

Журнал физической химии, 2021, T. 95, № 8, стр. 1200-1210
Роль неорганического фосфата в фотогенерации пероксида водорода в водных растворах производных аденина при 77 К
Т. А. Лозинова a, *, А. В. Лобанов b, c, Е. Н. Дегтярев a, b, О. Н. Бржевская a, А. В. Ландер d
a Российская академия наук, Институт биохимической физики им. Н.М. Эмануэля
Москва, Россия
b Российская академия наук, Федеральный исследовательский центр химической физики им. Н.Н. Семенова
Москва, Россия
c Российский экономический университет имени Г.В. Плеханова
Москва, Россия
d Российская академия наук, Институт теории прогноза землетрясений и математической геофизики
Москва, Россия
* E-mail: taloz@mail.ru
Поступила в редакцию 11.10.2020
После доработки 14.12.2020
Принята к публикации 14.12.2020
Аннотация
Выполнено сравнительное исследование влияния неорганического фосфата (Pi) на образование пероксида водорода в водных растворах 2 × 10–4 M производных аденина (AX) – аденина, аденозина и аденозин-5'-дифосфата, облученных при 77 K ближним УФ в диапазонах длин волн λ = 260–400 и 290–460 нм. Установлено, что выход H2O2 в облученных образцах увеличивается при добавлении 5 × 10–4 M Pi; выход H2O2, как правило, возрастает в присутствии NaCl; 10-кратное увеличение [Pi] приводит лишь к умеренному возрастанию [H2O2]. Показано, что влияние облучения на выход H2O2 зависит как от AX, так и от соотношения [NaCl] и [Pi]. Полученные данные сопоставлены с результатами измерений спектров ЭПР облученных растворов перед размораживанием. Обсуждены возможные механизмы фотоинициируемых процессов образования H2O2 в исследованных системах.
Ранее было показано, что при определенных условиях замораживания водных растворов производных аденина (AX, где X = H для аденина (A), X = рибоза для аденозина (Ado), X = рибозо-5'-дифосфат для аденозин-5'-дифосфата (ADP)) под действием облучения ближним УФ при 77 K происходит довольно интенсивное образование пероксида водорода (H2O2), определяемое при размораживании образцов [1–6]. Облучение в тех же условиях растворов тимина и производных других оснований нуклеиновых кислот (исследовали гуанозин-5'-монофосфат и цитидин) практически не приводит к образованию H2O2 [1, 2]. Таким образом, вероятно, что среди производных различных оснований нуклеиновых кислот AX могут служить основными фотоиндуцируемыми источниками образования H2O2.
В цитированных работах было рассмотрено влияние на выход H2O2 состава среды и условий облучения. На основании сопоставления результатов с оценками общего количества парамагнитных продуктов в облученных образцах при 77 K, полученных методом ЭПР, а также состава сигналов ЭПР (определяемого путем их компьютерного моделирования), был сделан вывод о существовании двух различных путей фотоиндуцируемого образования H2O2 в замороженных растворах AX. Путь I преимущественно реализуется при относительно высоком содержании AX (1 × 10–3 M) в растворах AX + 0.1 M NaCl и, соответственно, относительно высоких интенсивностях суммарных сигналов ЭПР, регистрируемых в области g ≈ 2.00 (сигналы S). Интегральная интенсивность сигналов S (IntS) в этих случаях и относительное содержание в них сигналов пероксильных радикалов ${\text{O}}_{2}^{{ - {\kern 1pt} \bullet }}$ и ${\text{HO}}_{2}^{ \bullet }$ достаточно велико. (Высокая доля данных пероксильных радикалов в фотоиндуцируемых в наших системах суммарных сигналах ЭПР при 77 K была показана ранее [7].) Предположено, что этот путь, по-видимому, может состоять во взаимодействии радикалов ${\text{O}}_{2}^{{ - {\kern 1pt} \bullet }}$ и ${\text{HO}}_{2}^{ \bullet }$, с участием воды, возможно, в процессе размораживания образцов [1, 2].
Путь II проявляется при низких IntS сигналов ЭПР (низкие концентрации AX (2 × 10–4 M), отсутствие NaCl, ограничение интенсивности облучения (Ihν) и минимальной длины волны возбуждения λmin от ~260 до ~290 нм). Этот путь приводит к увеличению выхода H2O2 при падении IntS сигналов ЭПР и, соответственно, без существенного выхода пероксильных радикалов в окружающую среду. Мы предполагаем, что он тесно связан с самоассоциативными свойствами AX [3–6]. Сопоставление результатов определения выхода H2O2 в облученных растворах после размораживания и данных ЭПР в соответствующих облученных растворах при 77 K указывает на существенность и, возможно, предпочтительность этого пути.
В [6] было показано увеличение выхода пероксида водорода в облученных ближним УФ с λmin ~ ~ 260 нм при 77 K образцах AX (2 × 10–4 M) при внесении небольшого количества неорганического фосфата (5 × 10–4 M) в растворы на фоне присутствовавшего в растворах 0.1 M NaCl. Этому увеличению сопутствовало падение IntS сигналов ЭПР в облученных растворах, наблюдавшееся при 77 K. В случае Ado подобное влияние фосфата (Pi) показано также при [Pi] = 5 × 10–5 M. Цель данной работы – более подробное исследование влияния среды и полосы облучения на данный эффект.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использовали препараты A, Ado и ADP фирмы “Serva”, В качестве неорганического фосфата (Pi) использовали соль NaH2PO4 ⋅ 2H2O класса “extra pure”, остальные реактивы – класса “х.ч.”. Для приготовления растворов применяли воду, очищенную на установке Millipore. Эксперименты выполняли в слабокислой среде (pH 6.6), доведение pH растворов до этой величины производили добавлением HCl и NaOH. В исследованных растворах [AX] = 2 × 10–4 M, [NaCl] 0, 0.05 и 0.1 М.
Обpазцы замоpаживали в тефлоновыx контейнерах быстрым погpужением в жидкий азот (77 К) и оcвобождали непоcpедcтвенно пеpед оcвещением. Облучение пpоизводили pтутной лампой cвеpxвыcокого давления ДPШ-1000 cо cветофильтpами УФC-5 (стандартное значение 40%-го пpопуcкания в диапазоне 260–400 нм) и ФС-6 (стандартное значение 40%-го пpопуcкания в диапазоне 290–470 нм). Интенcивноcть оcвещения Ihν ограничивалась до ~0.6 от максимальной интенсивности источника облучения пpи помощи pешеток, калибpованныx на cпектpофотометpе, аналогично [3–5]. Это было связано с тем, что эффективность фотопродукции H2O2 (α), определяемая как отношение количества образующегося пероксида [H2O2] к IntS, существенно снижается при увеличении Ihν [3, 4].
Образцы облучали при 77 К в кварцевом дьюаре. Время облучения составляло 16 мин. Спектры ЭПР регистрировали при 77 К на ЭПP-cпектpометpе Bruker EMX-8 и, в ряде случаев, – на изготовленном в лаборатории ЭПР-спектрометре (частота 9.5 ГГц), использовавшемся в работах [1–7]. Мощность СВЧ-излучения составляла W = 200 мкВт, амплитуда модуляции – 2 Гс. Результаты, полученные на различных приборах, приведены с учетом пересчета на соответствующие параллельные измерения в идентичных образцах, приводившиеся в наших предыдущих работах. Регистрация спектров ЭПР на спектрометре Bruker EMX-8, обладающем более высокой чувствительностью, чем лабораторный ЭПР-спектрометр, позволила приблизительно оценить соотношение интегральных интенсивностей довольно слабых сигналов фотосенсибилизируемых атомов ${{{\text{H}}}^{ \bullet }}$ (дублет c pаcщеплением ~507 Гc, обычно не накладывающийcя на cигналы S) и сигналов S. Соответствующие измерения выполняли при W = 10 мкВт и пересчитывали с учетом соответствующих коэффициентов (табл. 1).
Таблица 1.
Результаты определения H2O2, IntS и IntH в спектрах ЭПР образцов AX, облученных со светофильтром УФС-5, Ihν = 0.6
| Систе-ма | [NaCl], M |
[Pi], M |
A | Ado | ADP | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [H2O2] | IntS | α | [${{{\text{H}}}^{ \bullet }}$] | [H2O2] | IntS | α | [${{{\text{H}}}^{ \bullet }}$] | [H2O2] | IntS | α | |||
| I | 0 | 0 | 12 18# |
62 ~0# |
0.19 – |
10 – |
55 65# |
91 41# |
0.60 1.54# |
7 – |
– 41# |
– 48# |
– 0.85# |
| II | 0.05 | 0 | 19 | 256 | 0.07 | – | 130 | 444 | 0.29 | 33 | 40 | 380 | 0.10 |
| III | 0.1 | 0 | 31 27# 28## |
304 489# 469## |
0.10 0.06# 0.06## |
12 – – |
190 109# 141## |
401 409# 448## |
0.47 0.27# 0.31## |
21 – – |
– 46# 41## |
– 228# 268## |
– 0.20# 0.15## |
| IV | 0 | 0.0005 | 32 | 84 | 0.38 | 1 | 260 | 75 | 3.47 | 4 | 13 | 117 | 0.11 |
| V | 0.05 | 0.0005 | 49 | 114 | 0.43 | 5 | 400 | 230 | 1.74 | – | 52 | 154 | 0.34 |
| VI | 0.1 | 0.0005 | 43 274## |
146 122## |
0.29 2.25## |
10 – |
490 435## |
– 184## |
– 2.36## |
– – |
160 158## |
138 152## |
1.16 1.04## |
| VII | 0 | 0.005 | 46 | 88 | 0.52 | 14 | 305 | 79 | 3.86 | 24 | 42 | 428 | 0.10 |
| VIII | 0.05 | 0.005 | 44 | 207 | 0.21 | 20 | 525 | 278 | 1.89 | 27 | 160 | 174 | 0.92 |
| IX | 0.1 | 0.005 | 39 | 129 | 0.30 | 9 | 575 | 308 | 1.87 | 30 | 180 | 140 | 1.29 |
Методика оценки вклада различных компонент в сигналы S, выполнявшаяся на основании построения модельного сигнала, идентична описанной в [7] и применявшейся во всех цитированных выше работах. Основными составляющими (“базисными сигналами”), вносящими вклад в регистрируемые спектры облученных растворов AX, являются сигналы пероксильных радикалов – ${\text{O}}_{2}^{{ - {\kern 1pt} \bullet }}$ и ${\text{HO}}_{2}^{ \bullet }$, электрондефицитных радикалов аденина ${{{\text{A}}}^{ \bullet }}$; радикалов рибозы ${\text{R}}{{{\text{i}}}^{ \bullet }}$ в Ado и ADP, (локализованных преимущественно на C5'); C8OH-аддуктов аденина – ${\text{AO}}{{{\text{H}}}^{ \bullet }}$; сигналы ${\text{Cl}}_{2}^{{ - {\kern 1pt} \bullet }}$; (наблюдаемые в присутствии NaCl в замораживаемых растворах), сигналы стабилизированных в матрице электронов e– и радикалов ${\text{NO}}_{2}^{ \bullet }$ (пpиcутcтвие которых в cпектpаx обуcловлено, по-видимому, вероятными пpимеcями нитpатов в компонентаx pаcтвоpов). В присутствии в растворах добавок Pi попытка внесения в базисную систему сигналов фосфатных радикалов и радикалов ${\text{O}}{{{\text{H}}}^{ \bullet }}$, вносящих существенный вклад в анализируемые спектры при более высоких концентрациях реагентов [8], при используемых концентрациях оказалась безуспешной.
После записи спектров ЭПР облученные образцы хранили в течение суток в жидком азоте (77 K) вплоть до определения H2O2. Определение Н2О2 проводили спектрально-иодометрическим методом [9], аналогично выполнявшемуся в цитированных выше работах [1–6]. С этой целью образцы после фотолиза размораживали при комнатной температуре, для чего каждый образец помещали в отдельную пробирку объемом 5 мл. После таяния образцов к ним добавляли 1 мл Н2SO4 (0.2 М), вытесняли растворенный кислород продувом углекислым газом, после чего смешивали с 2 мл деаэрированного 5%-го водного раствора иодида калия и вновь продували СО2. Выделение иода, образующего с избытком иодид-аниона комплексный анион ${\text{I}}_{3}^{ - }$, регистрировали методом спектрофотометрии (λmax = 351 нм, ε = = 26 400 М–1 см–1). Точность определения концентрации пероксида водорода <1 мкМ. Параллельно с облученными образцами в каждом из опытов выполняли определение H2O2 в контрольных, не облучавшихся образцах, хранившихся то же время при 77 К. Пероксид водорода в этих образцах практически отсутствовал ([H2O2] < < 1 мкМ).
Среднеквадратичная ошибка определения H2O2 в облученных образцах составляет ~24%; среднеквадратичная ошибка определения IntS составляет ~33%. Количество параллельных образцов в различных опытах варьировало от 3 до 7.
РЕЗУЛЬТАТЫ
Результаты определения количества H2O2 в размороженных образцах AX, облученных при 77 К с светофильтрами УФС-5 и ФС-6, приведены в табл. 1, 2. Там же представлены результаты определения интегральных интенсивностей сигналов ЭПР в этих образцах перед размораживанием (IntS). Во всех исследованных системах выход H2O2 увеличивается при добавлении 5 × 10–4 M Pi в соответствующие растворы как в случае облучения со светофильтром УФС-5, так и при ограничении полосы возбуждения до λmin ~ 290 нм (светофильтр ФС-6). В присутствии 0.05 M NaCl максимальное увеличение выхода H2O2 при внесении в растворы 5 × 10–4 M Pi наблюдалось в растворах Ado и составляло 3.1–2.8 раза при облучении образцов с указанными светофильтрами, соответственно.
Таблица 2.
Результаты определения H2O2, IntS и IntH в спектрах ЭПР образцов AX, облученных со светофильтром ФС-6, Ihν = 0.6
| Систе-ма | [NaCl], M |
[Pi], M |
A | Ado | ADP | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [H2O2] | IntS | α | IntH | [H2O2] | IntS | α | IntH | [H2O2] | IntS | α | |||
| I | 0 | 0 | 9 25# |
79 ~0# |
0.11 – |
7 – |
20 85# |
42 47# |
0.48 1.83# |
– – |
– 52# |
– ~0# |
– – |
| II | 0.05 | 0 | 19 | 86 | 0.22 | 1 | 110 | – | – | – | 47 | 110 | 0.43 |
| III | 0.1 | 0 | 23 34# |
61 80# |
0.38 0.29# |
30 – |
110 65# |
159 107# |
0.69 0.61# |
10 – |
– 48# |
– 80# |
– 0.63# |
| IV | 0 | 0.0005 | 32 | 67 | 0.48 | 4 | 190 | 73 | 2.60 | 9 | 14 | 65 | 0.22 |
| V | 0.05 | 0.0005 | 42 | 68 | 0.62 | 15 | 305 | 196 | 1.56 | 8 | 66 | 83 | 0.80 |
| VI | 0.1 | 0.0005 | 31 | 75 | 0.41 | 11 | 430 | – | – | – | 180 | 73 | 2.47 |
| VII | 0 | 0.005 | 35 | 148 | 0.24 | 8 | 255 | 61 | 4.18 | 10 | 48 | 108 | 0.44 |
| VIII | 0.05 | 0.005 | 28 | 134 | 0.21 | 6 | 450 | 220 | 2.04 | 16 | 136 | 62 | 2.19 |
| IX | 0.1 | 0.005 | 52 | 148 | 0.35 | 12 | 405 | 111 | 3.65 | 15 | 110 | 62 | 1.77 |
| Примечание. Время облучения 16 мин. Приведены средние значения результатов, полученных в параллельных опытах; α = = [H2O2], мкМ/ IntS, условные единицы. # – данные [4]. | |||||||||||||
10-кратное увеличение [Pi] приводит лишь к относительно слабому возрастанию выхода H2O2. (При этом максимальное увеличение выхода H2O2 в 3.1–2.1 раза наблюдалось в растворах ADP + 0.05 M NaCl при облучении со светофильтрами УФС-5 и ФС-6, соответственно, что согласуется с высокой оценкой константы ассоциации ADP с Pi [6].)
Необходимо отметить существенное расхождение между результатами влияния Pi при его низком содержании (5 × 10–4 M) на фоне 0.1 М NaCl на выход H2O2 при облучении образцов A со светофильтром УФС-5, полученными в [6] и в данной работе. Оценка выхода H2O2 в [6] превышает приводимое в этой работе значение в ~7 раз. Наблюдаемые в обеих работах величины IntS сигналов ЭПР близки. В связи с этим в данных условиях было произведено два независимых эксперимента, средние значения выхода H2O2 в которых оказались достаточно близки (47 и 38 мкМ), так же, как и оценки IntS сигналов ЭПР. Таким образом, результат определения выхода H2O2 в растворах A, приведенный в [6], по-видимому, ошибочен (в табл. 1 выделен курсивом). Следует заметить, что приготовление образцов в [6] и в данной работе осуществлялось различными операторами. Представляется вероятным, что результаты экспериментов могут существенно зависеть от процесса приготовления растворов для замораживания – времени нахождения смешанного раствора в жидком состоянии и интенсивности перемешивания как исходного раствора, так и в ходе доведения pH до требуемой величины (pH 6.6). Принципиальная возможность влияния способа приготовления растворов на результаты опытов отмечена в [10].
Наблюдавшееся в [6] снижение IntS сигналов ЭПР в облученных со светофильтром УФС-5 растворах AX при добавлении 5 × 10–4 M Pi на фоне 0.1 М NaCl, сопровождающееся увеличением выхода H2O2 в растворах при их размораживании, подтверждено в данной работе для 0.05 М NaCl (высокие IntS). Это, по нашему мнению, может свидетельствовать об определяющей роли пути II в рассматриваемых системах [6]. Однако, данный эффект не всегда заметен при небольших величинах IntS. Нам представляется, что описанные выше результаты могут объясняться совокупностью четырех факторов.
1. Влияние процесса замораживания растворов. Известно, что замораживание водных растворов ароматических органических молекул приводит к их агрегации [11, 12]. Очевидно, что степень агрегации молекул AX в замораживаемых растворах должна существенно зависеть от их исходного состояния – состава раствора (содержания относительно “нейтральных” составляющих, в данных экспериментах – присутствия солей NaCl, Pi) и степени возможной самоассоциации основных (фоточувствительных) компонентов. Показано, что в присутствии неорганических солей происходит дезагрегация этих соединений ([11, 12] и ссылки в этих работах). Принципиальное влияние процесса замораживания водных растворов тимина на выход свободных радикалов в случае γ- облучения при 77 K продемонстрировано в [13].
2. Самоассоциация AX. Абсолютные значения констант самоассоциации AX ($K_{{{\text{sa}}}}^{{{\text{AX}}}}$) в жидких водных растворах, получаемые различными методами и, следовательно, в различных диапазонах [AX], могут различаться на порядки [14]. Тем не менее, их последовательность для рассматриваемых нами соединений, в достаточной степени согласуется, по крайней мере, в случае образования димерных ассоциатов ($K_{{{\text{sa2}}}}^{{{\text{AX}}}}$). Эта последовательность, по литературным данным, по-видимому, такова: $K_{{{\text{sa}}}}^{{{\text{Ado}}}}$ > $K_{{{\text{sa}}}}^{{{\text{ADP}}}}$ [15] и $K_{{{\text{sa2}}}}^{{{\text{Ado}}}}$ > $K_{{{\text{sa2}}}}^{{\text{A}}}$ [16]. (Дополнительные ссылки на литературные данные приведены в [3].)
Последовательность изменения фотопродукции H2O2 в растворах AX при одинаковых условиях облучения (в отсутствие Pi) в целом совпадает с последовательностью изменения констант самоассоциации AX. Для [AX] = 2 × 10–4 M в присутствии 0.1 М NaCl выход H2O2 снижается в ряду Ado > ADP > A: 0.7–1.3/0.3/0.2 при облучении со светофильтром УФС-5 и 0.4–0.8/0.3/0.2 при облучении с ФС-6 (Результаты нормированы на среднее значение выхода H2O2 в растворах Ado + + 0.1 М NaCl, облученных со светофильтром УФС-5, полученное по измерениям в [4–6] и в данной работе (147 мкМ).) При этой же нормировке в отсутствие NaCl выход H2O2 в растворах Ado/ADP/A составляет: 0.4/0.3/0.1 и 0.1–0.6/0.4/ 0.1–0.2 при облучении образцов со светофильтрами УФС-5 и ФС-6, соответственно.
3. Дезагрегирующее действие солей. Дезагрегирующее действие ряда солей в замораживаемых водных растворах AX продемонстрировано в [11, 12] и ссылках в этих работах. Показано, что в качестве дезагрегирующих агентов соли Pi менее эффективны, чем NaCl [11]. Из AX наиболее “стойким” по отношению к дезагрегирующим агентам показан нуклеотид (AMP).
4. Комплексообразование AX с Pi. В условиях проведенных в данной работе экспериментов (pH 6.6) в растворах присутствуют преимущественно две ионных формы Pi – H2PO$_{4}^{ - }$ и HPO$_{4}^{{2 - }}$ (pK1 ~ 2.1, pK2 ~ 7.2 [17]) в соотношении ~ 4 : 1. Результаты расчета соотношения различных ионных форм Pi в зависимости от кислотности раствора приведены в [18].
При pH 6.6 в водных растворах преимущественно содержатся только нейтральные формы A и Ado (${\text{p}}K_{1}^{{{\text{A}}{\text{.Ado}}}}$ ~ 4.2–3.5 и ${\text{p}}K_{2}^{{{\text{A}}{\text{.Ado}}}}$ ~ 9.8–12.5, соответственно [19]). В случае ADP значение ${\text{p}}K_{2}^{{{\text{ADP}}}}$ (депротонирование гетероцикла) лежит в тех же пределах, что и ${\text{p}}K_{1}^{{{\text{A}}{\text{.Ado}}}}$; величина ${\text{p}}K_{1}^{{{\text{ADP}}}}$ (первичное депротонирование фосфатного фрагмента) лежит в более кислой области. При pH 6.6 в водном растворе преимущественно содержатся формы ADP2– и ADP3– (${\text{p}}K_{3}^{{{\text{ADP}}}}$ = 7.2, депротонированные по фосфатным группам [17, 20]). (Дальнейшее депротонирование ADP происходит по рибозной группе (${\text{p}}K_{4}^{{{\text{ADP}}}}$ > 12.5), что согласуется с ${\text{p}}K_{2}^{{{\text{Ado}}}}$ ~ 12.5 [19, 20]).
Возможное комплексование AX с Pi показано в [6] для A и ADP. Однако, этот эффект наблюдался в 0.05 М цитратном буфере (pH 5.6). В отсутствие достаточного количества противоионов (в отсутствие буфера либо NaCl) в растворах ADP, даже при доведении pH растворов ADP + Pi до использовавшейся в предыдущих работах величины (pH 6.6) количества ионов Na+, по-видимому, не вполне достаточно для предотвращения отталкивания отрицательно заряженных фосфатных групп ADP и Pi при [Pi] = 5 × 10–4 M. На фоне 0.05 М NaCl увеличение выхода H2O2 при [Pi] = = 5 × 10–4 M в ряду A : Ado : ADP составляет 2.6–2.2 : 3.1–2.8 : 1.3–1.4 (светофильтры УФС-5–ФС-6). Десятикратное увеличение [Pi] приводит в этих же условиях к заметному изменению этого соотношения: 0.9–0.7 : 1.3–1.5 : 3.1–2.1 (табл. 1, 2).
Влияние полосы облучения неоднозначно и, по-видимому, зависит как от AX, так и от соотношения [NaCl] и [Pi] (табл. 1, 2). Показано, что в жидких водных растворах в условиях преимущественного стэкинг-взаимодействия AX наблюдаются гипохромный эффект в области λ ≈ 260 нм и некоторое возрастание оптического поглощения при λ > 290 нм [21, 22] (Большее количество ссылок приведено в [4].) Хотя приводимые в цитируемых работах результаты получены на аналогах динуклеотидов [18] и олигомере дезоксиаденозина (dA)20 [22], они, по-видимому, вполне применимы к нашим системам, особенно в случае возможного преимущественного образования димерных ассоциатов [16, 23]. Константы самоассоциации ароматических органических молекул заметно возрастают при снижении температуры растворов. В [24] для растворов пурина показано увеличение Ksa в ~2.7 раза при снижении температуры раствора от 26 до 5°С. Нами наблюдалось в [25] увеличение оптического поглощения в области λ ~ 300–325 нм в водном растворе ADP при его охлаждении до ~4○С.
В отсутствие Pi в случае Ado на фоне 0.1 М NaCl падение выхода H2O2 при облучении образцов со светофильтром ФС-6 по сравнению с результатами облучения со светофильтром УФС-5 практически совпадает с данными [4] (~40% от выхода при облучении с УФС-5). Падение выхода H2O2 при облучении со светофильтрами ФС-6/УФС-5 в присутствии Pi несколько меньше и соизмеримо при [Pi] = 5 × 10–4 и 5 × 10–3 M (12–24% от выхода при облучении с УФС-5) вплоть до использования максимальных [NaCl] и [Pi] (табл. 1, 2).
В случае ADP как в отсутствие Pi, так и при низкой концентрации Pi наблюдается превышение выхода H2O2 при облучении со светофильтром ФС-6 по сравнению с результатами облучения со светофильтром УФС-5. Это качественно совпадает с результатами [4]. Однако при [Pi] = = 5 × 10–3 M в присутствии NaCl наблюдается, наоборот, значительное падение выхода H2O2 при облучении со светофильтром ФС-6 по сравнению с результатами облучения со светофильтром УФС-5 (табл. 1, 2), достигающее 39% при максимальной величине [NaCl]. Таким образом, в этом случае, по-видимому, могут играть существенную роль вторичные взаимодействия между фотоинициируемыми интермедиатами.
Результаты оценки относительного содержания различных компонент сигналов S в растворах Ado и ADP (одинаковые компоненты, используемые при моделировании спектров ЭПР) приведены в табл. 3, 4. Во всех доступных для сравнения в данной работе случаях отношение содержания компонент ${{{\text{A}}}^{ \bullet }}$ к ${\text{R}}{{{\text{i}}}^{ \bullet }}$ при облучении образцов со светофильтром УФС-5 оказывается существенно ниже в растворах ADP, чем в соответствующих растворах Ado. Этот эффект, по-видимому, отсутствует в растворах, облученных со светофильтром ФС-6.
Таблица 3.
Результаты анализа спектров ЭПР и определения концентрации H2O2 в образцах Ado | ADP, облученных со светофильтром УФС-5, Ihν = 0.6, время облучения 16 мин, Per – суммарное относительное содержание пероксильных радикалов ${\text{O}}_{2}^{{ - {\kern 1pt} \bullet }}$ и ${\text{HO}}_{2}^{ \bullet }$
| Система | [NaCl], M | [Pi], M | Per | ${{{\text{A}}}^{ \bullet }}$ | ${\text{R}}{{{\text{i}}}^{ \bullet }}$ | ${\text{AO}}{{{\text{H}}}^{ \bullet }}$ | ${\text{Cl}}_{2}^{{ - {\kern 1pt} \bullet }}$ | IntS | [H2O2], мкМ |
|---|---|---|---|---|---|---|---|---|---|
| I | 0 | 0 | 0.24 | – | 0.23 | – | 0.27 | – | 0.15 | – | – | – | 91 | – | 55 | – |
| II | 0.05 | 0 | 0.52 | 0.45 | 0.19 | 0.03 | 0.10 | 0.38 | 0.11 | 0.07 | 0.03 | 0.07 | 444 | 380 | 130 | 40 |
| III | 0.1 | 0 | 0.54 | – | 0.18 | – | 0.13 | – | 0.07 | – | 0.04 | – | 401 | – | 190 | – |
| IV | 0 | 0.0005 | 0.42 | 0.45 | 0.25 | 0.03 | 0.03 | 0.38 | 0.02 | 0.09 | – | – | 75 | 117 | 260 | 13 |
| V | 0.05 | 0.0005 | 0.63 | 0.57 | 0.26 | 0.15 | 0.00 | 0.14 | 0.02 | 0.00 | 0.02 | 0.07 | 230 | 154 | 400 | 52 |
| VI | 0.1 | 0.0005 | – | 0.45 | – | 0.16 | – | 0.12 | – | 0.12 | – | 0.06 | – | 138 | 490 | 160 |
| VII | 0 | 0.005 | 0.28 | 0.46 | 0.19 | 0.00 | 0.18 | 0.31 | 0.15 | 0.09 | – | – | 79 | 428 | 305 | 42 |
| VIII | 0.05 | 0.005 | 0.44 | 0.31 | 0.05 | 0.00 | 0.22 | 0.33 | 0.08 | 0.07 | 0.03 | 0.14 | 278 | 174 | 525 | 160 |
| IX | 0.1 | 0.005 | 0.45 | 0.51 | 0.04 | 0.00 | 0.22 | 0.19 | 0.06 | 0.06 | 0.08 | 0.02 | 308 | 140 | 575 | 180 |
Таблица 4.
Результаты анализа спектров ЭПР и определения концентрации H2O2 в образцах Ado | ADP, облученных со светофильтром ФС-6, Ihν = 0.6
| Система | [NaCl], M | [Pi], M | Per | ${{{\text{A}}}^{ \bullet }}$ | ${\text{R}}{{{\text{i}}}^{ \bullet }}$ | ${\text{AO}}{{{\text{H}}}^{ \bullet }}$ | ${\text{Cl}}_{2}^{{ - {\kern 1pt} \bullet }}$ | IntS | H2O2 |
|---|---|---|---|---|---|---|---|---|---|
| I | 0 | 0 | 0.54 | – | 0.14 | – | 0.15 | – | 0.12 | – | – | 42 | – | 20 | – |
| II | 0.05 | 0 | – | 0.54 | – | 0.26 | – | 0.00 | – | 0.11 | – | 0.03 | – | 110 | 110 | 47 |
| III | 0.1 | 0 | 0.45 | – | 0.06 | – | 0.14 | – | 0.16 | – | 0.09 | – | 159 | – | 110 | – |
| IV | 0 | 0.0005 | 0.33 | 0.50 | 0.03 | 0.07 | 0.38 | 0.28 | 0.09 | 0.09 | – | 73 | 65 | 190 | 14 |
| V | 0.05 | 0.0005 | 0.48 | 0.66 | 0.12 | 0.16 | 0.24 | 0.04 | 0.01 | 0.00 | 0.02 | 0.05 | 196 | 83 | 305 | 66 |
| VI | 0.1 | 0.0005 | – | 0.51 | – | 0.00 | – | 0.28 | – | 0.00 | – | 0.09 | – | 73 | 430 | 180 |
| VII | 0 | 0.005 | 0.24 | 0.46 | 0.16 | 0.00 | 0.03 | 0.29 | 0.23 | 0.08 | – | 61 | 108 | 255 | 48 |
| VIII | 0.05 | 0.005 | 0.64 | 0.39 | 0.00 | 0.14 | 0.00 | 0.05 | 0.00 | 0.25 | 0.02 | 0.00 | 220 | 62 | 450 | 136 |
| IX | 0.1 | 0.005 | 0.43 | 0.39 | 0.05 | 0.04 | 0.13 | 0.17 | 0.10 | 0.10 | 0.08 | 0.05 | 111 | 62 | 405 | 110 |
| Примечание. Время облучения 16 мин. Приведены средние значения результатов, полученных в параллельных опытах; Per – суммарное относительное содержание пероксильных радикалов ${\text{O}}_{2}^{{ - \, \bullet }}$ и ${\text{HO}}_{2}^{ \bullet }$. | |||||||||
В растворах ADP (но не Ado) в ряде случаев наблюдается увеличение выхода H2O2 при облучении со светофильтром ФС-6 по сравнению с результатами облучения образцов со светофильтром УФС-5 (табл. 3, 4, системы II, IV, V, VI, VII). Хотя причины наблюдаемого различия не выяснялись, можно предполагать определяющую роль в нем фотоиндуцируемого поглощения первичных интермедиатов наблюдаемых фотопроцессов.
Оценки коэффициентов экстинкции свободных радикалов, фиксируемых методом ЭПР при 77 K и их возможных предшественников (по литературным данным), приведены в [4]. В присутствии NaCl наибольшими коэффициентами экстинкции (ε) в области 280 ≥ λ ≤ 300 нм обладают свободные радикалы ${\text{C}}{{{\text{l}}}^{ \bullet }}$ и ${\text{Cl}}_{2}^{{ - {\kern 1pt} \bullet }}$, фиксируемые при 77 K методом ЭПР в наших работах (ε ~ 3000 и ~1200 М–1 см–1, соответственно, при λ = 280 нм). Оптическое поглощение радикалов ${\text{Cl}}_{2}^{{ - {\kern 1pt} \bullet }}$ увеличивается при λ > 300 нм, поглощение ${\text{C}}{{{\text{l}}}^{ \bullet }}$ падает. Следует отметить также возможность поглощения света в области λ ∼ 280 нм в замороженных водных растворах первоначально образующимися свободными радикалами ${\text{O}}{{{\text{H}}}^{ \bullet }}$, не связанными или слабо связанными водородными связями с молекулами воды [26].
Оптическое поглощение других свободных радикалов, обусловленных образованием ${\text{C}}{{{\text{l}}}^{ \bullet }}$ и ${\text{Cl}}_{2}^{{ - {\kern 1pt} \bullet }}$, и возможных интермедиатных радикалов ${\text{ClO}}{{{\text{H}}}^{{ - {\kern 1pt} \bullet }}}$ [27] увеличивается при λ > 310 нм (ссылки в [4]). Максимальное поглощение этих радикалов наблюдается в области λ ~ 350 нм, включающей интенсивную линию излучения ртутной лампы при λ ~ 366 нм и пропускаемой обоими использовавшимися светофильтрами. В доступных для сравнения случаях при облучении со светофильтром УФС-5 относительное содержание радикалов ${\text{Cl}}_{2}^{{ - {\kern 1pt} \bullet }}$ в растворах ADP несколько выше, чем в растворах Ado, за исключением системы с максимальным содержанием Pi и NaCl (табл. 3).
Ряд реакций, провоцируемых образованием радикалов ${\text{C}}{{{\text{l}}}^{ \bullet }}$ в водных растворах при комнатных температурах (по литературным данным), а также, вероятно, при отжиге облученных при 77 K образцов (использованном в [1]), приведен в цитируемой работе. В частности, распад возможных интермедиатных радикалов ${\text{ClO}}{{{\text{H}}}^{{ - {\kern 1pt} \bullet }}}$ может приводить к образованию радикалов ${\text{O}}{{{\text{H}}}^{ \bullet }}$ и тем самым влиять на выход H2O2. Неизвестно, однако, в какой мере приведенные в [1] реакции, тем более под действием дополнительного облучения, могут осуществляться при 77 K.
Столь же направленные различия в выходах H2O2 в растворах Ado и ADP при облучении со светофильтрами УФС-5 и ФС-6, по-видимому, существуют и в растворах, не содержащих NaCl (системы IV, VII). Можно предположить, что эти различия связаны с отмеченным выше возможным оптическим поглощением свободных радикалов ${\text{O}}{{{\text{H}}}^{ \bullet }}$, не связанных или слабо связанных с молекулами H2O, в области λ ~ 280 нм [26].
Следует отметить, что образование H2O2, как под действием видимого света, так и под действием инфракрасного облучения в полосах поглощения растворенного в воде O2 наблюдалось в отсутствие фотосенсибилизаторов в воде при комнатной температуре [28]. Использовавшиеся в наших работах светофильтры (УФС-5, ФС-6) пропускают свет с λ > 700 нм, что не позволяет полностью исключать из рассмотрения возможность предполагаемых в этой работе процессов, обусловленных образованием 1O2. Однако, выход H2O2 в отсутствие фотосенсибилизаторов в этой работе при лазерном облучении с λ ≈ 633 нм и даже с λ ≈ ≈ 1260 нм не превышает 20 нМ, что вообще значительно ниже чувствительности использовавшегося нами метода определения [H2O2].
Приведенные результаты, полученные в отсутствие в замораживаемых растворах Pi либо при низкой концентрации Pi, подтверждают ранее сделанный вывод о существенности пути II в рассматриваемых процессах. Введение небольших количеств Pi (до 5 × 10–5 M в случае Ado [6]), таким образом, существенно влияет на состояние растворов AX перед замораживанием, и микроструктура водных растворов, по-видимому, – основной фактор, влияющий на фотопродукцию H2O2 в наших системах.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Хотя основным механимом ассоциации AX предполагается стэкинг-взаимодействие мономеров [14–16, 21, 22], отмечается существенность физико-химических факторов среды (pH, состав растворов, присутствие ионов металлов, диапазоны исследуемых концентраций AX и добавок) [15, 23]. Перечисленные факторы влияют на конформацию мономеров, связывание их (в том числе, посредством водородных связей) с соседними молекулами, включая, безусловно, и молекулы растворителя. В случае полимеров эта конфигурация “оказывает решающее влияние на конформацию, которую принимает макромолекула при заданных физических условиях и которая, в свою очередь, связана с надмолекулярной структурой” [29].
Известно существование в водных растворах кластеров (наноассоциатов [30]). Ссылки на существование различных типов кластерных структур (от димерных до октамерных и более высоких порядков) приведены в [31, 32]. В замороженных водных растворах доминируют гексамерные кластеры в различных конформациях.
Размеры наноассоциатов воды в жидких водных растворах варьируются от 1–10 нм до 200 [33] и даже тысяч нм [30] в зависимости от состава растворов. Их структура существенно зависит от растворенных веществ, в том числе – NaCl [30, 33]. Возможное влияние биомолекул на структуру водных кластеров отмечалось ранее (например, в [34]). В образовании водных кластеров определяющую роль, по-видимому, играет способность молекул воды образовывать водородные связи с соседними молекулами [33]. Протоны, участвующие в образовании водородных связей, расположены между двумя атомами O (собственной и соседней молекул H2O). Водородная связь между атомами H и O носит отчасти ковалентный характер (10%, [35]). Состояние, когда протон оказывается приближенным к соседнему атому кислорода, типично для границ раздела (вода–газ, вода–твердое тело) [32]. Представляется вероятным, что поверхность ассоциатов AX также можно рассматривать как некую поверхность раздела.
В [36] (и ссылках в этой работе) продемонстрировано, что поверхность льда может быть как источником, так и стоком ориентационных дефектов Бьеррума [37]. Кроме того, она способна аккумулировать возможные ионные дефекты структуры льда – как гидрониум-, так и гидроксид-ионы [36]. В [38] сделано заключение, что, несмотря на расхождение экспериментальных результатов с теоретическими оценками, при pH > 3 суммарный заряд поверхности воды на границе с воздухом отрицателен. Предполагается, что структура поверхностного слоя воды / льда оказывает существенное влияние на протекание в них химических реакций вследствие зависимости от нее энергетических характеристик идентичных молекул, находящихся в этом слое [36].
Необходимо заметить, что все указанные в [36] особенности поверхностного слоя льда проявляются при температурах в диапазоне ~ 110–150 K. Однако, как при приготовлении исследовавшихся образцов, так и при их размораживании после облучения, данный температурный интервал неизбежно проходится.
Особенность поверхностного слоя воды и ассоциатов AX, по-видимому, может играть важную роль в фотопродукции H2O2 в замороженных водных растворах. Расстояние между стэкинг-ассоциированными молекулами AX (~3.4 Å [20]) позволяет, в принципе, внедрение в них одномолекулярного слоя H2O (размер молекулы H2O ~ 3 Å), а также – молекул O2 (размер ~3 Å [39]). В присутствии NaCl это допускает возможность внедрения в ассоциаты ионов Na+ (размер ионного радиуса ~1 Å) однако не позволяет внедрения ионов Cl– (размер ионного радиуса ~1.8 Å) [40]. В то же время в [36] показано, что ионы Na+ имеют тенденцию к проникновению внутрь замораживаемого раствора, тогда как ионы Cl–, напротив, склонны оставаться на границе. Таким образом, в присутствии NaCl возможность зарядового различия между пограничным слоем AX и внешней водной средой достаточно неопределенна.
В [41] приведены результаты анализа изменения редокс-потенциала жидкой воды под действием УФ-облучения, влияния растворенных в воде газов и влияния длительного времени хранения образцов после облучения. Показано, что пpиcутcтвие pаcтвоpенныx газов в воде и cильно pазбавленныx водныx cpедаx может cущеcтвенно влиять на cвойcтва растворов, а cтpуктуpная и динамичеcкая упоpядоченноcть cиcтемы pаcтет cо вpеменем, прошедшим после облучения жидких образцов.
Первоначальный продукт реакций, образующийся под действием УФ-облучения – атомы ${{{\text{H}}}^{ \bullet }}$ [41], наблюдался методом ЭПР при 77 K в присутствии Pi (0.5 М, pH 4.5) [42]. Значительно большая интенсивность сигналов ${{{\text{H}}}^{ \bullet }}$ показана в присутствии фотоиндуцируемого донора электрона (e–) триптофана (Trp), λ > 240 нм и возможных “промежуточных” акцепторов e–, включая ионы фосфата [43, 44]. При pH 5.0, [Trp] = 5 × 10–4 M, и [KNO3] (конечный акцептор e–) = 0.01 M показана практически линейная зависимость увеличения выхода ${{{\text{H}}}^{ \bullet }}$ и продукта восстановления конечного акцептора – радикалов ${\text{NO}}_{2}^{ \bullet }$ от [Pi] [42]. Конcтанта cкоpоcти взаимодейcтвия e– с ионами ${\text{NO}}_{3}^{ - }$ в жидких водных растворах составляет k > 9 × × 109 М–1 c–1 [45]. Взаимодействие e– с ионами ${\text{NO}}_{3}^{ - }$ приводит к разнообразным процессам, однако, преимущественно не приводящим к образованию ${{{\text{H}}}^{ \bullet }}$ [46].
Таким образом, вероятно, что увеличение выхода ${{{\text{H}}}^{ \bullet }}$ в рассматриваемой системе обусловлено, в основном, взаимодействием фотоиндуцируемого электрона с ионами Pi:
(1)
${{{\text{е}}}^{--}} + {{{\text{Н}}}_{{\text{2}}}}{\text{РО}}_{4}^{ - } \to {{{\text{Н}}}_{{\text{2}}}}{\text{РО}}_{4}^{{2 - {\kern 1pt} \bullet }} \to {{{\text{H}}}^{ \bullet }} + {\text{НРО}}_{4}^{{2 - }}.$Среднее расстояние между фотоактивируемым донором e– (Trp) и молекулами акцептора Н2РО$_{4}^{ - }$ при его концентрациях 0.05–0.5 М составляет от ~16 до 7.5 Å, что значительно меньше возможных средних расстояний между фотоиндуцируемым донором e– AX и этим акцептором в наших работах. При [Pi] = 5 × 10–4 M среднее расстояние между молекулами AX и молекулами Pi составляет ~75 Å. В данной работе при такой концентрации Pi показано увеличение фотопродукции H2O2 в 2.7 раза в растворах A и в 4.7 раза в растворах Ado по сравнению с растворами, не содержавшими добавок Pi (при облучении со светофильтром УФС-5, табл. 1).
В присутствии более слабого конкурирующего реагента, взаимодействующего с фотоиндуцируемым e– (0.1 M NaCl; k < 106 М–1 c–1 [48]) увеличение выхода H2O2 при включении в растворы Pi снижается. В этой работе при добавлении в растворы 5 × 10–4 M Pi наблюдаемое увеличение выхода H2O2 составляет для A – в 1.4 раза, для Ado – в 2.6 раза. В [6] для Ado показано увеличение выхода H2O2 на фоне 0.1 M NaCl в присутствии 5 × 10–5 M Pi (в 2.3 раза). В этом случае среднее расстояние между молекулами Ado и Pi может составлять уже ~160 Å.
Таким образом, существуют основания для предположения о том, что наблюдаемое нами увеличение выхода H2O2 при внесении в замораживаемые растворы AX неорганического фосфата в умеренных концентрациях ([Pi] = 5 × 10–4 M и ниже) связано в значительной степени с изменением структуры замораживаемых водных растворов. Изменение кластерной структуры водных растворов в присутствии Pi и, вероятно, возможное изменение состояния поверхностного слоя как на границах растворителя с агрегатами AX, так и, возможно, с “внутриагрегатными” слоями растворителя, предположенное в [6], по-видимому, могут играть значительную роль в увеличении фотопродукции H2O2 в наших системах в присутствии различных добавок.
Ранее отмечалось, что в ряде случаев выход пероксида водорода в рассматривавшихся системах может заметно превосходить растворимость O2 в водных и водно-солевых системах [6] и ссылки в этой работе. Это может, в частности, объясняться адсорбцией O2 на поверхности образцов в процессе их приготовления к облучению. Однако, оценка количества спинов в спектрах ЭПР образцов Ado + 0.05 M NaCl + 5 × 10–3 M Pi (система VIII), облученных со светофильтром УФС-5, дает содержание в них ~2.8 × 1014 спинов. Это означает, что определяемое количество молекул H2O2 в тех же образцах превышает количество свободных радикалов в них, регистрируемое перед их размораживанием в ~300 раз. Таким образом, возникает вопрос о возможном участии H2O в наблюдаемых процессах.
Возможные свободнорадикальные механизмы, предполагавшиеся нами для фотоиндуцируемого увеличения выхода H2O2 в присутствии различных добавок, приведены в цитируемых работах. В частности, в условиях, когда основным путем образования H2O2 может быть путь II, предполагаются реакции:
(2)
${\text{HO}}_{2}^{ \bullet } + {{{\text{H}}}^{ \bullet }} \to {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}}$(3)
${\text{O}}{{{\text{H}}}^{ \bullet }} + {\text{O}}{{{\text{H}}}^{ \bullet }} \to {{{\text{H}}}_{{\text{2}}}}{{{\text{O}}}_{2}}.$Рис. 1.
Примеры спектров ЭПР, регистрируемых при 77 К в облученных растворах Ado (2 × 10–4 M), Ihν = 0.6, thν = 16 мин; а – светофильтр УФС-5, W = 200 мкВт, б – то же при W = 10 мкВт, в – спектры ЭПР, регистрируемые при W = 200 мкВт в образцах, облучениых со светофильтром ФС-6. На рис. (а) и (в) экспериментальные спектры ЭПР – серые линии, модельные – обычно накладывающиеся на них тонкие черные линии. Слева римскими цифрами показаны номера систем, соответствующие табл. 1–4. В квадратных рамках – выход H2O2 в мкМ в соответствующих системах; (*) – сигнал дьюара, в спектрах (б), зарегистрированных при низкой мощности W, этот сигнал удален вычитанием; в моделировавшихся спектрах включен в систему базисных сигналов.
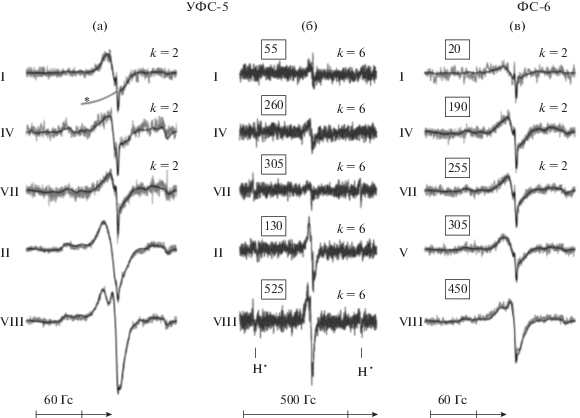
Не исключено, что определенную роль в образовании H2O2 в рассматриваемых системах могут играть процессы, связанные с переносом энергии. Обращает на себя внимание близость оптических характеристик AX и воды (полос возбуждения флуоресценции и спектров флуоресценции).
При облучении водных растворов AX при 77 K светом с λ ~ 260 нм наблюдается флуоресценция с λmax ≈ 348–362 нм, отнесенная на счет эксимеров AX [11], что в целом соответствует результатам, полученным в жидких растворах (dA)20 (λmax ≈ ≈ 362 нм) [21]. Положение максимума коротковолновой флуоресценции воды, возбуждаемой светом в области λ ~ 260 нм, варьирует в пределах 345–360 нм [49, 50].
При облучении растворов (dA)20 в UVA-диапазоне (возбуждение при λ ~ 330 нм) максимум флуоресценции наблюдается при 420 нм [21]. Максимум длинноволновой флуоресценции воды, возбуждаемой светом с λ ~ 310 нм, расположен при 410–425 нм [49, 50]. Вследствие большой ширины полос регистрируемые спектры флуоресценции (dA)20 и воды хорошо перекрываются (ширина их на полувысоте максимума составляет ~100 нм [21, 50]).
В [50] показано, что интенсивность эмиссионных спектров воды существенно зависит от присутствия в ней O2. Предположено, что наблюдаемые отличия в эмиссионных спектрах, регистрируемых в присутствии O2, обусловлены образованием нанопузырьков газа и адсорбцией содержащихся в следовых количествах примесей на водно-пузырьковых границах растворов.
В [51] продемонстрировано существенное влияние микрогетерогенности среды, обусловленной, в частности, образованием микропузырьков растворенного O2 на наблюдаемые спектры флуоресценции. При этом наблюдалось отсутствие однозначного соответствия между степенью очистки воды и параметрами ее флуоресценции; причем как интенсивность длинноволновой флуоресценции воды, так и положение ее максимума существенно зависело от условий приготовления растворов. Таким образом, несмотря на принципиальную возможность процессов переноса энергии в рассматриваемых системах, предсказать их направленность без проведения специальных исследований не представляется возможным.
Системы, содержащие микро/нанопузырьки O2, безусловно, можно отнести к разряду “организованных сред” [52, 53]. “Организованные среды – это прозрачные, оптически однородные растворы, в которых в основной массе растворителя присутствуют наноразмерные системы, образующие собственную нанофазу”. Эти среды во многом подобны мицеллярным растворам, они гомогенны и однофазны в макромасштабе, но микрогетерогенны и двухфазны на наноуровне [52]. Соответствующие микровключения могут считаться микро / нанореакторами [53] (и ссылки в цитируемых работах).
Приведенные в данной работе исследования выполнены с использованием ЭПP-cпектpометpа Bruker EMX-8 ЦКП “Новые материалы и технологии” ИБХФ РАН.
Список литературы
Лозинова Т.А., Лобанов А.В., Ландер А.В. // Журн. физ. химии. 2015. Т. 89. № 8. С. 1329.
Лозинова Т.А., Лобанов А.В., Ландер А.В. // Там же. 2016. Т. 90. № 11. С. 1739.
Лозинова Т.А., Лобанов А.В., Ландер А.В. // Там же. 2017. Т. 91. № 12. С. 2146.
Лозинова Т.А., Лобанов А.В., Ландер А.В. // Там же. 2018. Т. 92. № 10. С. 1653.
Лозинова Т.А., Лобанов А.В., Ландер А.В. // Там же. 2019. Т. 93. № 5. С. 757.
Лозинова Т.А., Лобанов А.В., Ландер А.В., Бржевская О.Н. // Там же. 2020. Т. 94. № 2. С. 306.
Лозинова Т.А., Ландер А.В. // Там же. 2014. Т. 88. № 1. С. 120.
Лозинова Т.А., Ландер А.В. // Биофизика. 2013. Т. 58. № 3. С. 445.
Лобанов А.В., Рубцова Н.А., Веденеева Ю.А., Комиссаров Г.Г. // Докл. РАН. 2008. Т. 421. № 6. С. 773.
Лобышев В.И. // Рос. хим. журн. 2007. Т. 51. № 1. С. 107.
Kleinwachter V. // Collection Czechoslov. Chern. Cornrnun. 1972. V. 37. P. 1622.
Physico-chemical Properties of Nucleic Acids / Ed. by Duchesne. London: Academic Press Inc., 1973. P. 119–142.
Szajdzinska-Pietek E., Bednarek J., Plonka A. et al. // Res. Chem. Intermed. 2001. V. 27. № 9. P. 937.
Beshnova D.A., Lantushenko A.O., Davies D.B., Evstigneev M.P. // J. Chem. Phys. 2009. V. 130. P. 165105.
Scheller K.H., Hofstetter F., Mitchell P.R. et al. // J. Am. Chem. Soc. 1981. V. 103. № 2. P. 247.
Morcillo J., Gallego E., Peral F. // J. Mol. Struct. 1987. V. 157. P. 353.
Мецлер Д. Биохимия. Т. 1. М.: Мир, 1980. 497 с. (D.E. Metzler. Biochemistry. Academic Press. New York San Francisco London. 1977.)
Бржевская О.Н., Дегтярев Е.Н., Левин П.П. и др. // Докл. АН. 2005. Т. 405. С. 259.
Краткая химическая энциклопедия. Т. 1 / Под ред. И.Л. Кнунянца, Г.Я. Бахаровской, А.И. Бусева и др. М.: Советская энциклопедия, 1961. С. 31.
Зенгер В. Принципы структурной организации нуклеиновых кислот. М.: Мир, 1987. 583 с. (Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag New York, 1984).
Browne D.T., Eisinger J., Leonard N.J. // J. Am. Chem. Soc. 1968. V. 90. P. 7302.
Banyasz A., Vaya I., Changenet-Barret P. et al. // J. Am. Chem. Soc. 2011. V. 133. P. 5163.
Peral F., Gallego E. // Biophys. Chem. 2000. V. 85. P. 79.
Van Holde K.E., Rossetti G.P. // Biochemistry. 1967. V. 6. № 7. P. 2189.
Лозинова Т.А. Фотохимическая модель синтеза аденозинтрифосфата: Дис. …канд. физ.-мат. наук. М.: МГУ, 1989.
Ghormley J.A., Hochanadel C.J. // J. Phys. Chem. 1971.V. 75. № 1. P. 40.
Kläning U.K., Wolff T. // Ber. Bunsenges. Phys. Chem. 1985. V. 89. P. 243.
Гудков C.В., Каpп О.Э., Гаpмаш C.А. и др. // Биофизика 2012. Т. 57. № 1. С. 5.
Инфракрасная спектроскопия полимеров / Под ред. И. Деханта (Пер. с нем.). М.: Химия, 1976. 472 с.
Коновалов А.И., Рыжкина И.С. // Изв. АН. Сер. хим. 2014. № 1. С. 1.
Raghavaiah P. Supramolecular Assembles of Amine-Based Compounds in Combination with Different Counter Systems Ranging from Inorganic Anions, a Heteropoly Anion and Substituted Organic Acids: A Thesis Submitted In partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy. Department of Chemistry Goa University Taleigao Plateau Goa 403 206 India. 2007. 190 p.
Ignatov I., Mosin O. // Journal of Health, Medicine and Nursing. 2014. V. 6. P. 50.
Sedlak M., Rak D. // J. Phys. Chem. B. 2013. V. 117. P. 2495.
Cole D.R., Herwig K.W., Mamontov E., Larese J.Z. // Reviews in Mineralogy & Geochemistry. 2006. V. 63. P. 313.
Isaacs E.D., Shukla A., Platzman P.M. et al. // J. Phys. Chem. Solids. 2000. V. 61. P. 403.
Park S.-C., Moon E.-S., Kang H. // Phys. Chem. Chem. Phys. 2010. V. 12. P. 12000.
Головин Ю.А. // СОЖ. 2000. Т. 6. № 9. С. 66.
Chaplin M. // Water. 2009. V. 1. P. 1.
Полинг Л., Полинг П. Химия. М.: Мир, 1978. 683 с. (L. Pauling, P. Pauling. Chemistry. W. H. Freeman & Co., San Francisco. 1975.)
Бугаенко Л.Т., Рябых С.М., Бугаенко А.Л. // Вестн. Моск. ун-та. Сер. 2. Химия. 2008. Т. 49. № 6. С. 363.
Belovolova L.V., Vinogradov E.A., and Glushkov M.V. // Phys. Wave Phenom. 2013. V. 21. № 3. P. 183.
Неделина О.С., Бржевская О.Н., Дегтярев Е.Н. и др. Проблемы зарождения и эволюции биосферы / Под ред. Э.М. Галимова. М.: Книжный дом “ЛИБРОКОМ”, 2008. С. 179.
Неделина О.С., Бржевская О.Н., Дегтярев Е.Н., Зубков А.С. // Докл. АН. 2012. Т. 442. № 4. С. 501.
Неделина О.С., Бржевская О.Н., Дегтярев Е.Н. и др. Проблемы зарождения и эволюции биосферы / Под ред. Э.М. Галимова. М.: КРАСАНД, 2013. С. 347.
Chen R., Avotinsh Y., Freeman G.R. // Can. J. Chem. 1994. V. 72. P. 1083.
Пикаев А.К. Современная радиационная химия. Радиолиз газов и жидкостей. М.: Наука, 1986. 440 с.
Левин П.П., Бpжевcкая О.Н., Неделина О.C. // Изв. АН. Cеp. xим. 2007. № 7. С. 1277.
Thomas J.K., Gordon S., Hart E.J. // J. Phys. Chem. 1964. V. 68. № 6. P. 1524.
Lobyshev V.I., Shikhlinskaya R.E., Ryzhikov B.D. // J. Mol. Liq. 1999. V. 82. P. 73.
Vallée P., Lafait J., Mentré P. et al. // J. Chem. Phys. 2005. V. 122. P. 114513.
Беловолова Л.В., Глушков М.В., Виноградов Е.А. // 2009. Сборник избранных трудов V Международного конгресса “Слабые и сверхслабые поля и излучения в биологии и медицине”. СПб., 2009. С. 10. www.biophys.ru/archive/congress2009/pro-p10.pdf
Штыков С.Н. Люминесцентный анализ / Под ред. Г.И. Романовской (Научн. Совет РАН по аналит. химии. Проблемы аналитической химии. Т. 19). М.: Наука. 2015. С. 127.
Штыков С.Н. // Журн. аналит. химии. 2002. Т. 57. № 10. С. 1018.
Дополнительные материалы отсутствуют.
Инструменты
Журнал физической химии