моль | Eerr, кДж/моль | r(S1–O5), Å | rerr, Å |
|---|---|---|---|---|
| MP2 | ||||
| aug-cc-pVDZ | 25.01 | 19.98 | 1.900 | 0.134 |
| aug-cc-pVTZ | 35.87 | 9.12 | 1.803 | 0.037 |
| aug-cc-pVQZ | 36.88 | 8.11 | 1.779 | 0.013 |
| aug-cc-pV5Z | 37 | 7.98 | 1.770 | – |
| DPLNO-CCSD(T) | ||||
| aug-cc-pVDZ | 31.79 | 13.2 | – | – |
| aug-cc-pVTZ | 42.72 | 2.27 | – | – |
| aug-cc-pVQZ | 44.68 | 0.31 | – | – |
| aug-cc-pV5Z | 44.99 | – | – | – |
| cc-pVDZ-F12 | 17.01 | 27.98 | – | – |
По результатам, представленным в табл. 3, можно сделать вывод, что при учете растворителя погрешность практически всех методов снижается. Для методов связанных кластеров достаточным становится использование трехэкспоненциального базисного набора. Исключение составляет метод DLPNO-CCSD(T)-F12D, погрешность которого, наоборот, сильно возросла. Методы теории возмущений по-прежнему не обеспечивают требуемую точность расчета.
Результаты оценки геометрических и энергетических параметров, полученные методами теории функционала плотности, для растворителя приведены в табл. 4.
Таблица 4.
Оценка погрешности методов теории функционала плотности (COSMO, 100% H2SO4)
| Функци-онал | aug-cc-pVDZ | aug-cc-pVTZ | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔE1 | Eerr | r(S1–O5) | rerr | ΔE1 | Eerr | r(S1–O5) | rerr | |
| кДж/моль | Å | кДж/моль | Å | |||||
| PBE | 19.42 | 25.56 | 1.941 | 0.175 | 25.82 | 19.16 | 1.884 | 0.118 |
| M06-L | 24.69 | 20.29 | 1.906 | 0.140 | 28.07 | 16.92 | 1.831 | 0.065 |
| B3LYP | 28.59 | 16.4 | 1.888 | 0.122 | 35.07 | 9.92 | 1.830 | 0.064 |
| PBE0 | 39.65 | 5.34 | 1.845 | 0.079 | 42.85 | 2.14 | 1.787 | 0.021 |
| M06 | 39.75 | 5.24 | 1.839 | 0.073 | 38.96 | 6.02 | 1.788 | 0.021 |
| M06-2X | 45.93 | –0.94 | 1.815 | 0.049 | 48.23 | –3.25 | 1.762 | –0.004 |
| LC-BLYP | 37.48 | 7.5 | 1.821 | 0.054 | 41.68 | 3.31 | 1.775 | 0.009 |
| ωB97x | 39.59 | 5.4 | 1.828 | 0.062 | 44.35 | 0.63 | 1.781 | 0.014 |
| B2T-PLYP | 30.64 | 14.34 | 1.871 | 0.105 | 39.57 | 5.42 | 1.799 | 0.033 |
Для методов теории функционала плотности также наблюдается тенденция к снижению ошибки энергии при переходе к растворителю. Для трехэкспоненциального базисного набора погрешность в пределах допустимой достигается при использовании функционалов PBE0, M06-2X, LC-BLYP и ωB97x. Однако увеличение погрешности на функционале M06-2X при переходе к более широкому базису также может рассматриваться в пользу скорее случайного попадания ошибки в требуемый диапазон.
Таким образом, имеется один функционал, удовлетворяющий условиям адекватности как для вакуума, так и для растворителя – ωB97x, который был использован для дальнейших расчетов.
Следующий этап – расчет геометрии (рис. 2) и энергии переходного состояния, а также отдельных частиц ${\text{HSO}}_{3}^{ + }$ и H2O (табл. 5).
Рис. 2.
Геометрия переходного состояния (TS), рассчитанная методом MP2/aug-cc-pV5Z в вакууме (a) и в растворителе (б).
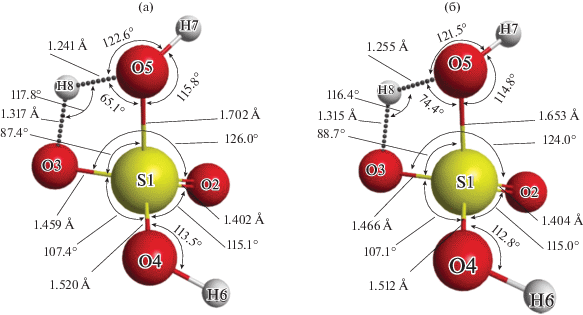
Таблица 5.
Энергетические характеристики реакций (1)–(3), кДж/моль
| ΔEi | Вакуум | 0% SO3 | 5% SO3 | 15% SO3 |
|---|---|---|---|---|
| ωB97x/aug-cc-pVTZ | ||||
| ΔE≠ | 183.83 | 175.23 | 175.09 | 175.06 |
| ΔE1 | 66.15 | 44.35 | 43.94 | 43.86 |
| ΔE2 | 163.6 | 151.96 | 152.01 | 152.02 |
| ΔE3 | 229.75 | 196.32 | 195.95 | 195.88 |
| MP2/aug-cc-pV5Z | ||||
| ΔE≠ | 177.40 | 170.02 | 169.87 | 169.85 |
| ΔE1 | 57.63 | 37.03 | 36.61 | 36.54 |
| ΔE2 | 156.99 | 146.39 | 146.44 | 146.46 |
| ΔE3 | 214.63 | 183.42 | 183.05 | 183.0 |
| DLPNO-CCSD(T)/aug-cc-pV5Z | ||||
| ΔE≠ | 188.87 | 180.01 | 179.87 | 179.84 |
| ΔE1 | 69.08 | 44.99 | 44.57 | 44.50 |
| ΔE2 | 156.98 | 149.23 | 149.30 | 149.31 |
| ΔE3 | 226.06 | 194.22 | 193.87 | 193.80 |
Погрешность метода ωB97x/aug-cc-pVTZ при расчете энергий ΔE≠ и ΔE2 в вакууме оказалась несколько выше требуемой (5.04 и 6.62 кДж/моль), однако погрешность расчета суммарной энергии ΔE3 (3.69 кДж/моль) не превышает допустимой. Метод MP2/aug-cc-pV5Z не обеспечивает требуемой точности расчетов, кроме оценки ΔE2. Суммарная энергия образования катиона ${\text{HSO}}_{3}^{ + }$, рассчитанная с использованием функционала ωB97x (229.75 кДж/моль), достаточно хорошо коррелирует с экспериментальными данными (225 ± ± 18 кДж/моль). В работе [3] приведены следующие значения энергии ΔE≠ газофазной реакции, кДж/моль:
которые указывают на занижение данной энергии при моделировании с использованием функционала B3LYP и метода теории возмущений, хотя и неплохом согласии этих энергий между собой при использовании одинакового базисного набора.Учет растворителя снижает энергетические барьеры реакции изомеризации (ΔE≠) на ~9 кДж/моль, реакции отрыва молекулы воды (ΔE2) на 18 (CCSD(T)) и 12 (ωB97x) кДж/моль, суммарной реакции (ΔE3) на 32 (CCSD(T)) и 33.5 (ωB97x) кДж/моль. Однако изменение концентрации SO3 в растворителе мало влияет на энергетические характеристики, для всех характеристик разность энергии при варьировании концентрации составляет менее 0.5 кДж/моль.
Для метода связанных кластеров расчет гессиана не проводился, и термодинамические характеристики оценивались через электронную энергию CCSD(T) и термохимические поправки по методу MP2. Оценка термодинамических характеристик исследуемых реакций приведена в табл. 6 и 7.
Таблица 6.
Тепловые эффекты реакций (1)–(3), 298 K
| ΔHi | Вакуум | 0% SO3 | 5% SO3 | 15% SO3 |
|---|---|---|---|---|
| ωB97x/aug-cc-pVTZ | ||||
| ΔH≠ | 172.24 | 163.21 | 162.89 | 162.87 |
| ΔH1 | 67.81 | 46.75 | 46.31 | 46.25 |
| ΔH2 | 155.02 | 141.45 | 141.54 | 141.52 |
| ΔH3 | 222.83 | 188.20 | 187.85 | 187.77 |
| MP2/aug-cc-pV5Z | ||||
| ΔH≠ | 165.49 | 157.88 | 157.76 | 157.75 |
| ΔH1 | 58.94 | 40.70 | 40.56 | 40.50 |
| ΔH2 | 147.81 | 134.80 | 134.57 | 134.55 |
| ΔH3 | 206.75 | 175.51 | 175.14 | 175.05 |
| DLPNO-CCSD(T)/aug-cc-pV5Z | ||||
| ΔH≠ | 176.96 | 167.86 | 167.76 | 167.74 |
| ΔH1 | 70.39 | 48.66 | 48.52 | 48.45 |
| ΔH2 | 147.79 | 137.64 | 137.44 | 137.40 |
| ΔH3 | 218.18 | 186.30 | 185.95 | 185.85 |
Таблица 7.
Изменение энергии Гиббса для реакций (1)–(3), 298 K
| ΔGi | Вакуум | 0% SO3 | 5% SO3 | 15% SO3 |
|---|---|---|---|---|
| ωB97x/aug-cc-pVTZ | ||||
| ΔG≠ | 175.22 | 167.53 | 167.26 | 167.18 |
| ΔG1 | 66.53 | 47.98 | 47.71 | 47.60 |
| ΔG2 | 108.41 | 93.12 | 93.18 | 93.14 |
| ΔG3 | 174.94 | 141.11 | 140.89 | 140.74 |
| MP2/aug-cc-pV5Z | ||||
| ΔG≠ | 168.56 | 162.44 | 162.38 | 162.39 |
| ΔG1 | 57.07 | 41.11 | 42.16 | 42.10 |
| ΔG2 | 101.45 | 87.94 | 86.68 | 86.65 |
| ΔG3 | 158.52 | 129.05 | 128.83 | 128.75 |
| DLPNO-CCSD(T)/aug-cc-pV5Z | ||||
| ΔG≠ | 180.03 | 172.42 | 172.38 | 172.38 |
| ΔG1 | 68.52 | 49.07 | 50.11 | 50.05 |
| ΔG2 | 101.43 | 90.78 | 89.54 | 89.50 |
| ΔG3 | 169.96 | 139.85 | 139.65 | 139.55 |
Согласно данным табл. 6, учет термодинамических характеристик несколько снижает энергию активации реакции (1) (ΔH≠) и суммарный тепловой эффект (ΔH3), однако расчетные данные по-прежнему коррелируют с экспериментальными.
Сравнение значений изменения энергии Гиббса и энтальпии реакции (табл. 6, 7) показывает, что энтропийный фактор оказывает существенное влияние на реакцию (2) (и соответственно суммарную реакцию), для реакции (1) он незначителен.
Энтропия суммарной реакции положительна, следовательно, увеличение температуры должно смещать равновесие в сторону образования катиона ${\text{HSO}}_{3}^{ + }$. Для оценки данного влияния была рассчитана величина ΔG3 в диапазоне температур 25–100°С (298–373 K) (рис. 3).
Рис. 3.
Изменение свободной энергии Гиббса суммарной реакции (ΔG3) в вакууме (a) и в растворителе (0% SO3) (б), рассчитанное методом DFT/ωB97x/aug-cc-pVTZ.
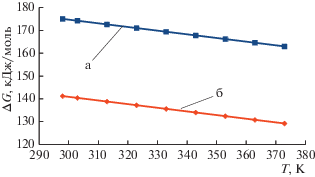
Наибольшие значения константы равновесия суммарной реакции для выбранного диапазона температур равны 1.6 × 10–23 (вакуум) и 8.3 × 10–19 (растворитель), что крайне мало для обеспечения сколь-нибудь заметной концентрации катиона ${\text{HSO}}_{3}^{ + }$ при данных температурах.
Учет влияния растворителя в явном виде [28] показывает, что в олеуме частицы ${\text{HSO}}_{3}^{ + }$ существуют преимущественно в виде ассоциатов ${\text{HSO}}_{3}^{ + }$ ⋅ H2SO4 (точнее HO3S⋅⋅⋅O–SO3H2, что можно рассматривать и как протонированную форму пиросерной кислоты). Отсюда можно сделать вывод, что катион ${\text{HSO}}_{3}^{ + }$ в свободном виде при температурах ниже 100°С не будет оказывать какого-то заметного влияния на процессы сульфирования, несмотря на то, что он – более активный сульфирующий агент по сравнению с SO3 [29].
Таким образом, проведено сравнение адекватности методов теории функционала плотности и теории возмущений для исследования реакции изомеризации катиона ${{{\text{H}}}_{{\text{3}}}}{\text{SO}}_{4}^{ + }$, выбран наиболее точный метод (ωB97x/aug-cc-pVTZ). Методами CCSD(T)/aug-cc-pV5Z, MP2/aug-cc-pV5Z и ωB97x/aug-cc-pVTZ проведено исследование реакции образования катиона ${\text{HSO}}_{3}^{ + }$ в вакууме и при неявном учете растворителя по модели COSMO.
Произведен расчет термодинамических характеристик реакции образования катиона ${\text{HSO}}_{3}^{ + }$ и показано, что при температурах ниже 100°С его концентрация как в вакууме, так и в растворителе слишком мала, для того, чтобы рассматривать его как истинный сульфирующий агент.
Список литературы
Weller M., Overton T., Armstrong F., Rourke J. Inorganic Chemistry. Oxford University Press, 2018. 943 p.
Bruckner R. Advanced Organic Chemistry: Reaction Mechanisms. Academic Press, 2001. 636 p.
Pommerening C.A., Steven M.B., Lee S.S. // J. Phys. Chem. A. 1999. V. 103. № 9. P. 1214. https://doi.org/10.1021/jp984104w
Sinha R., Chiavarino B., Fornarini S. et al. // J. Phys. Chem. Letters. 2010. V. 1. № 11. P. 1721. https://doi.org/10.1021/jz100458q
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865. https://doi.org/10.1103/PhysRevLett.77.3865
Zhao Y., Truhlar D.G. // J. Chem. Phys. 2006. V. 125 (19). P. 194101. https://doi.org/10.1063/1.2370993
Lee, C., Yang W., Parr R.G. // Phys. Rev B. 1988. V. 37. P. 785. https://doi.org/10.1103/PhysRevB.37.785
Perdew J.B., Ernzerhof M., Burke K. // J. Chem. Phys. 1996. V. 105 (22). P. 9982. https://doi.org/10.1063/1.472933
Zhao Y., Truhlar D.G. // Theor Chem Acc. 2006. V. 120 (1–3). P. 215. https://doi.org/10.1007/s00214-007-0310-x
Tawada Y., Tsuneda T., Yanagisawa S. et al. // J. Chem. Phys. 2004. V. 120. P. 8425. https://doi.org/10.1063/1.1688752
Najibi A., Goerigk L. // J. Chem. Theory Comput. 2018. V. 14. P. 5725.
Karton A., Tarnopolsky A., Lamère J.-F. et al. // J. Phys. Chem. A. 2008. V. 112. P. 12868. https://doi.org/10.1021/jp801805p
Grimme S., Ehrlich S., Goerigk L. // J. Comput. Chem. 2011. V. 32. P. 1456. https://doi.org/10.1002/jcc.21759
Grimme S., Antony J., Ehrlich S., Krieg H. // J. Chem. Phys. 2010. V. 132. P. 154104. https://doi.org/10.1063/1.3382344
Grimme S. // J. Comput. Chem. 2006. V. 27. P. 1787. https://doi.org/10.1002/jcc.20495
Kendall R.A., Dunning T.H., Harrison R.J. // J. Chem. Phys. 1992. V. 96. P. 6796. https://doi.org/10.1063/1.462569
Woon D.E., Dunning T.H. // Ibid. 1993. V. 98. P. 1358. https://doi.org/10.1063/1.464303
Riplinger C., Pinski P., Becker U. et al. // Ibid. 2016. V. 144. P. 024109. https://doi.org/10.1063/1.4939030
Hättig C., Schmitz G., Kossmann J. // Phys. Chem. Chem. Phys. 2012. V. 14(18). P. 6549. https://doi.org/10.1039/c2cp40400a
Peterson K.A., Adler T.B., Werner H.-J. // J. Chem. Phys. 2008. V. 128 (8). P. 84102. https://doi.org/10.1063/1.2831537
Klamt A., Schüürmann G. // J. Chem. Soc. Perkin Trans. 1993. V. 2. P. 799. https://doi.org/10.1039/P29930000799
Warlafen G. // J. Chem. Phys. 1964. V. 40. P. 2326. https://doi.org/10.1063/1.1725511
Klamt A., Eckert F., Arlt W. // Ann. Rev. Chem. and Biomol. Engng. 2010. V. 1. No 1. P. 101. https://doi.org/10.1146/annurev-chembioeng-073009-100903
Neese F. // WIREs Comput Mol Sci. 2017. V. 8 (1). P. e1327. https://doi.org/10.1002/wcms.1327
Bode B.M., Gordon M.S. // J. Mol. Graphics Mod. 1998. V. 16 (3). P. 133. https://doi.org/10.1016/S1093-3263(99)00002-9
Givan A., Loewenschuss A., Nielsen K.J., Rozenberg M. // J. Mol. Struct. 2007. V. 830(1). P. 21. https://doi.org/10.1016/j.molstruc.2006.06.027
Arstila H., Laasonen K., Laaksonen A. // J. Chem. Phys. 1998. V. 108 (3). P. 1031. https://doi.org/10.1063/1.475496
Degtyarev A.A., Rostova D.P. // Butlerov Comm. 2020. V. 62. № 4. P. 51. https://doi.org/10.37952/ROI-jbc-01/20-62-4-51
Degtyarev A.A., Osetrov A.Yu., Rostova D.P. // Ibid. 2020. V. 63. № 8. P. 64. https://doi.org/10.37952/ROI-jbc-01/20-63-8-64
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии