

Химическая физика, 2020, T. 39, № 4, стр. 19-30
Молекулярная и кристаллическая структура бинарных фторидов золота
Ш. Ш. Набиев 1, *, Л. А. Палкина 1
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
* E-mail: Nabiev_SS@nrcki.ru
Поступила в редакцию 19.05.2019
После доработки 10.06.2019
Принята к публикации 20.06.2019
Аннотация
Представлены и обобщены наиболее достоверные данные по структуре индивидуальных фторидов золота (I, III, V–VII) в различных агрегатных состояниях, в том числе в условиях матричной изоляции, а также в экстремальных (сверхвысокие давления) условиях. Исследованы колебательные спектры бинарных фторидов золота в разных степенях окисления. Особое внимание уделено изучению спектров аномально сильной льюисовской кислоты AuF5. На основании анализа совокупности спектроскопических, электронографических и рентгеноструктурных данных, а также данных квантовохимических расчетов сделаны предположения о строении молекул AuFk (k = 4–7) и AumFn (m = 2, 3; n = 6, 9) в конденсированном состоянии.
1. ВВЕДЕНИЕ
Изучение фторидов и оксофторидов металлов второго и третьего переходных рядов в высших степенях окисления относится к одному из наиболее интересных разделов химии фтора [1]. Интерес к ним обусловлен высокой реакционной способностью, не уступающей молекулярному фтору, а также сильными комплексообразующими свойствами [2, 3]. Комплексы высших фторидов и оксофторидов переходных металлов обладают высокой окислительной способностью и превосходят фтор по ряду эксплуатационных характеристик, что позволяет использовать их в качестве фторокислителей в реакциях с неорганическими и органическими веществами [1].
Среди высших фторидов металлов третьего переходного ряда одним из наименее изученных является AuF5 [4], первое сообщение о синтезе которого появилось в начале 70-х годов прошлого столетия [5]. Пентафторид золота в отличие от других фторидов переходных металлов обладает аномально высокой химической активностью, которая проявляется в том, что при Т = 273 К он окисляет элементарный Xe по крайней мере до XeF4 [6, 7]. При этом средняя энергия связи Au–F в AuF5 (≈54 ккал/моль) незначительно отличается от соответствующей величины в AuF3 (≈61 ккал/моль) [4, 8].
В данной работе представлены и обобщены наиболее достоверные данные по структуре индивидуальных фторидов золота (I, III, V–VII) в различных агрегатных состояниях, в том числе в условиях матричной изоляции, а также в экстремальных (сверхвысокие давления) условиях. Исследованы колебательные спектры фторидов золота в разных степенях окисления. Особое внимание уделено изучению спектров аномально сильной льюисовской кислоты AuF5. На основании анализа совокупности спектроскопических, электронографических и рентгеноструктурных данных, а также данных ab initio расчетов сделаны предположения о строении молекул AuFk (k = 4–7) и AumFn (m = 2, 3; n = 6, 9) в конденсированном состоянии.
2. СТРУКТУРНЫЕ ОСОБЕННОСТИ ФТОРИДОВ ЗОЛОТА
2.1. Фториды золота (I)
Возможность существования термодинамически нестабильного фторида золота (I) в газовой фазе была экспериментально доказана с использованием метода масс-спектрометрии [9]. Авторами работы [10] зарегистрирован микроволновый (МВ) спектр фторида золота(I), полученный методом лазерной абляции на поверхности металлического Au в присутствии F2. По результатам анализа МВ-спектра была определена вращательная постоянная Ве = = 7924.83328 МГц и постоянная колебательно-вращательного взаимодействия αе = 56.02704 МГц, что, в свою очередь, позволило определить равновесную длину связи Au–F во фториде золота(I): Re = 1.918449(5) Å.
В работе [11] на основе приближений Хартри–Фока (ХФ), второго порядка теории возмущений Меллера–Плессета (МР2), связанных кластеров (СК), теории функционала плотности (B3LYP) были изучены геометрические параметры мономера AuF и димера Au2F2 (рис. 1). Согласно проведенным расчетам длина связи Au–F в AuF составляет 1.964 Å (ХФ), 1.973 Å (B3LYP), 1.944 Å (MP2) и 1.954 Å (СК), что несколько выше соответствующей величины, определенной из МВ-спектра AuF [10].
Рис. 1.
Геометрическая структура AuF, димера Au2F2, основного (C2v) и переходного (D3h) состояний AuF3 и структура димера Au2F6 (D2h).
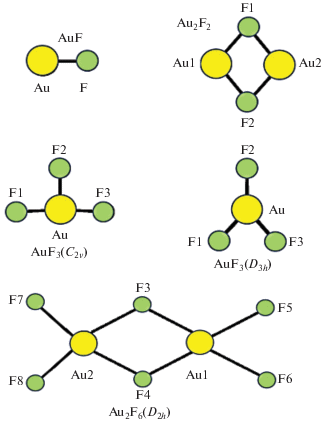
Для димера Au2F2 длина связи Au–F больше: 2.268 Å (ХФ), 2.261 Å (B3LYP), 2.215 Å (MP2) и 2.229 Å (СК). Значение угла ∠ (F–Au–F) лежит в пределах 93.7–101.7°, расстояние F∙∙∙F равно 3.215 Å (ХФ) и 3.151 Å (МР2), а расстояние Au∙∙∙Au составляет 2.856 Å (СК), 3.008 Å (МР2) и 3.051 Å (B3LYP). Столь короткий контакт Au∙∙∙Au в Au2F2 свидетельствует о наличии аурофильного взаимодействия. Понятие “аурофильности” введено относительно недавно [12] и определяется как “непредсказуемое” взаимодействие между атомами Au даже при электронной конфигурации c “замкнутой оболочкой” и эквивалентных зарядах. По силе это взаимодействие соответствует энергии Н-связи (≤50 кДж/моль), в некоторых случаях превышает ее и способно влиять на конформацию комплексов и их кристаллическую упаковку [11].
2.2. Фториды золота (III)
Молекулярная геометрия мономера AuF3 и димера Au2F6 экспериментально определена с помощью метода газовой электронографии (ЭГ), а также ab initio расчетов [11]. Геометрические параметры AuF3 и Au2F6 в газовой (паровой) фазе приведены в табл. 1. Полученные ЭГ данные, а также результаты расчетов показали, что структура основного состояния (1A1) AuF3 имеет симметрию C2v (рис. 1) с одной короткой (R(Au–F2) = = 1.893 ± 0.012 Å) и двумя более длинными (R(Au–F3) = R(Au–F4) = 1.913 ± 0.008 Å) связями Au–F. При этом AuF3 в состоянии 1A1 обладает Т‑образной конфигурацией с ∠(F2–Au–F3) = = 94.4°. Такая необычная геометрия реализуется из-за проявления эффекта Яна–Теллера и, как следствие, искажения тригонально-бипирамидальной (ТБП) структуры с симметрией D3h, в которой две экваториальные позиции заняты неподеленными электронными парами.
Таблица 1.
Геометрические параметры мономеров AuF3 и димеров Au2F6 в газовой (паровой) фазе (T = 600 K)
| Параметр* | Значение |
|---|---|
| Мономер AuF3 (C2v) | |
| ΔR ≡ R(Au1–F3) – R(Au1–F2) | 0.020 |
| R(Au1–F2) | 1.893 ± 0.012 |
| R(Au1–F3) | 1.913 ± 0.008 |
| R(F2∙∙∙F3) | 2.950 ± 0.057 |
| R(F3∙∙∙F4) | 3.748 ± 0.029 |
| ∠(F2–Au–F3) | 94.4 ± 1.9 |
| ∠(F3–Au–F4) | 157.0 ± 4.1 |
| Димер Au2F6 (D2h) | |
| ΔR ≡ R(Au1–F3)b – R(Au1–F5)t | 0.1570.002 |
| R(Au1–F5)t | 1.876 ± 0.006 |
| R(Au1–F3)b | 2.003 ± 0.007 |
| R(Au1∙∙∙Au2) | 3.082 ± 0.006 |
| ∠(F3–Au1–F4) | 92.1 ± 1.0 |
| ∠(F5–Au1–F6) | 80.4 ± 1.6 |
Димер Au2F6 имеет плоскую конфигурацию симметрии D2h (рис. 1), в которой атомы золота, образующие слегка искаженную плоскую квадратную координацию, соединены F-мостиками. Согласно данным ЭГ, а также результатам ab initio расчетов [11], длина концевой связи Au1–F5 близка к таковой в мономере AuF3: 1.876 Å (ЭГ), 1.895 Å (B3LYP) и 1.873 Å (MP2); длина мостиковой связи Au1–F3 больше: 2.033 Å (ЭГ), 2.059 Å (B3LYP) и 2.03 Å (MP2); $\angle $(F5–Au1–F6) близок прямому: 92.1° (ЭГ), 89.7° (B3LYP), 89.3° (MP2), а ∠(F3–Au1–F4) = 80.4° (ЭГ), 78.8° (B3LYP).
Кристаллическая структура монокристаллического AuF3, который кристаллизуется в гексагональную конфигурацию (пространственная группа P6122-D62), исследовалась в работе [13]. Было показано, что кристаллическая структура AuF3 очень своеобразна и представляет собой спиралевидную полимерную цепь с F-мостиками; связь между отдельными цепями осуществляется посредством более слабых F-мостиков. Каждый атом золота образует достаточно прочные связи с двумя концевыми и двумя мостиковыми F-атомами, что приводит к реализации плоской квадратной конфигурации AuF4; каждый из блоков AuF4 соединен между собой F-мостиками, в результате чего образуется спиральная цепочечная структура. Длина связей R(Au–F1) = R(Au–F2) = 1.868 Å, R(Au–F3) = R(Au–F4) = 1.998 Å, R(Au–Fb) = 2.761 Å, ∠ (Au–F3–Au) = 119.3°. Структурные параметры мономеров AuF3 в кристаллическом состоянии представлены в верхней части табл. 2.
Таблица 2.
Экспериментальные и расчетные структурные параметры мономера AuF3 и димера (AuF5)2 в кристаллическом состоянии
| Параметр* | Экспериментальные данные | Теоретические расчеты** | |
|---|---|---|---|
| рентгеноструктурный анализ | электронография | ||
| Мономер AuF3 | |||
| R(Au–F1), R(Au–F2) | 1.868(3) | 1.893 ± 0.012 | 1.902 |
| R(Au–F3), R(Au–F4) | 1.998(2) | 1.913 ± 0.008 | 1.911 |
| R(Au–Fb) | 2.761(3) | – | – |
| R(F2∙∙∙F3) | – | 2.950 ± 0.057 | – |
| R(F3∙∙∙F4) | – | 3.748 ± 0.029 | – |
| ∠(Au–F3–Au) | 119.3(2) | – | – |
| ∠(F2–Au–F3) | – | 94.4 ± 4.0 | 94.6 |
| Димер (AuF5)2 | |||
| R(Au–F1), R(Au–F4) | 1.891(6) | 1.901(5) | 1.925 |
| R(Au–F2), R(Au–F3) | 2.103(5) | 2.031(5) | 2.060 |
| R(Au–F5), R(Au–F6) | 1.854(6) | 1.875(6) | 1.884 |
| ∠(F2–Au–F3) | 78.4(2) | 80.1(5) | 78.0 |
| ∠(F1–Au–F4) | 178.5(3) | 181.0(11) | 178.7 |
| ∠(F5–Au–F6) | 87.0(3) | 92.3(17) | 88.8 |
* См. примечание к табл. 1. ** Усредненные данные, полученные в результате ab initio расчетов с использованием методов ХФ, MP2, CК и B3LYP.
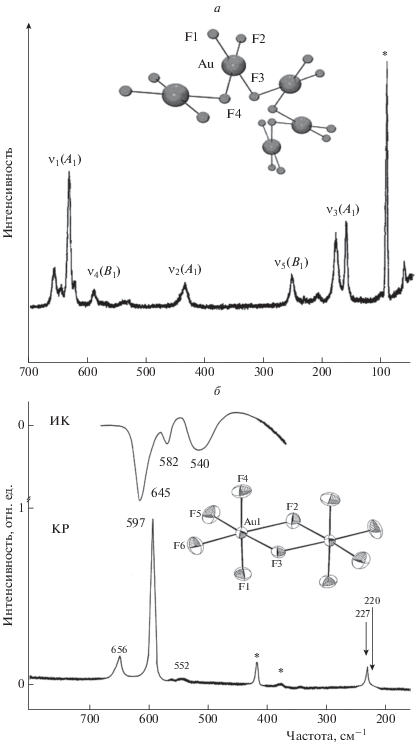
Молекула AuF3 (С2v) имеет шесть нормальных колебаний с типами симметрии Гvib = 3A1 + 2В1 + В2, активных как в ИК-спектре, так и в спектре комбинационного рассеяния (КР). В колебательном спектре AuF3 наблюдается шесть полос со следующими частотами: ν1(Аl) = 632 см–1, ν2(Аl) = 439 см–1, ν3(А1) = 182, 164 см–1, ν4(B1) = 594 см–1, ν5(B1) = = 254 см–1, ν6(В2) = 206 см–1. Вид спектра КР AuF3 в твердой фазе представлен на рис. 2а. Расщепление линий при 650 и 200 см–1 связано с наличием различно ориентированных блоков AuF4 в решетке AuF3, с действием кристаллического поля, а также с проявлением эффектов внутренней асимметрии [14].
Рис. 2.
Спектр КР AuF3 (а) и колебательный спектр AuF5 (б) в кристаллической фазе. Звездочкой обозначены линии сапфира (материал КР-ячейки). На вставках приведен вид кристаллической структуры AuF3 (вверху) и AuF5 (внизу).
Молекула Au2F6 (точечная симметрия D2h) характеризуется 18 нормальными колебаниями с типами симметрии Гvib = 4Ag + B1g + B2g + 3B3g + Au + + 2B1u + 3B2u + 3B3u, из которых колебания с симметрией Ag, B1g, B2g и B3g активны в спектре КР, а колебания с симметрией B1u, B2u и B3u – в ИК-спектре. Расчетные колебательные частоты Au2F6 имеют следующие значения: ν1(Ag) = 644 см–1,ν2(Ag) = = 486 см–1, ν3(Ag) = 230 см–1, ν4(Ag) = 121 см–1, ν5(Au) = 29 см–1, ν6(B1g) = 200 см–1, ν7(B1g) = 626 см–1, ν8(B1g) = 636 см–1, ν9(B1u) = 471 см–1, ν10(B1u) = = 217 см–1, ν11(B2g) = 172 см–1, ν12(B2u) = 636 см–1, ν13(B2u) = 471 см–1, ν14(B2u) = 125 см–1, ν15(B3g) = = 416 см–1, ν16(B3u) = 230 см–1, ν17(B3u) = 172 см–1, ν18(B3u) = 72 см–1.
У большинства молекул типа X2Y6 (точечная симметрия D2h) частоты ν1, ν8, ν12, ν16 принадлежат в основном валентным колебаниям концевых групп XY2. Значения этих частот выше, чем частот ν2, ν6, ν13, ν17, которые относятся преимущественно к колебаниям мостиковых групп ${{{\text{X}}}_{{\text{2}}}}{\text{Y}}_{{\text{2}}}^{'}{\text{.}}$
2.3. Фториды золота (V)
Полученные к сегодняшнему дню результаты по изучению структуры фторида золота(V) различными методами не дают однозначного ответа на вопрос о строении молекулы AuF5 как в газовой (паровой), так и в твердой фазах. Электронографические исследования [15] показали, что в паровой фазе при Т ≈ 500 К AuF5 состоит в основном из димеров Au2F10 (D2h) и тримеров Au3F15 (D3h) в соотношении ≈80 : 20. В более поздней работе [16], в которой изучалась структура твердого и газообразного AuF5, было показано, что он имеет структуру димера Au2F10 с гексакоординированным атомом Au и восьмигранную геометрию расположения F-атомов вокруг каждого атома Au. Был точно определен состав газообразного AuF5 – смесь димера и тримера в соотношении 82 : 12. В табл. 3 приведены структурные параметры Au2F10 и Au3F15 в газовой (паровой) фазе.
Таблица 3.
Геометрические параметры димеров Au2F10 и тримеров Au3F15 в газовой (паровой) фазе (Т ≈ 500 К) *
| Параметр** | Значение | |
|---|---|---|
| димер Au2F10 (D2h) | тример Au3F15 (D3h) | |
| ∆R ≡ R(Au–Fa) – R(Au–Ft) | 0.067(16) | – |
| R(Au–Fa)** | 1.889(9) | – |
| R(Au–Ft) | 1.822(8) | – |
| R(Au–Fb) | 2.030(7) | – |
| ∠(Fa–Au–Fa) | 181.0(1.1) | 193.1(3.2) |
| ∠(Ft–Au–Ft) | 93.3(1.7) | 75.3(6.5) |
| ∠(Fb–Au–Fb) | 80.1(0.5) | 115.7(1.1) |
| ∠(Au–Fa–Au) | 99.9(0.5) | 124.3(1.1) |
* См. примечание к табл. 1. ** Fa – атом фтора в аксиальной связи Au–F.
Анализ мессбауэровских спектров ряда простых и комплексных фторидов Аu(III) и Au(V) показал, что самые низкие значения изомерного сдвига δ зарегистрированы для фторидов AuF3 и AuF5 [17]. Эту особенность можно объяснить увеличением ковалентности связи Аu–F при переходе от ${\text{AuF}}_{4}^{ - }$ к AuF3 и от ${\text{AuF}}_{6}^{ - }$ к AuF5, обуславливающим увеличение электронной плотности на ядре. Аномально низкое значение δ шестикоординационного AuF3 можно объяснить образованием по оси z дополнительных π-связей за счет электронов атомных dzx- и dxy-орбиталей Au, более локализованных на атомах фтора по сравнению с делокализованными d-электронами в случае восьмикоординационного ${\text{AuF}}_{4}^{ - }.$ В то же время в соединениях Au(V) степень делокализации можно считать практически неизменной. Хотя и нет строгой зависимости δ от структурных параметров, эта величина чувствительна к изменению координации атома Au.
На основании рентгенографических данных порошка AuF5 было сделано предположение [18], что наиболее предпочтительной структурой AuF5 можно считать закрученную в спираль полимерную цепочку (винтовая ось шестого порядка), которая состоит из координационных полиэдров – искаженных октаэдров AuVF6. Эти полиэдры соединены между собой в цис-положении мостиковыми Au–F–Au-связями: …–F–AuF4–F–AuF4–F–… (симметрия C2v), причем расстояние Au∙∙∙Au равно 4.24 Å, а длина связей Au–Fb должна быть не менее 2.12 Å. Авторами работы [18] отмечалось, что надежное определение структуры AuF5 возможно только после получения рентгеноструктурных данных для кристаллического пентафторида золота.
Такие данные были получены в более поздней работе [16], авторами которой было обнаружено, что пентафторид золота в кристаллической фазе имеет схожую со структурой AuF5 в паровой фазе димерную, слегка искаженную структуру симметрии D2h, в которой молекулы AuF5 соединены между собой экваториальными F-мостиками. При этом длина связей R(Au–Fax) = 1.891, 1.901 Å; R(Au–Feqv) = 1.854, 1.874 Å, R(Au–Fb) = 2.013, 2.030 Å.
О реализации димерной структуры кристаллического AuF5 могут свидетельствовать результаты квантовохимических расчетов [16, 19]: R(Au–Fax) = = 1.965 Å, R(Au–Feqv) = 1.925 Å, R(Au–Fb) = 2.077 Å (МР2) [16]; R(Au–Fax) = 1.887 Å, R(Au–Feqv) = = 1.846 Å, R(Au–Fb) = 2.024 Å (B3LYP) [19].
На основании анализа совокупности экспериментальных и теоретических данных по структуре фторидов золота, а также предположительного описания геометрического строения AuFn (n = 1, 3, 5), представленных в настоящей работе, для монокристаллического AuF5 предпочтение отдается реализации именно димерной D2h-структуры. Структурные параметры (AuF5)2 (симметрии D2h) в кристаллическом состоянии представлены в нижней части табл. 2.
Особенностью колебательных спектров AuF5 в твердой фазе является тот факт, что они по своим параметрам кардинально отличаются от спектров других пентафторидов металлов [14]. Так, в ИК-спектре AuF5 наблюдаются три полосы: вблизи 645, 582, 540 см–1, а в спектре КР – две линии с частотами при 656, 597 см–1 и линия вблизи 227 см–1 с плечом при 220 см–1 (рис. 2б). Частоты в области 660–590 см–1 можно отнести к валентным, а частоты вблизи 220 см–1 – к деформационным колебаниям AuF5 в составе (AuF5)2. Полосы при 540 (ИК-спектр) и 550 см–1 (КР-спектр) можно отнести к колебанию мостиковых связей. Такое отнесение не противоречит данным рентгеноструктурного анализа и свидетельствует в пользу полимерной структуры AuF5 в твердой фазе. Анализ спектра КР AuF5 в растворе HF дает основание предположить, что в этом случае он также имеет полимерную структуру, где каждый атом Au имеет октаэдрическое окружение, а соседние фрагменты AuF6 соединены F-мостиками в цис-положениях. Линия вблизи 540 см–1 может и не наблюдаться из-за низкой интенсивности колебаний F-мостиковых связей в спектрах КР жидких сред [20].
3. КВАНТОВОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ СТРУКТУРЫ ФТОРИДОВ ЗОЛОТА(V, VI И VII)
Изучению геометрической конфигурации фторидов золота методами квантовой химии посвящено относительно небольшое количество работ (см., например, работы [11, 12, 16, 19] и ссылки в них). Это связано с тем, что для многоэлектронных молекул, какими являются фториды золота, нахождение минимумов на потенциальных поверхностях энергии (ППЭ), определение строения молекул, расчет равновесных межъядерных расстояний и валентных углов, энергии химических связей и других параметров, сопряжено с заметными трудностями [21]. Только в последние годы с разработкой новых подходов, методик и алгоритмов квантовохимических расчетов [22] появились реальные возможности расчета структурных параметров и колебательных частот для фторидов золота(V, VI и VII).
Авторами работы [19] методами ХФ, МР2, СК, B3LYP были рассчитаны структурные параметры мономера, димера и тримера AuF5. Результаты расчетов показали, что для мономера AuF5 реализуется квадратно-пирамидальная конфигурация (C4v), которая может легко изменяться посредством структурной трансформации в ТБП-конфигурацию (высота барьера равна 9.5 кДж · моль–1). Длины связей в AuF5 (C4v) составили: R(Au–Fax) = 1.80 Å (ХФ), 1.888 Å (МР2), 1.867 Å (СК), 1.882 Å (B3LYP); R(Au–Feqv) = 1.858 Å; 1.80 Å (ХФ), 1.93 Å (МР2), 1.905 Å (СК), 1.921 Å (B3LYP).
Анион ${\text{AuF}}_{6}^{ - },$ как и ожидалось, имеет симметрию Oh при длинах связи Au–F, равных 1.939 Å. Небольшие отклонения от конфигурации идеального октаэдра, по-видимому, являются следствием заметных катион-анионных взаимодействий, характерных для большинства комплексных фторидов с участием сильных кислот Льюиса [2, 3, 7, 14].
Для изучения возможности получения степени окисления золота до Au(VI) и Au(VII), авторами работ [19, 23] были рассмотрены низшие уровни электронного состояния молекул AuF6 и AuF7. Результаты расчетов показали, что для молекулы AuF6 существуют локальные минимумы, соответствующие низкоспиновому дублету (D2h) и высокоспиновому секстету (Оh). Секстет лежит на 369.6 кДж · моль–1 выше основного состояния дублета. За счет эффекта Яна–Теллера в дублете наблюдается уменьшение длины связей (Au–F)ax (≈0.12 Å), а также небольшое нарушение симметрии в экваториальной плоскости, сопровождающееся структурной трансформацией D4h → D2h.
Экспериментальному исследованию возможности существования молекулы AuF7 посвящена работа [24]. Ее авторы представили AuF7 как реально существующее соединение Au(VII) лишь на основании наблюдавшейся в ИК-спектре одной полосы при 734 см–1. Однако, согласно оценкам из работы [25], AuF6 имеет огромное адиабатическое сродство к электрону (≈8.2–9.6 эВ), что делает практически невозможным получение степени окисления Au выше +5. Авторы работ [19, 26] смогли обнаружить только один минимум на ППЭ основного состояния синглета d4 молекулы AuF7 – пятиугольную бипирамиду (D5h) (рис. 3а) и ни одного устойчивого минимума для триплета и квинтета.
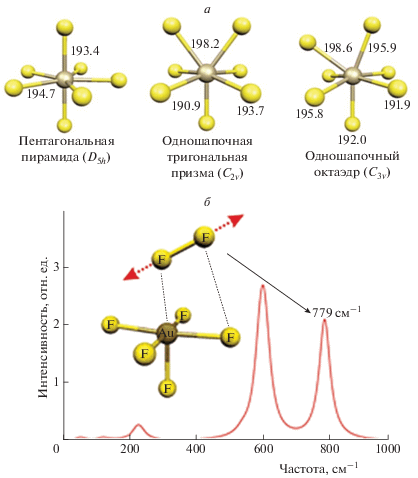
Модель VSEPR (Valence Shell Electron Pair Repulsion) [27] для систем типа XY7 предполагает реализацию еще двух координационных полиэдров в гепта-координатах [28]: одношапочную треугольную призму (C2v) и одношапочный октаэдр (C3v). Результаты расчетов [19, 26] показали, что структура одношапочной треугольной призмы молекулы AuF7 является переходным состоянием с мнимой частотой в 70.3i см–1. Оптимизация структуры одношапочного октаэдра (C3v) дает стационарную точку с двумя мнимыми частотами (50.7i и 37.0i см–1). Оптимизированные структуры для этих точек располагаются на 16.5 (C2v) и 17.2 кДж · моль–1 (C3v) выше минимума D5h.
Проведенные нами расчеты гармонических колебательных частот для фторидов Au(V), Au(VI) и Au(VII) показали, что AuF7 в конформации основного состояния D5h не имеет колебания с частотой (734 ± 3) см–1, на основе наличия которой в [24] был идентифицирован AuF7. Самые высокие расчетные значения частоты валентных колебаний для связей Au–F составили 634, 592 и 589 см–1 для D5h-, C2v- и C3v-структур соответственно. Самые высокие частоты, вычисленные для AuF6 (631 см–1), AuF5 (633 см–1) и (AuF5)2 (647 см–1), также заметно ниже, чем частота 734 см–1. Отметим, что вычисленные для AuF5 и (AuF5)2 частоты хорошо коррелируют с экспериментальными данными, полученными авторами [5, 6, 16, 17].
В работах [23, 26] сделано предположение, что наблюдаемая в [24] частота 734 см–1 принадлежит колебаниям аддукта AuF5 · F2, который более чем на 205 кДж · моль–1 стабильнее, чем AuF7. Результаты моделирования ИК-спектра AuF5 · F2 (рис. 3б) показали, что он сформирован слабыми полосами в области 50–250 см–1, а также двумя сильными полосами при 595 и 779 см–1. Полосы вблизи 50–250 и 595 см–1 могут быть отнесены к деформационным и валентному колебанию связей Au–F, а частота 779 см–1 – к колебанию связи F–F молекул F2 в составе AuF5 · F2. Отметим, что длина связи F–F в зависимости от координации молекул F2 в составе аддукта AuF5 · F2, находящихся в сильном поле AuF5, может увеличиваться. Это ведет к смещению частоты этой связи в область более низких значений на 30–40 см–1. По-видимому, именно эту частоту (734 см–1) наблюдали авторы работы [24].
4. ФТОРИДЫ ЗОЛОТА (IV И VI) ПРИ ВЫСОКИХ ДАВЛЕНИЯХ
Экстремальные условия, в частности высокие и сверхвысокие давления, широко используются как средство изучения природы веществ и происходящих в них явлений [29]. Эти условия применяются в качестве методов получения новых и обработки уже известных материалов [30]. Сверхвысокие давления в настоящее время активно используются в различных областях физики твердого тела, химии, геофизики, геохимии и др. [31, 32].
Для химии веществ, находящихся в условиях высокого давления, существен тот факт, что в этом случае происходит не только уменьшение межатомных расстояний и объема атомов, но и принципиальная перестройка электронных уровней [31, 33]. Вследствие этого вещество, находящееся в “сжатом” состоянии, может проявлять совершенно иные свойства, отличающиеся от присущих ему свойств при атмосферном давлении.
В работе [34] было показано, что окислительная способность фтора усиливается с ростом давления. Поэтому при условии устойчивости фторидов Au при высоких давлениях можно ожидать появления новых окислительных состояний золота, которые не проявляются при атмосферном давлении.
Недавно [35] с использованием методов теории функционала плотности и CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) [36] были изучены структура и устойчивость соединений AumFn (m = 1, n = 1–7 или m = 2, n = 1) при давлениях 1 атм. и 25, 50, 100, 200 ГПа. Устойчивость соединений AumFn определялась путем вычисления и сравнения значений энтальпии образования каждого соединения в диапазоне давлений ΔР = 0–200 ГПа (рис. 4а). Результаты расчетов [35] показали, что с ростом давления фториды Au2F, AuF2, AuF4 и AuF6 становятся стабильными, тогда как устойчивый при атмосферном давлении AuF5 разлагается на AuF4 и AuF6. Было также показано, что в диапазонах давления, характерных для стабильных фторидов AumFn (рис. 4б), можно проводить реакции их синтеза. Например, реакция AuF3 с Au при Р ≥ 13.3 ГПа дает AuF2, фторид AuF4 может быть синтезирован с использованием AuF3 и AuF5 в качестве прекурсоров при Р ≥ 13.5 ГПа, а AuF6 можно получить при более низких давлениях (Р ≥ 5.0 ГПа) смеси AuF5 и F2.
Рис. 4.
Устойчивость соединений AumFn (m = 1, n = 1–7 или m = 2, n = 1); a – энтальпии образования AumFn при разных давлениях: 1 – 1 атм, 2 – 25 Гпа, 3 – 50 Гпа (темные и светлые точки соответствуют устойчивым и неустойчивым соединениям AumFn); б – диапазоны давления для стабильных AumFn.
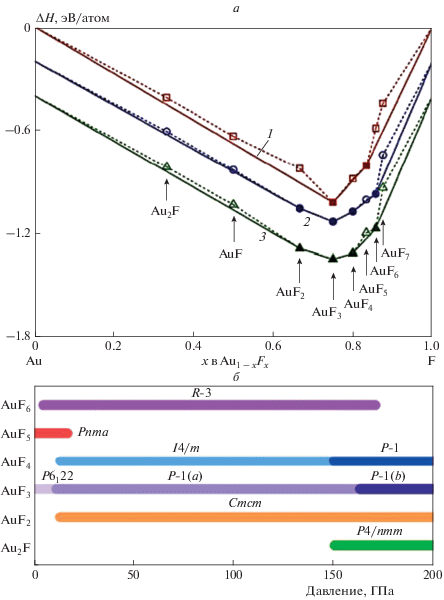
Соединения AumFn (m = 1, n = 1–7 или m = 2, n = 1) в условиях высокого давления проявляют также интересные структурные особенности. Так, с ростом давления структура AuF3 с пространственной группой P6122 трансформируется в структуру P-1. Обе структуры содержат квазиквадратные блоки AuF4; при этом с увеличением давления одиночные F-мостики, находящиеся в цис-положении, превращаются в двойные F-мостики.
Соединение AuF4 стабилизируется в тетрагональной структуре с пространственной группой I4/m при Р ≥ 13.5 ГПа, содержащей плоские квадратные молекулярные блоки. Примечательно, что ближайшее расстояние относительно атомов Au–Au при Р = 50 ГПа составляет 3.85 Å. Эта величина заметно превышает расстояние аурофильного взаимодействия, составляющее 3.0 Å и указывает на очень слабое взаимодействие между соседними атомами золота. Структура AuF5 с пространственной группой Pnma при P = 1.0 атм. состоит из димеров (AuF5)2 и становится нестабильной при Р ≥ 17.3 ГПа.
Устойчивость молекулы AuF6 прогнозируется в тригональной структуре (пространственная группа R-3), состоящей из октаэдрических блоков AuF6. Ближайшее расстояние относительно атомов Au–Au (4.22 Å) в этой структуре заметно больше, чем в AuF4. Кратчайшее расстояние F–F составляет 2.22 Å. Оно намного превышает длину ковалентной связи F–F (1.44 Å) [37] и указывает на то, что AuF6 является молекулярным кристаллом. По мнению авторов работы [35], учитывая большую величину электроотрицательности фтора (3.98) и особенности молекулярных кристаллов, степени окисления Au в AuF4 и AuF6 могут быть идентифицированы как (+4) и (+6) соответственно.
5. ФТОРИДЫ ЗОЛОТА В УСЛОВИЯХ МАТРИЧНОЙ ИЗОЛЯЦИИ
Метод низкотемпературной матричной изоляции (МИ) широко используется для синтеза соединений, неустойчивых при комнатной температуре, а также химически высокоактивных, токсичных и взрывоопасных веществ [38]. По спектрам молекул в условиях МИ могут быть определены их фрагменты, стехиометрия, симметрия и даже геометрические параметры [39].
В работах [40, 41] для синтеза фторидов золота в матрицах аргона, неона (Т = 4 К) и фтора (Т = 12 К) аналогично работе [10] использовался метод лазерной абляции. Результаты предварительных экспериментов в Ar-матрице (0.4% F2) показали [40], что в ИК-спектрах присутствуют полосы при 640.1, 575.1 и 474.7 см–1. При отжиге матрицы до Т = 30 К наблюдалась новая полоса при 646.1 см–1. Увеличение концентрации F2 в смеси до 4.0% привело к появлению дополнительной группы полос при 720, 690.1, 655.0, 610.6, 489.0 см–1 (рис. 5а). В экспериментах с использованием Ne-матрицы [41] (рис. 5б) наблюдались полосы при 720.0, 692.4, 664.8, 611.3 и 567.2 см–1, (полоса при 692.4 см–1 обладала наибольшей интенсивностью).
Рис. 5.
ИК-спектры продуктов реакций атомарного Au и F2 в условиях избытка Ar (а) и Ne (б) при Т = 4 К; вверху 1 – Au + F2 (4%) после 1 ч выдержки, 2 – после отжига до Т = 35 К, 3 – после облучения (λ > 220 нм), 4 – после отжига до Т = 40 К; внизу: 1 – Au + F2 (0.4%) после 1 ч выдержки, 2 – после 15 мин облучения λ > 530 нм, 3 – после 15 мин облучения при λ > 380 нм, 4 – после 15 мин облучения при λ = 240–380 нм, 5 – после отжига до Т = 10 К.
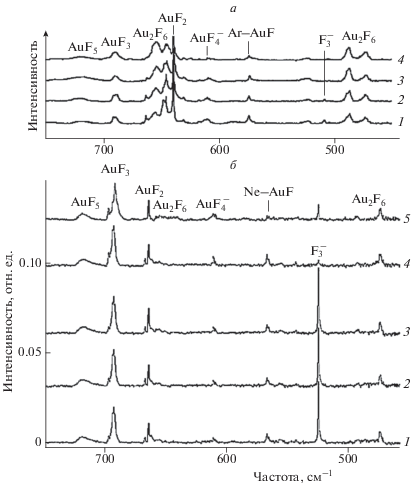
Перечисленные выше полосы в ИК-спектрах продуктов реакции атомов Au с F2 в условиях матричной изоляции отнесены к колебаниям Ng–AuF (Ng = Ne, Ar), AuF2, AuF3, ${\text{AuF}}_{4}^{ - },$ AuF5 и Au2F6. Так, частота 720 см–1 отнесена к валентному колебанию Au–F в мономере AuF5. Полосы при 567.2 и 575.1 см–1 в Ne- и Ar-матрицах, соответственно, принадлежат колебанию Au–F молекулы AuF в составе комплекса Ng–AuF. Интенсивные полосы при 640.1 (Ar) и 664.8 см–1 (Ne) относятся к антисимметричному колебанию связи Au–F молекулы AuF2. Результаты анализа сдвигов частоты Au–F в Ar- и Ne-матрицах указывают на отсутствие химической связи Ng–Au, наличие которой наблюдалась в случае молекулы AuF. Согласно результатам квантовохимических расчетов [41], частота антисимметричного колебания связи Au–F молекулы AuF2 составляет 647 см–1, что ниже экспериментального значения в матрице Ne (664.8 см–1). Средняя по интенсивности полоса при 690.1 см–1 (Ar) и наиболее интенсивная полоса при 692.4 см–1 (Ne) относятся к антисимметричному колебанию связи Au–F в AuF3.
Рост поглощения на частотах 611.3 (Ne) и 610.6 см–1 (Ar) после облучения Ar- и Ne-матриц УФ-излучением с λ > 380 и λ = 240–380 нм, соответственно, которое разрушает [F3]–-группу, дает основание предполагать, что причиной такого роста является образование аниона [AuF4]–, имеющего структуру плоского квадрата (симметрия D4h) [41]. При этом расчетная частота 617.2 см–1 отнесена к антисимметричному колебанию Au–F с типом симметрии Eu.
Группы полос поглощения при 655, 646 см–1 (Ar) и 655, 644 см–1 (Ne), а также при 489, 475 см–1 (Ar) и 494, 474 см–1 (Ne) могут быть отнесены соответственно к валентным и деформационным колебаниям связи Au–F в димере Au2F6. Однако, как уже отмечалось, для молекул типа Au2F6 концевые и мостиковые колебания трудно различимы. Поэтому можно предположить, что полосы при 646 и 474 см–1 относятся к колебаниям терминальной и мостиковой связей Au–F в димере Au2F6, а более интенсивные полосы при 655 и 489 см–1 – к колебаниям соответствующих связей в тримере Au3F9.
6. ЗАКЛЮЧЕНИЕ
Исследования молекулярной и кристаллической структуры бинарных фторидов золота на первых этапах были серьезно осложнены из-за их аномально высокой реакционной способности и, как следствие, трудностей в обращении с этими веществами. По мере развития технологий и появления новых конструкционных материалов структурные исследования фторидов золота вышли на новый качественный уровень. Результаты этих исследований показали, что фториды золота – это первые соединения, в которых степень окисления золота составляет (+5), а возможно, и (+7), и продемонстрировали примеры реализации редкой степени окисления Au, равной (+2).
В результате реакции атомов Au, полученных методом лазерной абляции, с F2 при избытке аргона и неона наряду с более тяжелым известным гомологом ArAuF получен новый гомолог NeAuF, характеризующийся сильной химической связью Nе–AuF. В ИК-спектрах аргоновой и неоновой матриц идентифицированы молекулы AuF2, AuF3, ${\text{AuF}}_{4}^{ - }$ и AuF5, при этом ИК-спектры AuF2 – это первое наблюдение бинарного фторида золота Au(II). Весьма интересным оказалось наличие в спектрах полос мостиковой и терминальных связей Au–F, отнесенных к колебаниям димера Au2F6.
Изучение фазовых диаграмм, структуры и электронных свойств бинарных фторидов Au при высоких давлениях показали, что AuF4 и AuF6 представляют собой стабильные молекулярные кристаллы со степенью окисления Au, равной (+4) и (+6) соответственно. Полученные результаты не только дополняют ряд промежуточных состояний окисления Au, но и свидетельствуют о возможности реализации для Au беспрецедентно высокой степени окисления. Детальный анализ электронных свойств бинарных фторидов золота свидетельствует о том, что ионные связи Au–F играют ключевую роль в определении их стабильности. Кроме того, для стабилизации AuF4 требуются меньшие давления, чем для стабилизации CuF4 или AgF4.
Отметим, что рассчитанные фазовые диаграммы устойчивости соединений AumFn (m = 1, n = 1–7 или m = 2, n = 1) при высоких давлениях могут оказаться весьма полезной “путевой картой” для будущего экспериментального синтеза указанных бинарных фторидов золота.
Список литературы
Synthetic Fluorine Chemistry / Eds. Olah G.A., Chambers R.D., Prakash G.K.S. N.Y.: Wiley InterScience, 1992.
Набиев Ш.Ш., Соколов В.Б., Чайванов Б.Б. // Успехи химии. 2014. Т. 83. № 12. С. 1135.
Brock D.S., Schrobilgen G.J., Žemva B. // Comprehensive Inorg. Chem. II. Eds. Reedijk J., Poepplemeier K. Oxford: Elsevier, 2013. V. 1. P. 755.
Puddephatt R.J. The Chemistry of Gold. N.Y.: Elsevier, 1978.
Leary K., Bartlett N. // J. Chem. Soc. Chem. Comm. 1972. № 15. P. 903.
Соколов В.Б., Прусаков В.Н., Рыжков А.В. и др. // Докл. АН СССР. 1976. Т. 229. С. 884.
Набиев Ш.Ш., Соколов В.Б. // Хим. физика. 2013. Т. 32. № 4. С. 15.
Mohr F. // Gold Bull. 2004. V. 37. № 3. P. 164.
Schroder D., Hrusak J., Tornieporth-Oetting I.C. et al. // Angew. Chem. Intern. Ed. 1994. V. 33. № 2. P. 212.
Evans C.J., Gerry M.C.L. // J. Amer. Chem. Soc. 2000. V. 122. № 7. P. 1560.
Reffy B., Kolonits M., Schulz A. et al. // J. Amer. Chem. Soc. 2000. V. 122. № 13. P. 3127.
Schmidbaur H., Schier A. // Chem. Soc. Rev. 2008. V. 37. № 9. P. 1931.
Žemva B., Lutar K., Jesih A. // J. Amer. Chem. Soc. 1991. V. 113. № 11. P. 4192.
Набиев Ш.Ш., Соколов В.Б., Чайванов Б.Б. // Кристаллография. 2011. Т. 56. № 5. С. 829.
Brunvoll J., Ischenko A.A., Sokolov V.B. et al. // Acta Chem. Scand. 1982. V. A36. № 9. P. 705.
Hwang I., Seppelt K. // Angew. Chem. Intern. Ed. 2001. V. 40. № 19. P. 3690.
Соколов В.Б., Циноев В.Г., Рыжков А.В. // Теорет. и эксперим. химия. 1980. Т. 16. № 3. С. 345.
Киселев Ю.М., Попов А.И., Горюнов А.В. и др. // ЖНХ. 1990. Т. 35. № 3. С. 611.
Riedel S., Kaupp M. // Inorg. Chem. 2006. V. 45. № 3. P. 1228.
Набиев Ш.Ш. // Изв. АН. Сер. хим. 1999. № 4. С. 715.
Mueller M. Fundamentals of Quantum Chemistry. N.Y.: Kluwer, 2001.
Lewars E. Computational Chemistry. Dordrecht: Springer, 2011.
Riedel S., Kaupp M. // Coord. Chem. Rev. 2009. V. 253. № 5–6. P. 606.
Тимаков А.А., Прусаков В.Н., Дробышевский Ю.В. // Докл. АН СССР. 1988. Т. 291. С. 125.
Craciun R., Picone D., Long R.T. // Inorg. Chem. 2010. V. 49. № 3. P. 1056.
Himmel D., Riedel S. // Inorg. Chem. 2007. V. 46. № 13. P. 5338.
Gillespie R.J. // Coord. Chem. Rev. 2008. V. 252. P. 1315.
Hoffmann R., Beier B., Muetterties E. // Inorg. Chem. 1977. V. 16. № 3. P. 511.
Recio J.M., Menendez J.M., de la Roza A.O. An Introduction to High-Pressure Science and Technology. N.Y.: CRC Press, 2016.
Алиев И.И., Коварский А.Л., Бучаченко А.Л. // Хим. физика. 2007. Т. 26. № 5. С. 11.
Blank V.D., Estrin E.I. Phase Transitions in Solids Under High Pressure. N.Y.: CRC Press, 2014.
Жаров А.А., Коновалова И.Б. // Хим. физика. 2016. Т. 35. № 7. С. 76.
Geballe Z., Liu H., Mishra A. // Angew. Chem. Intern. Ed. 2018. V. 57. № 3. P. 688.
Peng F., Botana J., Wang Y. // J. Phys. Chem. Lett. 2016. V. 7. № 22. P. 4562.
Lin J., Zhang S., Guan W. // J. Amer. Chem. Soc. 2018. V. 140. № 30. P. 9545.
Wang Y., Lv J., Zhu L. // Comput. Phys. Commun. 2012. V. 183. № 10. P. 2063.
Miao M. // Nature Chem. 2013. V. 5. № 10. P. 846.
Turner J., Poliakoff M. // Fresenius J. Anal. Chem. 1986. V. 324. № 8. P. 819.
Tretyakov Yu.D., Oleynikov N.N., Shlyakhtin O.A. Cryochemical Technology of Advanced Materials. Berlin-Heidelberg: Springer, 1997.
Wang X., Andrews L., Willmann K. et al. // Angew. Chem. Intern. Ed. 2012. V. 51. № 42. P. 10628.
Wang X., Andrews L., Brosi F. // Chem. Eur. J. 2013. V. 19. № 4. P. 1397.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика