

Химическая физика, 2022, T. 41, № 1, стр. 9-16
Коррекция механизма фотолиза аминоазобензола по данным кинетической пикосекундной спектроскопии
Ю. А. Михеев 1, *, С. М. Ломакин 1
1 Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
Москва, Россия
* E-mail: mik@sky.chph.ras.ru
Поступила в редакцию 09.12.2020
После доработки 17.05.2021
Принята к публикации 20.05.2021
- EDN: TVKKER
- DOI: 10.31857/S0207401X22010113
Аннотация
Исправлен механизм фотолиза красителя транс-аминоазобензола (t-AAB), возбуждаемого импульсным лазерным видимым (Vis) излучением (λex = 400 нм) и зондируемого методом пикосекундной абсорбционной спектроскопии. Учтено, что за поглощение света ответственны два типа фениламинильных катионов, принадлежащих ридимерам t-AAB2 (основное состояние красителя). Фотовозбуждение этих катионов приводит к расщеплению ридимеров t-AAB2 на мономерные франк-кондоновские (FC) пары двух видов. Один вид FC-пар состоит из нейтральных мономеров, которые релаксируют с потерей колебательного возбуждения и регенерацией t-AAB2. Другие FC-пары состоят из нейтрального и электронно-поляризованного мономера, в котором ридберговская 3s-орбиталь азогруппы занята парой электронов, возбужденных с sp2-орбиталей азогруппы. Нейтральные мономеры этих FC-пар регенерируют ридимеры t-AAB2, встречаясь в ходе самодиффузии, а электронно-поляризованные мономеры претерпевают изомеризацию в цис-AAB.
ВВЕДЕНИЕ
Строение и цветность аминоазобензольных красителей
Недавно опубликованы работы [1–7], являющиеся фактически началом реновации научных представлений о структурно-спектроскопических свойствах простых аминоазобензольных красителей, служивших в течение 100 лет модельными соединениями в науке о природе цветности органических соединений. До сих пор большинство публикаций по красителям на основе аминоазобензола (ААВ) базируется на той идее, возникшей на этапе становления оганической химии, что они в своем основном состоянии являются мономерами. Считается, например, что желто-оранжевая окраска аминоазобензола и его замещенных производных обусловлена электронным (e-) таутомером Q (Схема 1 ), имеющим хиноидное строение [8].
Схема 1
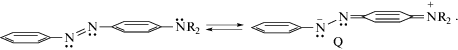
На Схеме 1 R = H в транс-аминоазобензоле (t‑AAB) и R = CH3 в диметиламиноазобензоле.
Между тем обобщение [1, 3–7] большого числа экспериментальных данных по спектроскопическим свойствам замещенных производных аминоазобензола в УФ/видимом (UV-Vis) диапазоне, находящихся в нейтральных и кислотных средах, показало неадекватность мономерно-хиноидной природы цветности (Схема 1 ). В реальности, согласно [1, 3–7], основным состоянием аминоазобензольных красителей являются ридберговские димеры (ридимеры), за окраску которых ответственны функционирующие в них хромогенные катионы фениламинильного типа (PhAT).
Межмономерная связь в ридимерах образуется в результате спаривания электронов, промотированных с sp2-орбиталей атомов N азогрупп аминоазокрасителей на ридберговские 3s-орбитали азогрупп. Азогруппа N=N каждого мономера выступает в качестве водородоподобного атома и служит квантовохимическим посредником между sp2-орбиталями атомов азота и собственной ридберговской 3s-орбиталью (R3s). Перекрывание этих орбиталей обеспечивает промотирование одного из sp2-электронов на R3s и способность образовывать ридберговские межмономерные связи (R3s–R3s), а также особенности структурных, спектроскопических, физико-химических свойств азобензола (AB) и его замещенных производных.
Опираясь на результаты работ [1, 3–7], можно заключить, что при синтезе красителя AAB2 и его R-производных происходит сборка молекул в ридимеры по Схеме 2 .
Схема 2
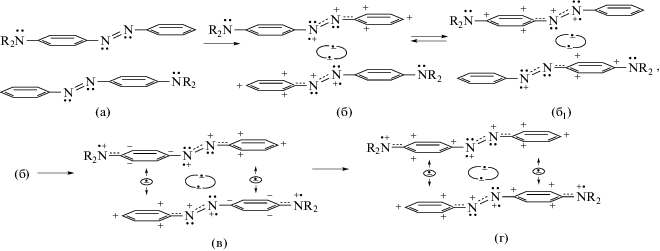
На Схеме 2 точки под атомами N обозначают электроны на sp2-орбиталях, точки над атомами N – электроны на pz-орбиталях. В ней показано, что молекулы красителя, встретившиеся в виде пар (2а), претерпевают трансформацию своего e‑состояния под влиянием образующейся ридберговской ковалентной связи ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$ (показана на схемах (2б), (2б1)). Здесь важно, что опреденная степень перекрывания sp2- и R3s-орбиталей создает условия не только для поочередного промотирования sp2-электронов с азотов азогруппы на R3s-орбиталь в локальном хромофоре –N=N–, но и для обратимого (волнового, ультрабыстрого) обмена электронами между sp2-орбиталями спаренных мономеров через связи ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$: (•+sp2) + + ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$ + (+•sp2) ↔ (+•sp2) + ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$ + (•+sp2). В данном e-обмене каждая из sp2-орбиталей, поочередно теряющая электрон и получающая положительный заряд (в схеме 2 он указан под атомами N), поляризует π-связь азогруппы, стягивая на свой атом N оба π-электрона азогруппы и оставляя на pz-орбитали соседнего азота положительный заряд. Этот заряд делокализуется с образованием PhAT за счет сопряжения в группах N+Ph+ – схема (2б) и N+Pħ+ – схема (2б1).
Согласно Схеме 2 образование ридимера генерирует ультрабыстрый e-обмен, вызывающий осцилляцию положительных зарядов между противолежащими PhAT-катионами N+Ph+ и N+Pħ+. В результате этого исходная пара (2а) электронейтральных мономеров трансформируется в равновесную смесь ридимерных e-таутомеров (2б) ↔ (2б1).
Отметим, что заряды в PhAT-катионах ${\text{P}}{{{\text{h}}}^{{\text{ + }}}}{\text{N}}_{{\centerdot \centerdot }}^{{\text{ + }}}$ и N+Pħ+ ≡ ${{{\text{R}}}_{2}}{\text{N}}_{{}}^{{\centerdot \centerdot }} - {\kern 1pt} {\text{P}}{{\hbar }^{{\text{ + }}}}{\text{N}}_{{\centerdot \centerdot }}^{{\text{ + }}}$ на схемах (2б), (2б1)) находятся преимущественно на атомах углерода колец в орто- и пара-положениях, как в бензильных и фениламинильных катионах. Сходным образом катионы PhAT имеют не только связывающие и разрыхляющие молекулярные π-орбитали (МО), но и нижние свободные орбитали (НСМО) с нулевой энергией. В PhAT-катионах переход электронов с высших занятых π-орбиталей (ВЗМО) происходит на НСМО при поглощении Vis-света (π → π*-переход) при 400 нм, обеспечивая им желто-оранжевый цвет.
Ридимеры (2б) обладают не только таутомерным e-обменом (2б) ↔ (2б1). В них каждая аминная группа способна подавать один из двух pz‑электронов в сопряженное с ней фениленовое Pħ-кольцо, преобразуя ридимер (2б) в (2в) с группами R2N•+Pħ–•. Получив электроны, кольца Pħ–• становятся донорами электронов для катионов Ph+N+ соседнего мономера, что служит причиной образования промежуточных комплексов с переносом заряда. На схеме (2в) стрелками обозначены характерные для КПЗ кулоновские и обменные взаимодействия между фениленовыми анионами Pħ–• и PhAT-катионами Ph+N+. В момент, когда электрон переносится с аниона Pħ–• группы R2N•+Pħ– на вакантную НСМО катиона Ph+N+, возникает катион R2N•+Pħ+ с вакантной НСМО.
Итоговый ридимер (2г) является e-таутомером ридимера (2б), но более прочным. В (2г) имеются не только ридберговская межмономерная связь ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$, но еще две одноэлектронных связи между противолежащими катионами PhAT обоих мономеров (по типу связи в катионе ${\text{H}}_{2}^{ + }$). Таким образом, фиксируются четыре катиона PhAT, обладающие Vis-поглощением в области 390–420 нм и раскрывающие физическую сущность ауксохромии, т.е. усиления желто-оранжевого цвета относительно незамещенного азобензола. При этом квантово-волновая компенсация зарядов в ридимерах (2г) обеспечивает им практическую электронейтральность.
В среде с невысокой кислотностью у тех же ридимеров наблюдается углубление окраски, связанное с протонированием ридимерных аминогрупп без распада ридимеров [1].
Схема 3

На Схеме 3 структуры (3а) и (3б) тоже являются следствием e-таутомерной ультрабыстрой переполяризации мономеров с осцилляцией положительных зарядов между атомами N в азогруппе за счет e‑обмена по схеме (•+sp2) + ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$ + (+•sp2) ↔ ↔ (+•sp2) + ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$ + (•+sp2). При этом не имеющие положительных зарядов кольца (отмечены на схеме (3а) светлыми треугольниками) периодически становятся катионами (кольца над черными треугольниками на схеме (3б)), повторяя ситуацию с e-таутомерами непротонированных ридимеров (Схема 2 , (2б) ↔ (2б1)).
Сочетание e-конфигураций (2б) ↔ (2б1) и (3а) ↔ ↔ (3б) позволило [1, 3–7] объяснить наличие у них характерных полос UV-поглощения (при 320 нм) и Vis-поглощения в области λ ≥ 500 нм на основе механизма Симпсона, показанного на примере азобензола [9]. Согласно [9] отсутствующая у алкилзамещенных бензола полоса UV-поглощения при 320 нм характерна для незамещенного AB. Появление этой UV-полосы у AB объясняется механизмом делокализации электронного возбуждения между фенильными группами, которые в работе [9] рассматриваются как резонаторы, не имеющие электронного π-сопряжения. Взятые в отдельности они имеют одинаковые энергетические (вырожденные) π*-состояния. Это вырождение снимается за счет расщепления π*-уровней смежных фенильных колец AB вследствие резонансного переноса энергии возбуждения, поэтому в спектрах молекул AB в дополнение к UV-полосе бензольного поглощения с νmax = 44 000 cм–1 (227 нм) появляется батохромно смещенная UV-полоса с νmax ≈ 32 000 cм–1 (313–320 нм) [9].
Сходные UV-полосы имеют дипротонированные ридимеры аминоазобензола ((AAB+H)2, Схема 3 ) из-за наличия в их незаряженных фенильных и фениленовых колец (отмечены светлыми треугольниками) [1, 3–7]. Между этими кольцами нет π-сопряжения по механизму Хюккеля, что превращает их в резонаторы с расщеплением π*‑уровней по механизму Симпсона [1, 3–7, 9].
Аналогичный механизм расщепления π*-уровней “по-Симпсону” имеет место и для катионов PhAT [1, 3–7]. Каждый из них индивидуально обладает Vis-полосами при 390–410 нм, а их бинарное сочетание (на Схеме 3 оно отмечено черными треугольниками) приводит к появлению у протонированных ридимеров аминоазобензола (AAB+H)2 Vis-полос при 500 нм. Такой же эффект наблюдается на Схеме 2 у непротонированных ридимеров (2б) и (2б1), но концентрация последних значительно меньше, чем ридимеров (2г), и полоса их слабого Vis-поглощения налагается на “хвост” Vis-полосы ридимера (2г) с λmax при 390–410 нм [5].
Тот факт, что образование ридимеров с их e-таутомерами (Схемы 2 и 3 ) продемонстрировано на большом числе производных аминоазобензола [1], заставляет обратиться к реновационному анализу экспериментальных данных по фотонике ААВ-красителей. Ниже представлена новая трактовка результатов, полученных методом пикосекундной абсорбционной спектроскопии (PAS) в работе [10] при лазерном импульсном возбуждении аминоазобензола. Одновременно раскрыто противоречие между кинетическим экспериментом [10] и его авторским описанием, даны адекватная кинетика и механизм процесса в аспекте ридимерной концепции.
Новое объяснение важных данных работы [10] в плане природы элементарных актов фотопроцессов позволяет надеяться, что аминоазобензольные красители (модельные соединения в теории цветности [8]) могут вызывать интерес исследователей и в плане практического применения, характерного, например, для бурно развивающейся биомедицинской оптики с созданием новых абсорбционных и флуоресцентных зондов, а также материалов для изучения и регуляции активности живых клеток и задач фотодинамической терапии [11–14].
ИНТЕРПРЕТАЦИЯ ДАННЫХ PAS АВТОРАМИ РАБОТЫ [10]
Авторы работы [10] преследовали цель с помощью методики PAS уточнить механизм транс → → цис (t → c)-фотоизомеризации аминоазобензола, традиционно считая его мономером t-AAB. Спектры транзитных (tr-) состояний генерировали, возбуждая раствор “мономерного t-AAB” в этаноле (и гептаноле) с концентрацией 1 мМ (T = = 296 K) на лазерной установке импульсами Vis-света с длиной волны λex = 400 нм и энергией меньше 200 мкДж/импульс при частоте повторения импульсов 1 Мгц. Для зондирования tr-состояниий на той же установке формировали пучки белого света (350–750 нм). Небольшую часть зондового пучка направляли в канал сравнения для получения разностных спектров. Пучки возбуждения и зондирования фокусировали под малым углом на кварцевую проточную ячейку. Зондирующие импульсы подавали с определенным временем задержки (Δti) после импульсов возбуждения. Итоговые спектры регистрировали в виде разности между спектрами Vis-поглощения tr-состояний и исходного аминоазобензола.
Приведенные на рисунке из работы [10] tr‑спектры (зависимости 1–4 разностной оптической плотности ΔD от λзонд) представляют собой сумму нескольких Vis-полос, интенсивность которых спадает в течение нескольких пикосекунд. Согласно расчетам из [10], наблюдаемый сразу после возбуждения (при Δt1 = 0.1 пс) спектр 1 (здесь и всюду ниже мы обсуждаем рисунок из работы [10]) представляет собой суперпозицию широкой полосы tr-поглощения в области 410–750 нм и полосы относительно долгоживущего отбеливания (bleachihg) с отрицательным значением ΔD в полосе поглощения основного состояния аминоазобензола (т.е. ридимеров t-AAB2 [1]) при 370–410 нм. Суммарное поглощение в области 450–750 нм (кривые 1–4) снижается в течение нескольких пикосекунд, причем наиболее быстро в интервале 600–750 нм.
Отмечается [10], что в области 400–450 нм идет сначала прирост ΔD (кривая 2), а затем снижение ΔD с переходом в отрицательную область. Отмеченный прирост ΔD в области 400–450 нм (кривая 2) авторы [10] не обсуждают, уделяя основное внимание кинетике затухания Vis-поглощения в области 450–750 нм.
По результатам расчетов установлено [10], что в спектральном интервале 600–725 нм снижение ΔD описывается суммой трех экспоненциальных функций ΔDi = Aiexp(–t/τi), принадлежащих Vis-полосам трех транзитных компонент с характеристическими временами релаксации τ1 = 0.2 пс, τ2 = 0.6 пс и τ3 = 1.9 пс соответственно. Результаты расчетов позволили также установить максимумы Vis-полос: компонента с τ1 = 0.2 пс имеет λmax = 625–650 нм, у компонент с τ2 = 0.6 пс и τ3 = = 1.9 пс λmax = 500–550 нм, причем спектральные формы Vis-полос двух последних компонент практически совпадают в ходе затухания.
Отмечается, что в интервале 450–600 нм компонента с τ1 = 0.2 отсутствует и затухание tr-поглощения описывается только двумя более медленными функциями: с τ2 = 0.6 пс и τ3 = 1.9 пс. В области 425–475 нм эти две компоненты налагаются на полосу отбеливания, возвратная эволюция которой соответствует моноэкспоненциальной функции с характеристической константой τ = 15 пс. В конце эволюционной релаксации остается раствор, обедненный формой t-AAB2 и обогащенный формой c-AAB, в которую превращается ≈25% мономеров t-AAB2. (Та же картина с транзитными Vis-полосами полностью воспроизводится в опытах с аминоазобензолом в гептаноле [10].)
Касаясь механизма (t → c)-изомеризации, авторы работы [10] традиционно постулируют, что имеют дело с мономерами t-AAB, которые при фотовозбуждении светом с λex = 400 претерпевают π → π*-переход S0 → S2. Состоянию S2 приписывают tr-полосу Vis-поглощения с λmax = 625–650 нм, быстро затухающую (с τ1 = 0.2 пс) за счет внутренней конверсии энергии возбуждения на низкий возбужденный уровень: S2 ~~→ S1. Уровень S1 связывают с n,π*-состоянием и считают, что слабая полоса n → π*-перехода в “мономере t‑AAB” должна находиться в той же Vis-области спектра (при 420–440 нм), к которой в литературе отнесли n → π*-полосу молекул t-AB. Состоянию S1 авторы [10] приписывают Vis-поглощение в виде широкой tr-полосы с λmax = = 525–550 нм. Последняя, согласно [10], затухает вследствие внутренней конверсии S1 ~~→ S0, идущей двумя разными путями: с τ2 = 0.6 пс и τ3 = = 1.9 пс, при участии (t → c)-изомеризации (конкретная природа этих путей не обсуждается).
КРИТИКА ИНТЕРПРЕТАЦИИ ДАННЫХ PAS ИЗ РАБОТЫ [10]
Сделанное в [10] описание (t → c)-фотоизомеризации аминоазобензола по механизму последовательно-параллельной реакции: S2 ~~→ S1, S1 ~~→ S0 (с константой скорости k2 = 1/τ2) и S1 ~~→ S0 (c k3 = 1/τ3), противоречит эксперименту. Действительно, согласно [10], транзитное Vis-поглощение в спектральном λ-интервале 410–750 нм затухает с самого начала по моноэкспоненциальному закону релаксации трех Vis-компонент с максимальными начальными скоростями и постоянными для каждой компоненты характеристическими временами: τ1 = 0.2 пс и τ2 = 0.6 пс, τ3 = 1.9 пс. Следовательно, изначально кинетика затухания этих компонент соответствует механизму параллельных реакций. При этом Vis-поглощение наиболее быстро распадающейся компоненты с τ1 = 0.2 пс (в области 600–725 нм) становится пренебрежимо низким после десяти периодов ее полураспада: 10T0.5 = 10(ln2)τ1 = 10 ∙ 0.693τ1 = 1.38 пс. Вместе с тем исчезновение быстрой компоненты не связано (в противоположность мнению авторов [10]) с ее превращением в компоненты с τ2 = 0.6 пс и τ3 = = 1.9 пс. Действительно, при последовательной реакции ее распад на начальной стадии должен был сочетаться с ростом ΔD медленных компонент, что противоречит их моноэкспоненциальной кинетике и, соотвественно, адекватному механизму реакций. Адекватный механизм, кроме того, должен учесть оставленный в [10] без обсуждения факт, что только распад быстрой компоненты с τ1 = 0.2 пс приводит к появлению промежуточного Vis-пичка в области 400–450 нм (кривая 2).
Отмеченное выше противоречие между авторской трактовкой механизма и реальной кинетикой Vis-полос обусловлено ее концептуальным противоречием факту ридимерного строения t‑AAB2. Теперь понятно [1, 3–7], что логика теоретических построений в [10] базировалась на ошибочном постулате о мономерном основном состоянии аминоазобензольных красителей, характеризующихся таким же n → π*-переходом, как и у t-AB. Опираясь на традиционное отнесение n → π*-перехода в t-AB к слабой полосе при 440 нм (которое, как показано в [1, 4–8], не соответствует действительности), авторы [10] постулировали, что n → π*-переход у “молекул t-AAB” тоже находится при 440 нм, располагаясь на длинноволновом крыле интенсивной полосы поглощения “t-AAB“ (λmax ~ 390 нм). Между тем слабая Vis-полоса t-AB, находящаяся при λmax = = 430–440 нм, принадлежит не n → π*, а π → π*-возбуждению катиона PhAT в одном из электронных таутомеров t-AB [3]. При этом, в противоположность традиционному представлению, n → π*-переходы в t-AB и ридимерах диметиламиноазобензола и диэтиламиноазобензола находятся в UV-области спектра при 300–330 нм [3–7], что естественно относится и к t-АAB2. Что же касается приписанного n → π*-переходу [10] небольшого усиления интенсивности на длинноволновом крыле Vis-полосы аминоазобензола в области λ > 420 нм, то оно обусловлено не n → π*-переходом, а некоторым увеличением содержания e-таутомеров (2б) и (2б1) Схемы 2 вследствие небольшого смещения равновесия (2г) → (2б) ↔ (2б1) в полярной спиртовой среде [5].
МЕХАНИЗМ ФОТОЛИЗА РИДИМЕРОВ t-AAB2
Согласно изложенному выше за поглощение света с λex = 400 нм ридимером t-AAB2 ответственны фениленовый, R2N•+PћN+, и фенильный, ${\text{P}}{{{\text{h}}}^{ + }}{\text{N}}_{{\centerdot \centerdot }}^{ + }$ катионы в его наиболее устойчивой форме (схема (2г), R = H). Соответствующие франк-кондоновские состояния (FC), возникающие при фотовозбуждении, претерпевают последующие трансформации по (Схема 4 ).
Схема 4
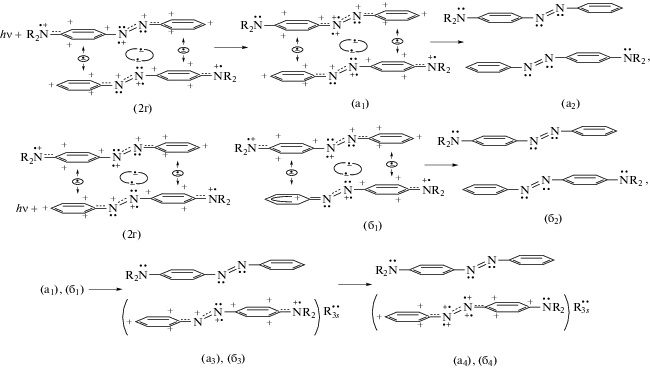
Согласно Схеме 4 фотовозбуждение ведет к разрыву ридберговских связей ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$ на стадии промежуточных FC-конфигураций (4а1) и (4б1). Не представленный на схеме (для экономии места) путь образования e-конфигурации (4а1) можно описать следующим образом. Сначала в группе ${{{\text{R}}}_{2}}{{{\text{N}}}^{{\centerdot + }}}{\text{P}}{{\hbar }^{ + }}{\text{N}}_{{\centerdot + }}^{{\centerdot \centerdot }}$ верхнего мономера (2г) электрон возбуждается с ВЗМО на НСМО. Вслед за этим идет e-переход с НСМО на pz-орбиталь атома N+ аминогруппы с образованием промежуточной структуры ${{{\text{R}}}_{{\text{2}}}}{{{\text{N}}}^{{\centerdot \centerdot }}}{\text{P}}{{\hbar }^{{\text{ + }}}}{\text{N}}_{{\centerdot {\text{ + }}}}^{{\centerdot \centerdot }}$ (на схеме не представлена для экономии места). Затем происходит перенос электрона на катион Pћ+ аминогруппы с pz-орбитали смежного с ним азота азогруппы ${\text{N}}_{{\centerdot + }}^{{\centerdot \centerdot }}.$ В итоге вместо исходной группы ${{{\text{R}}}_{{\text{2}}}}{{{\text{N}}}^{{\centerdot {\text{ + }}}}}{\text{P}}{{\hbar }^{{\text{ + }}}}{\text{N}}_{{\centerdot {\text{ + }}}}^{{\centerdot \centerdot }}$ появляется ${{{\text{R}}}_{{\text{2}}}}{{{\text{N}}}^{{\centerdot \centerdot }}}{\text{P}}{{\hbar }^{{\text{ + }}}}{\text{N}}_{{\centerdot {\text{ + }}}}^{{{\text{ + }}\centerdot }},$ и в азогруппе верхнего мономера (4а1) возникает атом ${\text{N}}_{{\centerdot + }}^{{ + \centerdot }}$ с двумя зарядами (на pz- и sp2-орбитали). Одновременно растет кулоновское возмущение ридберговской связи ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$, ведущее к ее разрыву.
e-Конфигурация (4б1) образуется при фотовозбуждении нижнего мономера (2г) путем перехода электрона с ВЗМО на НСМО в катионе Ph+N+ (Схема 4 ) и последующего e-переноса с НСМО на pz-орбиталь атома N+ азогруппы. При этом бензоидный секстет исходного катиона Ph+N+ трансформируется в катион Ph+=${{{\text{N}}}_{{\centerdot \centerdot }}}$ (типа катиона бензония [15]).
Согласно Схеме 4 на азогруппах мономеров исходной e-конфигурации (2г) заряды распределены поровну, а в FC-конфигурациях (4а1) и (4б1) – не поровну. Такая ситуация способствует их расщеплению на мономеры. При этом число зарядов на азогруппах в конфигурации (4а1) превышает их число на азогруппах в конфигурации (4б1), создавая более сильное кулоновское напряжение на азосвязи и ридберговской межмономерной связи. Это позволяет ожидать, что расщепление (4а1) будет более легким по сравнению с (4б1). Еще, согласно Схеме 4 , расщепление ридимерных FC-конфигураций (4а1) и (4б1) ведет к образованию гомогенных FC-пар (4а2) и (4б2), состоящих из нейтральных мономеров, а также гетерогенных FC-пар (4а4), (4б4) с нейтральным и поляризованным мономерами.
Гомогенные FC-пары появляются вследствие того, что разрыв ридберговских связей ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$ в ридимерах (4а1) и (4б1) приводит к возвращению обоих освободившихся электронов на исходные sp2-орбитали атомов N обоих мономеров, лишая sp2-орбитали зарядов: •+sp2 + e → :sp2. После этого катионы ${\text{P}}{{{\text{h}}}^{ + }}{\text{N}}_{{\centerdot \centerdot }}^{ + }$ и бензония восстанавливаются в нейтральные группы Ph${{{\text{N}}}_{{\centerdot \centerdot }}}$=, что приводит к нейтрализации зарядов на фениленовых группах путем захвата ими электронов из одноэлектронных связей. Появляющиеся при этом гомогенные (нейтральные) FC-пары (4а2) и (4б2) мономеров являются промежуточными состояниями (их последующее возвращение в основное состояние рассмотрено ниже).
Второй путь e-эволюции после разрыва ридберговской связи сочетает в себе образование мономерных пар (4а4) и (4б4), состоящих из нейтрального и e-поляризованного мономера. В этом случае разрыв связи ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$ в каждом из ридимеров (4а1) и (4б1) ведет к возврату только одного из двух освободившихся ридберговских электронов на sp2-орбиталь азогруппы с повышенным зарядом (верхние мономеры в конфигурациях (4а1) и (4б1)). В данном случае верхние мономеры в конфигурациях (4а1), (4б1) нейтрализуются вследствие возврата на их фениленовые кольца по электрону из одноэлектронных связей.
На этом же пути e-эволюции ((4а1), (4б1) → (4а3), (4б3)) вторая появившаясяся при разрыве связи ${{{\text{R}}}_{{3s}}}\centerdot - \centerdot {{{\text{R}}}_{{3s}}}$ орбиталь ${\text{R}}_{{3s}}^{\centerdot }$ (с одним электроном) сама захватывает электрон из оставшейся одноэлектронной связи. Таким образом появляются ридберговские орбитали с неподеленной e-парой ${\text{R}}_{{3s}}^{{\centerdot \centerdot }},$ которые стабилизируют на некоторое время поляризованные формы нижних мономеров (4а3), (4б3).
Далее в поляризованных мономерах (4а3) и (4б3) происходит обмен электронов с участием орбитали ${\text{R}}_{{3s}}^{{\centerdot \centerdot }}.$ Сначала ${\text{R}}_{{3s}}^{{\centerdot \centerdot }}$ отдает один электрон на pz-орбиталь положительно заряженного атома ${\text{N}}_{{\centerdot \centerdot }}^{ + }$ (левый азот в азогруппе), но снова захватывает электрон с sp2-орбитали того же атома. Это приводит к образованию атома ${\text{N}}_{{\centerdot + }}^{{ + \centerdot }}$ с двумя зарядами. Затем орбиталь ${\text{R}}_{{3s}}^{{\centerdot \centerdot }}$ вновь отдает электрон, но уже на pz-орбиталь заряженной аминогруппы, аннулируя катион Pћ+N+•R2, и вновь восстанавливаясь путем захвата pz-электрона с правого атома ${\text{N}}_{{ + \centerdot }}^{{\centerdot \centerdot }}$ азогруппы, переводя его в ${\text{N}}_{{\centerdot + }}^{{ + \centerdot }}.$ В результате появляются поляризованные мономеры с бинарными катионами PhAT и сильно
ослабленными связями  в азогруппе, чьи заряды уравновешиваются отрицательными зарядами ${\text{R}}_{{3s}}^{{\centerdot \centerdot }}$ (нижние мономеры в парах (4a4), (4б4)). Итогом описанных e-трансформаций являются FC-гетеропары из поляризованного и нейтрального
мономеров.
в азогруппе, чьи заряды уравновешиваются отрицательными зарядами ${\text{R}}_{{3s}}^{{\centerdot \centerdot }}$ (нижние мономеры в парах (4a4), (4б4)). Итогом описанных e-трансформаций являются FC-гетеропары из поляризованного и нейтрального
мономеров.
Относительно изложенной последовательности фотоиндуцированных электронных переходов (Схема 4 ) следует отметить, что она фактически отражает волновую природу электронов, которые перераспределяются, как сказано в [16] “последовательно по всей мезомерной системе связей”.
Механизм (t → c)-изомеризации
Предпочтение для (t → c)-изомеризации следует отдать не электронейтральным мономерам t-AAB, имеющим прочную двойную связь N=N, а мономерам с e-конфигурацией 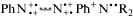 – это (4a4) и (4б4). В них сильно ослабленная азосвязь
– это (4a4) и (4б4). В них сильно ослабленная азосвязь 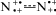 сочетается с сильным кулоновским отталкиванием положительно заряженных атомов азота.
Именно этим можно объяснить наличие наиболее быстро затухающей Vis-полосы с λmax = 625–650 нм (с характеристическим временем τ1 = 0.2 пс). Кроме того, только мономеры
сочетается с сильным кулоновским отталкиванием положительно заряженных атомов азота.
Именно этим можно объяснить наличие наиболее быстро затухающей Vis-полосы с λmax = 625–650 нм (с характеристическим временем τ1 = 0.2 пс). Кроме того, только мономеры 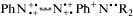 с бинарными хромогенными катионами PhAT ответственны за полосу Vis-поглощения в области
спектра с λmax = 625–650 нм. (Сходные Vis-полосы имеют дикатионы Ph+HN+–N+HPh+ протонированного азобензола и Ph+N+–N+Ph+ возбужденного AB [2, 3].)
с бинарными хромогенными катионами PhAT ответственны за полосу Vis-поглощения в области
спектра с λmax = 625–650 нм. (Сходные Vis-полосы имеют дикатионы Ph+HN+–N+HPh+ протонированного азобензола и Ph+N+–N+Ph+ возбужденного AB [2, 3].)
Именно (t → c)-изомеризация мономеров 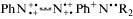 объясняет также и, “забытый“ авторами [10] факт, что исчезновение Vis-полосы с λmax = 625–650 нм нм сочетается с сопутствующим появлением пичка Vis-поглощения в области
400–450 нм. Это происходит из-за потери мономерами
объясняет также и, “забытый“ авторами [10] факт, что исчезновение Vis-полосы с λmax = 625–650 нм нм сочетается с сопутствующим появлением пичка Vis-поглощения в области
400–450 нм. Это происходит из-за потери мономерами 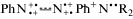 своего плоского трансизомерного строения и бинарно связанных (по механизму Симпсона)
хромогенов-резонаторов. В свою очередь, появляющиеся транзитные индивидуальные, не
способные резонировать по механизму Симпсона, катионы PhAT (Pћ+N+•R2 и Ph+N+) поглощают свет в области 400–450 нм [1–7] и в ходе изомеризации рекомбинируют с электронами ${\text{R}}_{{3s}}^{{\centerdot \centerdot }}$ орбиталей: (
своего плоского трансизомерного строения и бинарно связанных (по механизму Симпсона)
хромогенов-резонаторов. В свою очередь, появляющиеся транзитные индивидуальные, не
способные резонировать по механизму Симпсона, катионы PhAT (Pћ+N+•R2 и Ph+N+) поглощают свет в области 400–450 нм [1–7] и в ходе изомеризации рекомбинируют с электронами ${\text{R}}_{{3s}}^{{\centerdot \centerdot }}$ орбиталей: (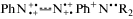 ) ${\text{R}}_{{3s}}^{{\centerdot \centerdot }}$ → → с-PhN=NPћNR2 ( c-AAB). Доля образующихся этим путем молекул c‑AAB должна, согласно Схеме 4
и в соответствии с экспериментом [10], составлять всего около четверти от числа исходных мономеров в составе t-AAB2, так как претерпеть изомеризацию способны только поляризованные мономеры
) ${\text{R}}_{{3s}}^{{\centerdot \centerdot }}$ → → с-PhN=NPћNR2 ( c-AAB). Доля образующихся этим путем молекул c‑AAB должна, согласно Схеме 4
и в соответствии с экспериментом [10], составлять всего около четверти от числа исходных мономеров в составе t-AAB2, так как претерпеть изомеризацию способны только поляризованные мономеры  .
.
Реакции нейтральных мономеров t-AAB
Нейтральные мономеры t-AAB образуются попарно ((4а2), (4б2)) и одиночно в гетеропарах (4а4) и (4б4). Их двойная связь N=N исключает (t → c)-изомеризацию, и для них остается только путь возвращения к ридимерам t-AAB2 (Схема 2 ). Образование ридимеров (4а2) и (4б2) из FС-пар должно идти значительно быстрее, чем из непарных мономеров t-AAB, которым для встречи и реакции по Схеме 2 необходимо пройти стадию самодиффузии. Мономеры t-AAB и c-AAB поглощают Vis-свет значительно слабее образующихся из них ридимеров (2г), и, скорее всего, их самодиффузия определяет релаксацию полосы “отбеливания“ (τ ≈ 15 пс).
В отличие от встречающихся мономеров t-AAB, FС-пары (4а2) и (4б2) вступают в релаксацию с определенной энергией колебательного (индекс “v”) возбуждения относительно термически равновесных пар (схема (2а)) основного состояния. При этом из FС-ридимеров (4а1) образуются гомопары (4а2)v с более высоким запасом колебательной энергии, чем образующиеся из ридимеров (4б1) гомопары (4б2)v. Поэтому процессы релаксации колебательно-возбужденных пар, ведущие к восстановлению ридимеров по схеме (2) , идут с разными скоростями. По-видимому, именно различие в получаемых для этих гомопар величинах колебательной энергии (Схема 4 ) отражается в неодинаковых значениях τ2 = 0.6 пс и τ3 = 1.9 пс [10], но при наличии у них одинаковых транзитных полос Vis-поглощения с λmax = 500–550 нм.
ЗАКЛЮЧЕНИЕ
На основе анализа экспериментальных данных работы [10] в рамках недавно созданной ридимерной концепции строения аминоазобензольных красителей исправлено представление о механизме появления и кинетике релаксации транзитных полос Vis-поглощения промежуточных продуктов импульсного лазерного фотолиза аминоазобензола. Показана несостоятельность традиционной мономерной азоидно-катионной (хиноидной) модели данного красителя для описания его фотоники. Представлен адекватный механизм фотопревращения, включающий в себя два параллельных пути фотовозбуждения ридимерных катионов фениламинильного типа, ведущих к расщеплению t-AAB2 на транзитные мономеры, один из которых ответствен за образование c-AAB и неполную регенерацию t-AAB2.
Список литературы
Михеев Ю.А., Ершов Ю.А. // ЖФХ. 2018. Т. 92. № 10. С. 1552; https://doi.org/10.1134/S0036024418100205
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // ЖФХ. 2015. Т. 89. № 2. С. 243.
Михеев Ю.А., Гусева Л.Н., Ершов Ю.А. // ЖФХ. 2015. Т. 89. № 11. С. 1773; https://doi.org/10.1134/S0036024415110138
Михеев Ю.А., Ершов Ю.А. // ЖФХ. 2018. Т. 92. № 8. С. 1251; https://doi.org/10.1134/S0036024418080174
Михеев Ю.А., Ершов Ю.А. // ЖФХ. 2020. Т. 94. № 1. С. 143; https://doi.org/10.1134/S0036024420010227
Михеев Ю.А., Ершов Ю.А. // ЖФХ. 2020. Т. 94. № 8. С. 1269; https://doi.org/10.1134/S0036024420080208
Михеев Ю.А., Ершов Ю.А. // ЖФХ. 2020. Т. 94. № 11. С. 1706; https://doi.org/10.31857/S0044453720110254
Гордон П, Грегори П. Органическая химия красителей. М.: Мир, 1987. С. 125.
Robin M. B., Simpson W.T. // J. Chem. Phys. 1962. V. 36. № 3. P. 580.
Hirose Ya., Yui H., Sawada Ts. // J. Phys. Chem. A. 2002. V. 106. № 13. P. 3067.
Татиколов А.С., Пронкин П.Г., Шведова Л.А. и др. // Хим. физика. 2019. Т. 38. № 12. С. 11; https://doi.org/10.1134/S0207401X19120185
Бердникова Н.Г., Донцов А.Е., Ерохина М.В. и др. // Хим. физика. 2019. Т. 38. № 12. С. 48; https://doi.org/10.1134/S0207401X19120045
Пронкин П.Г., Татиколов А.С. // Хим. физика. 2021. Т. 40. № 2. С. 3; https://doi.org/10.31857/S0207401X2102014X
Татиколов А.С. // Хим. физика. 2021. Т. 40. № 2. С. 11; https://doi.org/10.31857/S0207401X21020163
Стрейтвизер Э. Теория молекулярных орбит для химиков-органиков. М.: Мир, 1965. С. 217.
Хюккель В. Химическая связь. Критическое рассмотрение систематики, способов выражения и изображения в формулах. М.: Изд-во иностр. лит., 1960. С. 77.
Дополнительные материалы отсутствуют.
Инструменты
Химическая физика