

Известия РАН. Серия физическая, 2021, T. 85, № 2, стр. 270-275
Уточнение скоростей реакций пиролиза этана
О. И. Топор 1, *, А. А. Белов 1, 2, И. А. Федоров 1
1 Федеральное государственное бюджетное образовательное учреждение высшего образования
“Московский государственный университет имени М.В. Ломоносова”, физический факультет
Москва, Россия
2 Федеральное государственное автономное образовательное учреждение высшего образования
“Российский университет дружбы народов”
Москва, Россия
* E-mail: topor.oi15@physics.msu.ru
Поступила в редакцию 28.08.2020
После доработки 25.09.2020
Принята к публикации 28.10.2020
Аннотация
Проанализированы экспериментальные данные о скоростях реакций, описывающих пиролиз этана. Получена точность 1–25%, что существенно превосходит мировой уровень. Попутно проведен критический анализ существующих теоретических моделей скоростей химических реакций.
ВВЕДЕНИЕ
Важную роль в химической промышленности играют низшие олефины – этилен и пропилен. Они используются для производства полиэтилена, полипропилена, этанола, ацетона, фенола, лаков, растворителей и т.д. Основным способом получения олефинов является процесс термического разложения (пиролиза) углеводородов [1, 2] в ходе нефтепереработки. Простейшим таким процессом является термическое разложение этана. Этот процесс имеет большую практическую значимость.
Для решения этих задач (в частности, при разработке новых химических реакторов) широко применяется численное моделирование. При этом используют сложные газодинамические коды, в которые входят уравнения кинетики химических реакций. Результат такого моделирования сильно зависит от используемых данных по скоростям химических реакций.
Скорости реакций находят из экспериментальных измерений либо теоретических расчетов. Широкоупотребительные методы теоретического расчета имеют ряд слабых мест, их критика представлена ниже. Поэтому остановимся на экспериментальных измерениях.
Основными экспериментальными техниками являются проточные реакторы (для экзотермических реакций) и ударные трубы (для эндотермических реакций) [3]. В таких экспериментах получают значение скорости реакции K(T) при фиксированных значениях температуры и давления.
Как правило, по каждой реакции имеется много экспериментальных работ различных авторов, причем диапазон условий в них частично перекрывается, частично различается. Обычно каждый автор производит обработку своих измерений, используя аппроксимирующую формулу обобщенного аррениусовского типа
Здесь A, n, E – подгоночные параметры. Из-за неизбежных погрешностей эксперимента результаты различных авторов отличаются друг от друга, причем нередко значительно.Чтобы нивелировать индивидуальные отклонения отдельных экспериментов, применяют совместные обработки данных из большого количества разных источников. Такие обработки проводятся сотрудниками Национального института стандартов и технологий (NIST) [4, 5], NASA [6], университета Беркли [7] и публикуются в различных тематических изданиях (например, [8–11]). Однако в таких обработках остается неясным, с какой точностью получаются коэффициенты обобщенной формулы Аррениуса. Коэффициенты, даваемые разными коллективами, могут отличаться. В этих работах иногда делаются экспертные оценки точности полученных коэффициентов и коридоров достоверности полученных кривых, однако аккуратный статистический анализ отсутствует.
В работе [12] предложен метод совместной обработки таких экспериментальных данных, который впервые позволил помимо аппроксимаций находить статистически достоверные оценки их точности. Этот метод обеспечивает отличную точность совместной аппроксимации данных из различных источников.
В данной работе с использованием метода [12] проведена обработка экспериментальных данных по скоростям системы реакций, описывающей пиролиза этана. Получена точность 1–25%, которая значительно превосходит мировой уровень.
МОДЕЛИ СКОРОСТЕЙ РЕАКЦИЙ
Для квантово-механического расчета [13, 14] нужно задать потенциал взаимодействия реагирующих молекул. Однако аккуратное нахождение этого потенциала теоретическими методами практически невыполнимо. Поэтому применяют различные эмпирические псевдопотенциалы, которые содержат свободные параметры, подгоняемые под эксперимент. Предсказательная сила такого расчета невелика.
Другим широко распространенным подходом является теория переходного состояния. Мы проанализировали различные варианты этой теории, описанные в [15]. Приведем критику этих теорий.
Теория переходного состояния
Это название объединяет группу теорий, перечисленных далее. В них предполагается, что исходные реагенты соединяются в промежуточную молекулу (называемую также активной)
Активная молекула сталкивается с другими молекулами, в результате чего часть кинетической энергии столкновения переходит во внутренние степени свободы активной молекулы. Если энергия одной из степеней свободы активной молекулы оказывается достаточно большой (т.е. превышает высоту потенциального барьера), то эта степень свободы разрывается, и реализуется один из каналов химической реакции. При этом рассматривается и обратный процесс – потеря энергии активной молекулой при столкновениях с другими частицами.
Такое рассмотрение разумно для реакций между сложными молекулами, содержащими много атомов. Тогда энергия может долго переходить между степенями свободы активной молекулы. Однако именно для сложных молекул теории переходного состояния дают большое расхождение с экспериментом [15]. Причина в том, что для многоатомной активной молекулы сложно найти фактические степени свободы, поскольку она не может распасться на части произвольного состава. Кроме того, потенциал, соответствующий этим степеням свободы, точно неизвестен. Возможны только модели, содержащие подгоночные параметры.
Если же исходные реагенты малоатомные, то число степеней свободы в активной молекуле невелико. Поэтому реакция должна происходить практически в одно столкновение. При попытке это учесть, теория переходного состояния сильно занижает скорости реакций. Поэтому при отсутствии подгонок фактические границы применимости теории переходного состояния неясны.
Теория Линдемана
В рамках описанного выше подхода предполагается, что преодоление потенциального барьера активной молекулой возможно только за счет перекачки энергии из поступательного движения. При этом внутренние степени свободы активной молекулы не задействуются. В этой теории вводился подгоночный параметр – конечное времени жизни активной молекулы, то есть исключалась быстрая ее деактивация. В противном случае теория резко занижала скорость реакции [15].
Модификация Хиншельвуда
Модификация Хиншельвуда основана на теории Линдемана. Основным отличием было предположение о том, что энергия, требуемая для преодоления потенциального барьера, частично берется из внутренних степеней свободы активной молекулы. Это позволило отказаться от введения фиктивного времени жизни активной молекулы, но сделало модель чувствительной к выбору внутренних степеней свободы. Фактически их число оказывается новым подгоночным параметром. Известны случаи, когда расхождение этой теории с экспериментом оказывается большим [15].
Теория Слэтера
В теории Слэтера используется механизм столкновений Хиншельвуда–Линдемана. Активная молекула представляется в виде системы гармонических осцилляторов, которые не взаимодействуют друг с другом. Реакция происходит тогда, когда координата одного из осцилляторов достигает критического значения.
Эта модель наследует указанную выше проблему теории Хиншельвуда. Кроме этого, приближение гармонического осциллятора справедливо только вблизи минимума потенциала. По мере приближения к максимуму потенциального барьера все больше сказывается ангармонизм. Последний неизбежно приводит к обмену энергией между степенями свободы, что противоречит исходным предположениям теории.
Теория Райса–Рамспергера–Касселя
Теория Райса–Рамспергера–Касселя (РРК) также основана на механизме столкновений Хиншельвуда–Линдемана, но допускает обмен энергией между внутренними степенями свободы за счет введения ангармонического потенциала. Чтобы реакция произошла, активная молекула должна иметь достаточную энергию, причем эта энергия должна быть сконцентрирована в нужной части молекулы. Данная теория разработана как в классическом, так и в квантовом варианте.
Модель РРК, как и теория Хиншельвуда, чувствительна к эмпирическому выбору внутренних степеней свободы активной молекулы. Их число остается подгоночным параметром. Во-вторых, эта теория должна явно содержать константу связи различных внутренних степеней свободы. Эта величина также фактически является подгоночной.
Теория Маркуса–Райса
Теория Маркуса–Райса (РРКМ) считается наиболее употребительной. Она построена на основе модели РРК. В ней вводится дополнительная ступень реакции – так называемый активированный комплекс AB+
Активная молекула AB* имеет достаточную энергию, но эта энергия не сосредоточена в нужной степени свободы. В активированном комплексе AB+ произошло нужное перераспределение энергии. Данное состояние соответствует максимуму потенциального барьера между реагентами и продуктами. Фактически скорость реакции AB* → AB+ является еще одним подгоночным параметром (третьим по счету после двух параметров в модели РРК). Таким количеством параметров можно подогнать любые экспериментальные данные. Поэтому проверка этой теории на конкретных экспериментах ничего не доказывает.
СИСТЕМА РЕАКЦИЙ
Система реакций пиролиза этана была взята из [16]. Она приведена в табл. 1. Здесь звездочка (*) обозначает радикал (то есть наличие неспаренного электрона у атома ${\text{C}}$). Носителями цепи являются атомарный водород ${\text{H}}$ и метил CH3. Эта система реакций, с одной стороны, максимально проста, то есть включает минимально возможное число стадий. С другой стороны, она адекватно описывает пиролиз этана как при низких конверсиях, так и при высоких [16, 17].
Таблица 1.
Результаты обработки экспериментальных данных (пояснения см. в тексте)
| Реакция | ΔT, К | lg A | E, эВ | N | ΔK, % |
|---|---|---|---|---|---|
| ${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}} \to {\text{2CH}}_{{\text{3}}}^{{\text{*}}}$ | 777–2108 | 13.18 | 3.05 | 297 | 15 |
| ${\text{CH}}_{{\text{3}}}^{{\text{*}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}} \to {\text{C}}{{{\text{H}}}_{{\text{4}}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{5}}}^{{\text{*}}}$ | 162–1143 | 12.15 | 0.529 | 77 | 8 |
| ${{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{5}}}^{{\text{*}}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}\,{\text{ + }}\,{\text{H}}$ | 672–914 | 11.22 | 1.35 | 23 | 9 |
| ${\text{H}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}} \to {{{\text{H}}}_{{\text{2}}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{5}}}^{{\text{*}}}$ | 280–1480 | 14.09 | 0.410 | 89 | 4 |
| ${\text{H + }}{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} \to {{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{5}}}^{{\text{*}}}$ | 198–825 | 12.70 | 0.664 | 299 | 4 |
| ${\text{CH}}_{{\text{3}}}^{{\text{*}}}{\text{ + }}{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} \to {{{\text{C}}}_{{\text{3}}}}{\text{H}}_{{\text{7}}}^{{\text{*}}}$ | 353–502 | 8.40 | 0.319 | 23 | 3 |
| ${{{\text{C}}}_{{\text{3}}}}{\text{H}}_{{\text{7}}}^{{\text{*}}} \to {\text{CH}}_{{\text{3}}}^{{\text{*}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}$ | 538–805 | 9.12 | 0.95 | 24 | 25 |
| ${\text{2}}{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{5}}}^{{\text{*}}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}}$ | 203–343 | 11.86 | 0 | 5 | 1 |
| ${{{\text{C}}}_{{\text{3}}}}{\text{H}}_{{\text{7}}}^{{\text{*}}}{\text{ + }}{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} \to {{{\text{C}}}_{{\text{3}}}}{\text{H}}_{{\text{5}}}^{{\text{*}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{6}}}}$ | 300–2500 | 10.20 | 0.30 | – | 50 |
| ${\text{CH}}_{{\text{3}}}^{{\text{*}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} \to {\text{C}}{{{\text{H}}}_{{\text{4}}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{3}}}^{{\text{*}}}$ | 672–776 | 4.153 | 0.080 | 50 | 1 |
| ${\text{CH}}_{{\text{3}}}^{{\text{*}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{3}}}^{{\text{*}}} \to {\text{C}}{{{\text{H}}}_{{\text{4}}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{2}}}}$ | 300–900 | 13.39 | 0 | 19 | 15 |
| ${{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{3}}}^{{\text{*}}}\,{\text{ + }}\,{\text{H}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{2}}}}\,{\text{ + }}\,{{{\text{H}}}_{{\text{2}}}}$ | 213–298 | 13.47 | 0 | 8 | 5 |
| ${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} \to {}^{{\text{*}}}{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{4}}}^{{\text{*}}}$ | 400–1500 | 14.9 | 2.64 | – | 20 |
| $^{{\text{*}}}{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{4}}}^{{\text{*}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}} \to {\text{CH}}_{{\text{3}}}^{{\text{*}}}\,{\text{ + }}\,{{{\text{C}}}_{{\text{3}}}}{\text{H}}_{{\text{7}}}^{{\text{*}}}$ | 400–1500 | 16.16 | 2.21 | – | 25 |
| ${}^{{\text{*}}}{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{4}}}^{{\text{*}}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}$ | 900–1100 | 10.2 | 0 | – | 600 |
ОБРАБОТКА ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ
Мы проанализировали литературу по скоростям K перечисленных реакций. По всем реакциям, кроме четырех, есть прямые экспериментальные данные. Для этих реакций в табл. 1 стоит прочерк в графе N, содержащей количество экспериментальных точек.
Оказалось, что скорости реакций диссоциации C2H6 → ${\text{2CH}}_{{\text{3}}}^{{\text{*}}},$ ${{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{5}}}^{{\text{*}}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} + {\text{H,}}$ ${{{\text{C}}}_{{\text{3}}}}{\text{H}}_{{\text{7}}}^{{\text{*}}}$ → → ${\text{CH}}_{{\text{3}}}^{{\text{*}}} + {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}$ сильно зависят от давления. Простейший закон Аррениуса не позволяет это учитывать. Однако в прикладных задачах (например, при крекинге нефти) давление составляет от одной до нескольких атмосфер. В этих условиях указанные реакции ведут себя как мономолекулярные; то есть скорость расхода исходного реагента пропорциональна его концентрации, причем коэффициент пропорциональности не зависит от давления (т.е. от концентраций всех остальных молекул). В химической литературе этот случай называется пределом высоких давлений [15].
Поэтому по перечисленным реакциям мы собирали данные, относящиеся к максимальным давлениям, достигнутым в оригинальных работах. Обычно эти давления составляли от 0.2 до 4 атм. Для каждой реакции отобранные данные разумно согласуются друг с другом.
По каждой реакции было собрано от ~30 до ~300 точек для K(T). В пределах экспериментальных погрешностей эти точки укладываются на прямую линию в координатах 1/T–lg K; это соответствует n = 0 в (1). Пример такого графика для реакции ${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}} \to {\text{2CH}}_{{\text{3}}}^{{\text{*}}}$ приведен на рис. 1. По этой реакции имеется 297 экспериментальных точек. Видно, что они образуют несколько размытую “полосу”.
Рис. 1.
Реакция ${{{\text{C}}}_{2}}{{{\text{H}}}_{6}} \to 2{\text{C}}{{{\text{H}}}_{3}}.$ Точки – эксперименты (полный список ссылок приведен в [19]). Сплошная линия – линейная аппроксимация, пунктир – границы ее доверительного интервала (соответствуют одному стандартному уклонению).
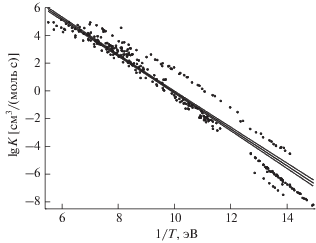
Эти данные были обработаны методом, предложенным в [12]. Напомним, что это метод совместной обработки, то есть аппроксимация строится для всего доступного массива данных, а не для точек каждой из работ в отдельности.
Метод заключается в аппроксимации полиномами, ортогонализованными на множестве экспериментальных точек с весами, соответствующими погрешностям этих точек. Для экзотермических реакций используется аппроксимация константой, для эндотермических – линейной функцией. Благодаря использованию ортогональных многочленов погрешности коэффициентов разложения не коррелированны между собой (тем самым, могут округляться независимо друг от друга). Это позволяет не только найти достоверную аппроксимацию, но и определить объективные оценки ее точности. Попутно уточняются оценки точности самих данных по отклонению от аппроксимирующей прямой.
УСРЕДНЕНИЕ ТЕОРЕТИЧЕСКИХ ОЦЕНОК
По четырем реакциям (реакция ${{{\text{C}}}_{{\text{3}}}}{\text{H}}_{{\text{7}}}^{{\text{*}}} + {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}$ → → ${{{\text{C}}}_{{\text{3}}}}{\text{H}}_{{\text{5}}}^{{\text{*}}} + {{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{6}}}}$ и три реакции с участием бирадикала ${\text{*}}{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{4}}}^{{\text{*}}}$) известны только теоретические оценки скорости. В зарубежной литературе эти реакции вовсе неосвещены. Данные по реакции ${\text{*}}{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{4}}}^{{\text{*}}} \to {{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}$ удалось найти только в работе [16]. Поскольку эта реакция, очевидно, является экзотермической, в качестве ее скорости можно взять предэкспоненциальный множитель в скорости эндотермической реакции ${{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}} \to {\text{*}}{{{\text{C}}}_{{\text{2}}}}{\text{H}}_{{\text{4}}}^{{\text{*}}}.$ Это довольно грубая оценка, но ее расхождение с данными из [16] укладывается в рамки погрешности, приведенные в этой работе. Поэтому такая оценка достаточно разумна.
Для других пар взаимно обратных реакций с участием радикалов такой подход неприменим, поскольку неспаренные электроны продуктов-радикалов могут находиться в высоковозбужденных состояниях. Поэтому такие реакции в обоих направлениях могут оказаться эндотермическими.
Для реакций, по которым имеются только теоретические формулы для скорости, эти формулы усредняются методом виртуального эксперимента [18]. Суть подхода заключается в следующем.
Для теоретических формул обычно приводятся диапазон температур, для которого они выводились, и некоторые оценки погрешности. Для каждой формулы разыграем достаточное количество (~30) случайных значений температуры в интервале ее применимости и вычислим значения K по этим формулам. Прибавим к этим значениям случайное возмущение в виде гауссовых случайных величин. Это возмущение имитирует случайную погрешность эксперимента. Оно имеют нулевое среднее и стандартное уклонение, равное оценке точности соответствующей теоретической формулы. Получим массив ∼30J “псевдоэкспериментальных” значений K(T), где J – количество теоретических формул. Этот массив обрабатывается тем же методом аппроксимации ортогональными полиномами. Выдаваемые при этом оценки доверительного интервала являются объективными оценками точности итоговых аппроксимаций.
БАЗА ДАННЫХ
Найденные коэффициенты аппроксимаций, а также оценки их точности приведены в табл. 1. Диапазон температур указан в кельвинах; предэкспоненциальный множитель A имеет размерность 1/с для мономолекулярных реакций и моль/(см3 ∙ с) для бимолекулярных реакций; энергия активации E дана в единицах электрон-вольт (1 эВ = 11604 К). Здесь N – число экспериментальных точек для каждой реакции, ΔK – средняя погрешность аппроксимации в указанном интервале температур.
Для тех реакций, по которым имеются экспериментальные данные, точность наших аппроксимаций составила от 1 до 25%. Подчеркнем, что эти оценки статистически достоверны и получены в результате математически строгой процедуры. Такую высокую точность удалось получить за счет совместной обработки большого количества данных.
Для реакций, по которым отсутствуют экспериментальные данные, полученная погрешность K(T) может интерпретироваться как оценка точности исходных теоретических моделей (как правило, различных вариантов теории переходного состояния). Видно, что эта точность заметно хуже, чем точность самих экспериментов.
В известных обзорах, рекомендации которых широко используются на практике, приводятся экспертные оценки точности аппроксимаций (см., например, [8]). Обычно они составляют 0.3–0.4 для lg K, т.е. 100–250%. Видно, что точность аппроксимаций, построенных в данной работе, кардинально превосходит мировой уровень.
Собранные экспериментальные данные со ссылками на оригинальные работы, а также построенные аппроксимации с оценками их точности и достоверности включены в базу данных ТЕФИС [19], разрабатываемую в Институте прикладной математики им. М.В. Келдыша РАН.
Работа поддержана РНФ (проект № 16-11-10001-П).
Список литературы
Мухина Т.Н., Барабанов Н.Л., Бабаш С.Е. и др. Пиролиз углеводородного сырья. М.: Химия, 1987.
Leatherd D.A., Purnell J.H. // Annu. Rev. Phys. Chem. 1970. V. 21. P. 197.
Davidson D.F., Hanson R.K. // Int. J. Chem. Kinet. 2004. V. 36. P. 510.
Westley F. Tables of recommended rate constants for chemical reactions occurring in combustion. National standard reference data series, NSRDS-NBS 67, 1980.
NIST Chemical kinetics database. Standard reference database 17-2Q98. Gaithersburg: NIST, 1998.
Burkholder J.B., Sander S.P., Abbatt J.P.D. et al. Chemical kinetics and photochemical data for use in atmospheric studies. Eval. No 18. Pasadena: JPL Publ., 2015.
http://www.me.berkeley.edu/gri_mech.
Baulch D.L. Bowman C.T., Cobos C.J. et al. // J. Phys. Chem. Ref. Data. 2005. V. 34. No 3. P. 757.
Ибрагимова Л.Б Смехов Г.Д., Шаталов О.П. // Физ.-хим. кин. в газ. динам. 2009. Т. 8. С. 1.
Mass U., Warnatz J. // Combust. Flame. 1988. V. 74. P. 53.
Miller J.A., Bowman C.T. // Progr. Energy Combust. Sci. 1989. V. 15. No 4. P. 287.
Белов А.А., Калиткин Н.Н. // ЖВМиМФ. 2020. Т. 60. № 7. С. 105.
Harris F.E. // Ann. Rev. Phys. Chem. 1972. V. 23. No 1. P. 415.
Murrell J.N., Carter S., Farantos S.C. et al. Molecular potential energy functions. N.Y.: Wiley, 1984.
Robinson P.G., Holbrook K.A. Monomolecular reactions. London, New York Sydney, Toronto: Wiley-interscience, 1972.
Nurislamova L.F., Stoyanovskaya O.P., Stadnichenko O.A. et al. // Chem. Prod. Proc. Model. 2014. V. 9. No 2. P. 143.
Snytnikov V.N. Snytnikov P.V., Amosov Yu.I. et al. // Kinet. Catalysis. 2010. V. 51. P. 10.
Belov A.A., Kalitkin N.N., Kozlitin I.A. // Fusion Engin. Design. 2019. V. 141. P. 51.
http://tefis.keldysh.ru.
Дополнительные материалы отсутствуют.
Инструменты
Известия РАН. Серия физическая