

Координационная химия, 2019, T. 45, № 6, стр. 372-377
Синтез и кристаллическая структура аквакомплексов цинка и кобальта с кукурбит[6]урилом
И. В. Андриенко 1, Е. А. Коваленко 1, *, И. Е. Кармадонова 1, 2, П. Е. Плюснин 1, 2, Д. Г. Самсоненко 1, 2, В. П. Федин 1, 2
1 Институт неорганической химии им. А.В. Николаева СО РАН
Новосибирск, Россия
2 Новосибирский государственный университет
Новосибирск, Россия
* E-mail: e.a.kovalenko@niic.nsc.ru
Поступила в редакцию 19.10.2018
После доработки 28.01.2019
Принята к публикации 30.01.2019
Аннотация
Супрамолекулярные комплексы [Zn(H2O)4(C36H36N24O12)](NO3)2 · 6.5H2O (I), [Zn(H2O)4(C36H36N24O12)]-(NO3)2 · 7H2O (II) и [Co(H2O)4(C36H36N24O12)](NO3)2 · 7H2O (III) получены при медленном (для I и III) и быстром (для II) охлаждении после кипячения водных растворов смеси солей соответствующих металлов и кукурбит[6]урила. По данным РСА (СIF files CCDC № 1862494 (I), 1862495 (II), 1862496 (III)) супрамолекулярные комплексы являются первыми примерами, где осуществляется прямая координация атомов цинка и кобальта к молекуле кукурбит[6]урила. Соединения I–III охарактеризованы методами РСА, ТГА, ИК-спектроскопии, элементного анализа.
Интерес к комплексам металлов с макроциклическими кавитандами (каликсаренами, циклодекстринами, кукурбит[n]урилами) обусловлен возможностью создания на их основе высокоорганизованных супрамолекулярных ансамблей, сочетающих органические и неорганические строительные блоки [1, 2].
Способность кукурбит[6]урила (СВ[6]) в водных растворах связывать ионы металлов была обнаружена Берендом еще в 1905 г. [3]. Наличие у CB[6] карбонильных групп, более поляризованных по сравнению со связями C–O в краун-эфирах или криптандах, приводит к более сильным взаимодействиям между кукурбитурилом и катионами металлов, чем с этими органическими макроциклами [4–11]. Молекула CB[6] построена из шести гликольурильных фрагментов, соединенных метиленовыми мостиками, и по форме напоминает бочку, в плоскости дна и крышки которой находятся атомы кислорода поляризованных карбонильных групп (порталы), способные или координировать ион металла, или образовывать водородные связи с аквакомплексами металлов. Комплексы металлов координируются атомами кислорода, расположенными в плоскости порталов CB[6], а не включаются в полость кавитанда. Таким образом, уникальное строение CB[6] позволяет, наряду со включением во внутримолекулярную полость органических молекул, образовывать супрамолекулярные аддукты или комплексы с широким рядом s-, p-, d- и f-элементов.
Несмотря на выполненные исследования в области взаимодействия CB[n] с комплексами переходных металлов, в этой химии остается много “белых пятен”. В частности, это относится к соединениям СВ[n] с Zn(II). Известен ряд супрамолекулярных архитектур, в которых тетраэдры [ZnCl4]2– занимают узлы при формировании упаковки так называемых “сотовых структур” [12–23]. В этих комплексах отсутствует прямое взаимодействие атомов цинка и кислорода порталов кукурбитурилов. Есть несколько примеров прямой координации атомов цинка к CB[5] [24, 25] и к замещенным СВ[6] [26, 27]. Для незамещенных СВ[6] таких примеров нет. Еще менее изучено взаимодействие CB[n] с солями Co(II). Описаны две структуры, где комплексы кобальта занимают полости, образованные различными соединениями кукурбит[n]урила с редкоземельными элементами [28–30]. Кроме того, известны примеры образования полиротаксанов на основе комплексов кобальта, органических лигандов и СВ[n] [31, 32], имеющих цепочечечное строение. Для СВ[8] получены соединения включения комплексов Co(II) с полиаминными лигандами [33–35].
В настоящей работе сообщается о синтезе, РСА и физико-химических свойствах трех новых супрамолекулярных комплексов кукурбит[6]урила с Zn(II) и Co(II) состава [Zn(H2O)4-(C36H36N24O12)](NO3)2 · 6.5H2O (I), [Zn(H2O)4-(C36H36N24O12)](NO3)2 · 7H2O (II) и [Co(H2O)4-(C36H36N24O12)](NO3)2 · 7H2O (III).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали реагенты Zn(NO3)2 · 6H2O и Co(NO3)2 · 6H2O марки “ч”. Кукурбит[6]урил синтезировали по описанной методике [36]. Синтезы проводили в стеклянных флаконах с завинчивающимися крышками. Анализы на С, H, N выполняли в аналитической лаборатории ИНХ СО РАН на приборе Euro EA 3000. ИК-спектры регистрировали на спектрофотометре Scimitar FTS 2000 (таблетки с KBr). Термогравиметрические (ТГ) измерения в атмосфере гелия проводили c использованием микротермовесов TG 209 F1 Iris® фирмы NETZSCH. Масса навески 10 мг, Al тигель, скорость потока газа 60 мл/мин, скорость нагрева 10 град/мин в интервале температур 20–400°С. Результаты экспериментов обрабатывали с использованием стандартного пакета программного обеспечения ProteusAnalysis [37].
Синтез I и II. 2 мл 1 М водного раствора цинка азотнокислого и 0.020 г декагидратакукурбит[6]урила (C36H36N24O12 · 10H2O, СВ[6] · 10H2O, 0.018 ммоль) нагревали до 100°С и выдерживали при этой температуре 48 ч. Объемные бесцветные кристаллы I в виде полиэдров получали при медленном охлаждении реакционной смеси до комнатной температуры в течение 2 сут. Выход 0.020 г (83% в расчете на СВ[6] · 10H2O). При быстром охлаждении реакционной смеси до комнатной температуры в течение 2 ч получали бесцветные кристаллы II в форме палочек. Выход 0.010 г (42% в расчете на СВ[6] · 10H2O). При извлечении из маточного раствора кристаллы соединений I и II трескаются и теряют кристалличность. Выполненные элементный анализ и ТГА, а также данные ИК-спектров для высушенных на воздухе образцов I и II совпадают.
| Найдено, %: | C 30.7; | H 4.2; | N 25.7. |
| Для C36H62N26O31Zn (в расчете на 9H2Oкристал) | |||
| вычислено, %: | C 30.4; | H 4.4; | N 25.6. |
ИК-спектр (ν, см–1): 3442, 3003, 2955, 2388, 2289, 1740, 1610, 1480, 1416, 1378, 1327, 1299, 1260, 1236, 1191, 1148, 1029, 967, 821, 800, 760, 675, 633, 455.
ТГА: потеря массы 8% при нагревании до 90°С соответствует удалению шести молекул кристаллизационной воды, на второй ступени потеря массы 10% при нагревании до 160°С – удалению трех молекул кристаллизационной и четырех молекул координационной воды.
Синтез III. 2 мл 1 М водного раствора кобальта азотнокислого и 0.020 г СВ[6] · 10H2O (0.018 ммоль) нагревали до 100°С и выдерживали при этой температуре 48 ч. Объемные розовые кристаллы III получали при охлаждении реакционной смеси до комнатной температуры в течение 2 сут. При извлечении из маточного раствора кристаллы трескаются и теряют кристалличность. Выход 0.010 г (42% в расчете на СВ[6] · 10H2O)
| Найдено, %: | C 31.1; | H 4.1; | N 25.9. |
| Для C36H60N26O30Co (в расчете на 8H2Oкристал) | |||
| вычислено, %: | C 31.0; | H 4.3; | N 26.1. |
ИК-спектр (ν, см–1): 3420, 3003, 2956, 2388, 2290, 1740, 1610, 1482, 1417, 1378, 1327, 1300, 1260, 1236, 1191, 1149, 1029, 970, 821, 800, 759, 676, 633, 454.
ТГА: потеря массы 5% при нагревании до 90°С соответствует удалению четырех молекул кристаллизационной воды, на второй ступени потеря массы 10% при нагревании до 160°С – удалению четырех молекул кристаллизационной и четырех молекул координационной воды.
РСА. Дифракционные данные для монокристаллов соединений I–III получены при 130 K на автоматическом дифрактометре Agilent Xcalibur, оснащенном двухкоординатным детектором AtlasS2 (графитовый монохроматор, λ(MoKα) = = 0.71073 Å, ω-сканирование). Интегрирование, учет поглощения, определение параметров элементарной ячейки проведены с использованием пакета программ CrysAlisPro [38]. Кристаллические структуры расшифрованы по программе SHELXT [39] и уточнены полноматричным МНК в анизотропном (за исключением атомов водорода) приближении (SHELXL) [40]. Позиции атомов водорода органических лигандов рассчитаны геометрически и уточнены по модели “наездника”. Катионы Zn(II) в структуре I разупорядочены по трем позициям (Zn(1), Zn(2) и Zn(3)) с относительными весами 0.79/0.12/0.09. Координационное окружение катионов Zn(II), располагающихся в минорных положениях (Zn(2) и Zn(3)), установить не удалось ввиду малой заселенности данных позиций. Катионы Zn2+ и Co2+ в структурах II и III разупорядочены каждый по четырем равновероятным позициям. Часть координационного окружения катионов Zn2+ и Co2+ также разупорядочена. Кристаллографические данные и детали дифракционных экспериментов I–III приведены в табл. 1.
Таблица 1.
Основные кристаллографические данные и параметры уточнения структур I–III
| Параметр | Значение | ||
|---|---|---|---|
| I | II | III | |
| Брутто-формула | C36H57N26O28.5Zn | C36H58N26O29Zn | C36H58N26O29Co |
| M, г/моль | 1375.44 | 1384.45 | 1378.01 |
| Сингония | Ромбическая | Ромбическая | Ромбическая |
| Пр. гр. | P212121 | Pnnm | Pnnm |
| a, Å | 11.5609(2) | 11.5797(3) | 11.5454(3) |
| b, Å | 16.1024(3) | 16.0615(4) | 16.0936(5) |
| c, Å | 29.5339(6) | 14.8855(4) | 14.7975(4) |
| V, Å3 | 5497.98(18) | 2768.51(12) | 2749.48(13) |
| Z | 4 | 2 | 2 |
| ρ(выч.), г/см3 | 1.662 | 1.661 | 1.664 |
| μ, мм−1 | 0.564 | 0.561 | 0.430 |
| F(000) | 2852 | 1436 | 1430 |
| Размер кристалла, мм | 0.24 × 0.12 × 0.09 | 0.32 × 0.26 × 0.19 | 0.24 × 0.20 × 0.19 |
| Область сканирования по θ, град | 3.27–25.68 | 3.38–25.68 | 3.38–29.06 |
| Диапазон индексов hkl | −14 ≤ h ≤15, −18 ≤ k ≤ 20, −37 ≤ l ≤ 32 | −15 ≤ h ≤ 14, −15 ≤ k ≤ 20, −13 ≤ l ≤ 19 |
−12 ≤ h ≤ 15, −16 ≤ k ≤ 22, −19 ≤ l ≤ 21 |
| Nhkl измеренных/независимых | 22 779/10 368 | 7426/2738 | 11 001/3302 |
| Rint | 0.0246 | 0.0142 | 0.0176 |
| Nhkl с I > 2σ(I) | 9253 | 2385 | 2808 |
| GOOF по F 2 | 1.046 | 1.069 | 1.063 |
| R-факторы (I > 2σ(I)) | R1 = 0.0587, wR2 = 0.1597 |
R1 = 0.0820, wR2 = 0.2390 |
R1 = 0.0742, wR2 = 0.2157 |
| R-факторы (по всем отражениям) | R1 = 0.0671, wR2 = 0.1666 |
R1 = 0.0894, wR2 = 0.2482 |
R1 = 0.0827, wR2 = 0.2250 |
| Остаточная электронная плотность (max/min), e/Å3 | 1.339/−0.395 | 0.702/−0.841 | 1.469/−0.646 |
Полные таблицы межатомных расстояний и валентных углов, координаты атомов и параметры атомных смещений для комплексов депонированы в Кембриджском банке структурных данных (№ 1862494 (I), 1862495 (II), 1862496 (III); http:// www.ccdc.cam.ac.uk/structures/), а также могут быть получены у авторов.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Соединения I–III получены при нагревании до 100°С смеси 1 М раствора нитрата цинка или кобальта с СВ[6] в течение 48 ч. При медленном охлаждении реакционной смеси в течение 48 ч (синтезы I и III) получены монокристаллы в виде полиэдров, по данным РСА имеющие состав [Zn(H2O)4(C36H36N24O12)](NO3)2 · 6.5H2O и [Co(H2O)4(C36H36N24O12)](NO3)2 · 7H2O соответственно. При быстром охлаждении реакционной смеси в течение 2 ч (синтез II) образуются монокристаллы в виде палочек, которые, по данным РСА, кристаллизуются в пр. гр. Pnnm (в отличие от P212121 для I) и имеют состав [Zn(H2O)4-(C36H36N24O12)](NO3)2 · 7H2O. Разный режим охлаждения реакционной смеси в случае соединений I и II с цинком приводит к образованию кристаллов разной формы. При медленном охлаждении кристаллизуется более упорядоченная фаза, в которой симметрия ниже. Быстрое охлаждение приводит к кристаллизации более разупорядоченной фазы, в которой симметрия выше.
При извлечении из маточного раствора кристаллы соединений I–III трескаются и теряют кристалличность. Кристаллы соединений I–III нерастворимы в воде, метаноле, спирте. По данным ТГА, проведенного на свежеприготовленных образцах, содержание воды в них выше, чем в образцах, отобранных для РСА.
В структуре комплекса I атом Zn(II) находится в октаэдрическом окружении, состоящем из двух атомов кислорода карбонильных групп молекулы СВ[6] (Zn–O 2.124(5) и 2.200(4) Å) и четырех атомов кислорода аквалигандов (Zn–O 2.026(5)–2.092(5) Å). Таким образом, формируется комплексный катион состава [Zn(H2O)4(CB[6])]2+ (рис. 1а).
Рис. 1.
Координационное окружение катиона Zn(II) в структурах I (а) и II (б). Эллипсоиды 50%-ной вероятности. Атомы водорода не показаны. Для II aльтернативные положения катионов Zn(II) и аквалигандов показаны пунктирными линиями.
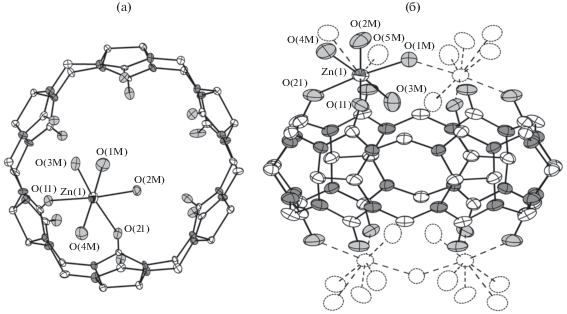
Кристаллы соединений II и III изоструктурны. Атомы Zn(II) в структуре II и Co(II) в III также находятся в искаженном октаэдрическом координационном окружении, состоящем из двух атомов кислорода карбонильных групп молекулы СВ[6] (Zn–O 2.103(3) и 2.315(3) Å для II; Co–O 2.075(3) и 2.308(3) Å для III) и четырех аквалигадов (Zn–O 1.864(8)–2.113(4) Å; Co–O 1.934(5)–2.097(3) Å). Центр молекулы СВ[6] располагается в частной позиции с симметрией 2/m, что приводит к разупорядочению катиона Zn2+ со своим координационным окружением по четырем равнозаселенным позициям (рис. 1б).
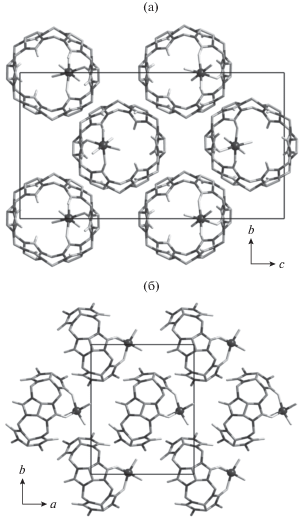
Катионы Zn2+ в структуре I разупорядочены по трем позициям (Zn(1), Zn(2) и Zn(3)), в то время как катионы Zn2+ и Co2+ в II и III соответственно разупорядочены каждый по четырем равновероятным позициям. Описанные выше комплексные катионы [M(H2O)4(CB[6])]2+ (M = Zn, Co) в структурах I–III соединены посредством водородных связей между аквалигандами, карбонильными группами молукул СВ[6] и молекулами кристаллизационной воды, образуя цепочки, параллельные кристаллографической оси a. Цепочки из соседних слоев смещены относительно друг друга на половину трансляции вдоль оси a (рис. 2). В пространстве между цепочками образуются каналы, направленные вдоль оси a, в которых располагаются нитрат-анионы. Нитрат-анионы, аквалиганды и молекулы кристаллизационной воды образуют единую разветвленную сетку водородных связей: O…O 2.67–2.98 Å для I, 2.63–3.01 Å для II и 2.54–3.00 Å для III.
Рис. 2.
Кристаллическая упаковка в структурах I (а) и II (б). Атомы водорода не показаны. Позиции катионов Zn(II) показаны темными шарами. Показано только одно из альтернативных положений катионов Zn(II) аквалигандов.
В ИК-спектрах комплексов I–III в области 3700–3100 см–1 наблюдаются полосы валентных колебаний O–H молекул кристаллизационной воды, в областях 3100–2980 см–1 – групп N–H и 2980–2890 см–1 – групп C–H, при 1743–1740 см–1 – колебания групп С=O СВ[6]. Полосы поглощения нитрат-анионов в спектрах комплексов проявляются при 1378 см–1.
Результаты ТГА соединений I–III показывают, что при скорости нагрева 10°С/мин молекулы воды удаляются в интервале 20–160°С в две стадии, процесс сопровождается эндоэффектами. Потеря массы на первой ступени при 90°С составляет 8% для I и II и 5% для III, что соответствует удалению шести и четырех молекул кристаллизационной воды соответственно. Суммарная потеря кристаллизационной и координационной воды на двух ступенях составляет 18 и 15%. Нагревание выше 250°С приводит к полному термическому разрушению (рис. 3).
Рис. 3.
ТГ-кривые для соединений: для Zn(H2O)4(C36H36N24O12)](NO3)2 · 9H2O (сплошная линия) и [Co(H2O)4(C36H36N24O12)](NO3)2 · 8H2O (пунктирная линия).
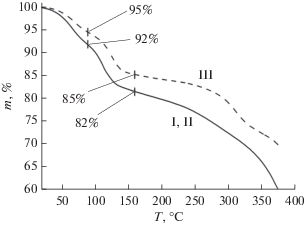
Исходя из данных элементного и термогравиметрических анализов состав поликристаллических образцов можно представить следующими формулами: [Zn(H2O)4(C36H36N24O12)](NO3)2 · 9H2O для соединений из синтеза I и II и [Co(H2O)4-(C36H36N24O12)](NO3)2 · 8H2O для соединения из синтеза III.
До настоящей работы был получен и структурно охарактеризован единственный комплекс цинка с производным СВ[6] [26], в котором наблюдается непосрественная координация карбонильных групп макроцикла к катиону Zn2+. Описано несколько соединений Zn(II) с кукурбит[5]урилом [24, 25]. Примеры комплексов с непосредственной координацией карбонильных групп кукурбитурилов к катионам Co2+ в литературе отсутствуют. Таким образом, соединения, полученные в настоящей работе – первые комплексы цинка(II) и кобальта(II) с нефункционализированным кукурбит[6]урилом.
Список литературы
Lee J.W., Samal S., Selvapalam N. et al. // Acc. Chem. Res. 2003. V. 36. P. 621.
Lagona J., Mukhopadhyay P., Chakrabarti S. et al. // Angew. Chem. Int. Ed. 2005. V. 44. P. 4844.
Behrend R., Meyer E., Rusche F. // Justus Liebigs Ann. Chem. 1905. V. 339. P. 1.
Freeman W.A. // Acta Crystallogr. B. 1984. V. 40. P. 382.
Герасько О.А., Самсоненко Д.Г., Федин В.П. // Успехи химии. 2002. Т. 71. С. 741.
Gerasko O.A., Sokolov M.N., Fedin V.P. // Pure Appl. Chem. 2004. V. 76. P. 1633.
Коваленко Е.А., Наумов Д.Ю., Федин В.П. // Коорд. химия. 2011. Т. 37. С. 139 (Kovalenko E.A., Naumov D.Y., Fedin V.P. // Russ. J. Coord. Chem. 2011. V. 37. P. 137. doi 10.1134/S1070328411010076).
Коваленко Е.А., Наумов Д.Ю., Федин В.П. и др. // Коорд. химия. 2012. Т. 38. С. 165 (Kovalenko E.A., Naumov D.Y., Fedin V.P. et al. // Russ. J. Coord. Chem. 2012 V. 38. P. 153. doi 10.1134/S1070328412020054).
Коваленко Е.А., Палаткина М.Ю., Самсоненко Д.Г. и др. // Коорд. химия. 2012. Т. 38. С. 395 (Kovalenko E.A., Palatkina M.Y., Samsonenko D.G. et al. // Russ. J. Coord. Chem. 2012. V. 38. P. 379. doi 10.1134/ S1070328412050041).
Lü J., Lina J.-X., Caoa M.-N. et al. // Coord. Chem. Rev. 2013. V. 257. P. 1334.
Герасько О.А., Коваленко Е.А., Федин В.П. // Успехи химии. 2016. Т. 85. № 8. С. 795.
Wei L.T., Zhang Y.Q., Zhou K.Zh. et al. // Inorg. Chim. Acta. 2016. V. 445. P. 1.
Wei L.T., Zhang Y.Q., Zhou K.Zh. et al. // Inorg. Chim. Acta. 2016. V. 445. P. 277.
Zhang Ch., Zhang Y.-Q., Xue S.-F. et al. // Inorg. Chim. Acta. 2016. V. 450. P. 258.
Zhao Y., Liang L.-L., Chen K. et al. // CrystEngComm. 2013. V. 15. P. 7987.
Qiu Sh.-Ch., Li Q., Zhang J. et al. // J. Incl. Phenom. Macrocycl. Chem. 2016. V. 86. P. 1.
Zhang D.-Q., Sun T., Zhang Y.-Q. et al. // Eur. J. Inorg. Chem. 2015. V. 318. P. 323.
Qiu Sh.-Ch., Li Q., Chen K. et al. // Inorg. Chem. Commun. 2016. V. 72. P. 50.
Li Q., Qiu Sh.-Ch., Zhang Y.-Q. et al. // RSC Adv. 2016. V. 6. P. 77805.
Li Q., Zhang Y.-Q., Zhu Q.-J. et al. // Chem. Asian J. 2015. V. 10. P. 1159.
Liang L.-L., Zhao Y., Zhang Y.-Q. et al. // CrystEngComm. 2013. V. 15. P. 3943.
Ji N.-N., Cheng X.-J., Zhao Yet al. // Inorg. Chem. 2014. V. 53. P. 21.
Xiao X., Chen K., Xue S.-F. et al. // J. Mol. Struct. 2010. V. 969. P. 216.
Cong H., Chen K., Wang Ch.-Z. et al. // Wuji Huaxue Xuebao. 2014. V. 30. P. 2839.
Liu J.-X., Long L.-Sh., Huang R.-B. et al. // Acta Crystallogr. E. 2005. V. 61. P. m1021.
Zhang Y.-Q., Zhen L.-M., Yu D.-H. et al. // J. Mol. Struct. 2008. V. 875. P. 435.
Qin X., Chen W.-J., Zhang Y.-Q. et al. // J. Mol. Struct. 2011. V. 996. P. 12.
Liang Zh.-Y., Chen H.-Y., Shan Ch.-Y. et al. // Polyhedron. 2010. V. 110. P. 125.
Liang L.-L., Zhao Y., Chen K. et al. // Polymers. 2013. V. 5. P. 418.
Limei Zh., Jiannan Zh., Yunqian Zh. et al. // Supramolecular Chem. 2008. V. 20. P. 709.
Wang Zh.-B., Zhao M., Li Y.-Zh. et al. // Supramolecular Chem. 2008. V. 20. P. 689.
Yi S., Captain B., Ottaviani M.F. et al. // Langmuir. 2011. V. 27. P. 5624.
Коваленко Е.А., Митькина Т.В., Герасько О.А. и др. // Коорд. химия. 2011. Т. 37 С. 163 (Kovalenko E.A., Mitkina T.V., Geras’ko O.A. et al. // Russ. J. Coord. Chem. 2011. V. 37. P. 161. https://doi.org/10.1134/ S1070328411020023).
Mitkina T.V., Sokolov M.N., Naumov D.Y. et al. // Inorg. Chem. 2006. V. 45. P. 6950.
Mitkina T.V., Zakharchuk N.F., Naumov D.Y. et al. // Inorg. Chem. 2008. V. 47. P. 6748.
Freeman W.A., Mock W.L., Shih N.-Y. // J. Am. Chem. Soc. 1981. V. 103. P. 7367.
NETZSCH Proteus Thermal Analysis. Vtersion 4.8.1. Bayern (Germany): NETZSCH-Gerätebau, 2005.
CrysAlisPro 1.171.38.41. Rigaku Oxford Diffraction. 2015.
Sheldrick G.M. // Acta Crystallogr. A. 2015. V. 71. P. 3.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия