

Координационная химия, 2022, T. 48, № 5, стр. 302-313
Синтез, строение и фотолюминесценция комплексов цинка(II) и серебра(I) с 2-(3,5-диметил-1H-пиразол-1-ил)-4,6-дифенилпиримидином
К. А. Виноградова 1, *, М. И. Рахманова 1, Е. Б. Николаенкова 2, В. П. Кривопалов 2, М. Б. Бушуев 1, Н. В. Первухина 1, Д. Ю. Наумов 1, С. А. Мартынова 1
1 Институт неорганической химии им. А.В. Николаева СО РАН
Новосибирск, Россия
2 Новосибирский институт органической химии им. Н.Н. Ворожцова
Новосибирск, Россия
* E-mail: kiossarin@mail.ru
Поступила в редакцию 03.11.2021
После доработки 26.11.2021
Принята к публикации 28.11.2021
- EDN: ZWZTRB
- DOI: 10.31857/S0132344X22050097
Аннотация
Получены комплексы [AgL2]PF6 (I) и [ZnLCl2] (II) (L = 2-(3,5-диметил-1H-пиразол-1-ил)-4,6-дифенилпиримидин) при взаимодействии хлорида цинка(II) или гексафторофосфата серебра(I) с L (мольноe соотношение M : L = 1 : 1 или 1 : 2) в органических средах. Строение комплексов определено по данным рентгеноструктурного анализа (CCDC № 2118498, 2118499). В обоих комплексах L координируется бидентатно-циклическим способом, координационный узел – искажeнный тетра-эдр. Координационный узел комплекса II образован одной координированной молекулой L и двумя хлорид-ионами (ZnN2Cl2), серебра(I) – двумя молекулами L (AgN4). Для соединений I, II исследованы фотолюминесцентные свойства в твердом состоянии. Соединение II демонстрирует флуоресценцию (1.3, 11 нс) в голубой области спектра (λмакс = 378 нм, квантовый выход 7.8%). Ма-ксимум полосы при 530 нм в спектре фотолюминесценции I имеет батохромный сдвиг на ~140–160 нм относительно максимума полос эмиссии L и II. Комплекс I демонстрирует белую флуоресценцию (1.6, 11 нс, квантовый выход 5.5%), которая происходит за счeт внутрилигандных переходов, возмущeнных координацией L к атому серебра. Исследована фотостабильность комплекса I при 300 и 80 K.
Соединения, излучающие белым светом, имеющим в координатах цветности Международной комиссии по освещению (CIE) значения около (0.33, 0.33), называются белыми люминофорами, а устройства на их основе – белоизлучающими светодиодами (wLED, white light-emitting diodes). Устройства, излучающие белый свет, находят важные приложения, например, наиболее широко используются в осветительных приборах. Исходя из этого, поиск и исследование новых соединений, обладающих эффективной белой фото- и электролюминесценцией, является важной задачей.
Выделяют два основных подхода к получению белых люминофоров – создание однокомпонентных и многокомпонентных эмиттеров. К однокомпонентным эмиттерам можно отнести органические и координационные соединения, которые имеютширокий спектр фотолюминесценции в видимой области, что приводит к белому излучению [1, 2]. Для создания многокомпонентных систем используют следующее методики: (1) допирование полимерных матриц органическими веществами, которые излучают в красной, зелeной и голубой областях спектра [2–5]; (2) нанесение друг на друга органических слоeв, излучающих красным, зелeным и синим [2]; (3) допирование металлорганических координационных полимеров красным, зелeным и голубым флуорофорами в нужном соотношении [6–9]. Одной из ключевых проблем использования многокомпонентных эмиттеров в осветительных устройствах является то, что разные флуорофоры зачастую имеют разную устойчивость и, как следствие, могут по отдельности выходить из строя (“выгорать”). Это, естественно, нарушает баланс компонентов и в целом белое свечение [10, 11]. Кроме этого, их существенным недостатком является и то, что цвет излучения может зависеть от напряжения [11]. Системы, в которых используется одно вещество как эмиттер белого света, лишены перечисленных недостатков, вследствие чего являются перспективными для возможных приложений.
На данный момент можно выделить следующие индивидуальные соединения, которые могут демонстрировать белую фотолюминесценцию: комплексы цинка(II) и кадмия(II) [12, 13], хотя для них более характерно всe же голубое/синее свечение [14–18], комплексы платины [19–22], редкие примеры комплексов лантанидов [23, 24], некоторые органические молекулы [25–27]. В последние годы появляются работы, в которых используются комплексные соединения монетных металлов – медь(I) [28, 29], золото(I) [30] и серебро(I) [31–33]. Изучены некоторые механизмы белой люминесценции, например координационные соединения платины(II) образуют эксиплексы при возбуждении, и за счeт практически одновременной эмиссии молекулы и эксиплекса получается белое свечение [10, 22].
В [32] одним из исследуемых соединений был ионный комплекс [Ag(Dppb)2]BF4 (Dppb = 1,2-бис(дифенилфосфино)бензол), который обладает широкой полосой эмиссии c максимумом при 526 нм в твeрдом состоянии. Этот комплекс был использован как единственное излучающее вещество в wLED устройстве в матрице из поли(винилкарбазола). Сконструированное устройство обладает более широкой полосой эмиссии, чем исходный комплекс – практически белым свечением (максимальная яркость 365 кд/м2 при 20 В). В [33] показана возможность получения белой эмиссии и конструирование wLED устройства на основе гибридного соединения серебра(I) [H2DABCO]- [Ag2X4(DABCO)] (DABCO = 1,4-диазабицикло[2.2.2]октан). Недавно нами было опубликовано исследование, в котором детально изучены два комплекса нитрата серебра(I) с 2-амино-4-фенил-6-метилпиримидином (Pym) [34]. При комнатной температуре в твeрдом состоянии один из описанных комплексов ([Ag3(Pym)2(H2O)0.55(NO3)3]) демонстрировал белое свечение.
В настоящей работе для синтеза комплексов серебра(I) мы тоже использовали лиганд на основе пиримидина, но при этом в положение 2 пиримидинового кольца вместо NH2-группы был введен пиразольный заместитель (схема 1 ). Полученное соединение, 2-(3,5-диметил-1H-пиразол-1-ил)-4,6-дифенилпиримидин (L), в отличие от ранее использованного Pуm, выступает преимущественно бидентатным лигандом, что будет способствовать получению не полимерных, а моноядерных соединений. Также представляло интерес синтезировать и исследовать фотолюминесцентные (ФЛ) свойства комплекса цинка(II) с лигандом L и оценить его перспективность для получения белой эмиссии.
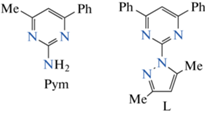
Схема 1 .
Цель настоящей работы − синтез комплексов серебра(I) и цинка(II) с лигандом L, установление кристаллического строения и детальное исследование их фотолюминесцентных свойств в твердом состоянии.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для синтеза комплексов использовали соединение L (CAS 202192-87-8), гексафтороборат серебра(I) (CAS 26042-63-7), хлорид цинка(II) (CAS 7646-85-7), хлористый метилен, этанол и ацетонитрил (все марки “х. ч.”), которые являются коммерчески доступными и применялись без дополнительной очистки. Для записи спектров поглощения и спектров ФЛ в растворе использовали хлористый метилен со степенью чистоты для ВЭЖХ. Несмотря на то что соединение L является коммерчески доступным, оно может быть синтезировано по реакции 2-гидразино-4,6-дифенилпиримидина (CAS 76071-58-4) и ацетилацетона (CAS 123-54-6) в подкисленном этанольном растворе по методикам, описанным в [35, 36].
Элементный анализ (C, H, N) образцов комплекса I выполняли на приборе vario MICRO cube с использованием стандартной методики. Элементный анализ (C, H, N) фторсодержащего комплекса II проводили по методике [37].
Для съемки спектров диффузного отражения (СДО) образцы комплексов смешивали с сульфатом бария в массовом соотношении 1 : 100. СДО образцов записывали на приборе UV-3101 PC Shimadzu. Спектры представлены как функция Кубелки–Мунка, F(R) = (1 − R)2/2R, где R − коэффициент диффузного отражения образца по сравнению с BaSO4. Оптические спектры в растворах записывали на спектрофотометре СФ-2000. Термогравиметрический (ТГ) анализ образцов выполняли на термоанализаторе NETZSCH TG 209 F1 Iris Thermo Microbalance. Рентгенофазовый анализ (РФА) образцов выполняли на дифрактометре Shimadzu XRD-7000 (CuKα-излучение, Ni-фильтр, щели Соллера 2.5°, щель расходимости 0.5°), оснащенном детектором DECTRIS MYTHEN2 R 1K.
ИК-спектры комплексов и лиганда L снимали на спектрофотометрах Scimitar FTS 2000 Fourier-spectrometer DIGILAB в диапазоне 4000–400 см –1 и Vertex 80 Bruker в диапазоне 600–100 см –1. Образцы растирали в агатовой ступке с сухим KBr или полиэтиленом и прессовали в таблетки. Отнесение полос ИК-спектров проводили путем сравнения с литературными данными [38, 39].
Для исследования стабильности комплекса серебра(I) при действии света использовали лазерный диод, излучающий свет длиной волны 405 нм (мощность 200 мВт). Образцы комплекса прессовали в таблетки с KBr, полученные таблетки облучали определенное время, после чего регистрировали ИК-спектры в диапазоне 600−300 см−1.
Синтез комплексa [AgL2]PF6 (I). К раствору L (60.0 мг, 0.184 ммоль) в 1.0 мл хлористого метилена добавляли раствор AgPF6 (23.2 мг, 0.0919 ммоль) в 3.0 мл ацетонитрила. Образовался бесцветный прозрачный раствор. При уменьшении объема раствора до ~2 мл начал выпадать белый осадок. Полученную смесь перемешивали около 30 мин при комнатной температуре. Осадок отфильтровывали на стеклянном пористом фильтре, промывали ацетонитрилом, высушивали на воздухе. Выход 57 мг (68%).
В течение недели в маточном растворе появлялись крупные, хорошо ограненные бесцветные кристаллы. По данным РСА, кристаллы имеют состав [AgL2]PF6.
Синтез поликристаллического образца комплекса [ZnLCl2] (II). К раствору лиганда L (40.2 мг, 0.123 ммоль) в 2 мл хлористого метилена добавляли раствор ZnCl2 (23.2 мг, 0.160 ммоль) в 2 мл этанола. Полученную прозрачную и бесцветную реакционную смесь перемешивали в течение 10 мин при нагревании (Т = 60°С), после чего образовался белый поликристаллический продукт. Полученный продукт перемешивали с маточным раствором в течение 2 ч при небольшом нагревании (Т = 35°С). Осадок отфильтровывали, промывали 2 мл хлористого метилена, высушивали на воздухе. Выход 42 мг (74%).
Синтез кристаллов [ZnLCl2] (II). К раствору лиганда L (50.0 мг, 0.153 ммоль) в 2 мл этанола добавляли раствор ZnCl2 (7.0 мг, 0.051 ммоль) в 2 мл этанола. Полученную прозрачную и бесцветную реакционную смесь перемешивали около 2 мин при нагревании (Т = 60°С), образовался белый поликристаллический продукт. Полученный продукт перемешивали с маточным раствором в течение 1 ч. Осадок отфильтровывали, промывали 1 мл этанола. Выход 23 мг (99%). Прозрачные, бесцветные призматические кристаллы появились в маточном растворе спустя 3 мес. По данным РСА, кристаллы имеют состав [ZnLCl2].
РСА монокристаллов соединений I и II выполнен при 150 K на дифрактометре Bruker D8 Venture, оснащенном детектором CMOS PHOTON III и микрофокусным источником IµS 3.0 (фокусирующие зеркала Монтеля, MoKα-излучение). Интенсивности отражений измерены методом φ‑ и ω-сканирования узких (0.5°) фреймов. Редукция данных проведена с помощью пакета программам APEX3 [40]. Структуры расшифрованы прямым методом и уточнены полноматричным МНК по F 2 в анизотропном приближении для неводородных атомов с использованием комплекса программ SHELXTL [41]. Атомы водорода органических лигандов локализованы из карт разностной электронной плотности и уточнены в приближении жесткого тела. Кристаллографические данные и детали дифракционного эксперимента приведены в табл. 1.
Таблица 1.
Кристаллографические данные и параметры уточнения структур I и II
| Параметр | Значение | |
|---|---|---|
| Эмпирическая формула | C42H36F6N8PAg | C21H18N4Cl2Zn |
| М | 905.63 | 462.66 |
| Сингония | Моноклинная | Моноклинная |
| Пр. группа | P21/c | C2/c |
| a, Å | 15.6308(3) | 23.992(2) |
| b, Å | 18.6760(4) | 14.014(1) |
| c, Å | 14.0639(2) | 16.239(3) |
| β, град | 111.582(1) | 131.286(2) |
| V, Å3 | 3817.7(1) | 4102.8(9) |
| Z | 4 | 8 |
| ρ(выч.), г/см3 | 1.576 | 1.498 |
| μ, мм–1 | 0.643 | 1.472 |
| F(000) | 1840 | 1888 |
| Размеры кристалла, мм | 0.12 × 0.08 × 0.08 | 0.15 × 0.10 × 0.04 |
| Диапазон углов θ, град | 1.401–26.373 | 1.920–25.708 |
| Число измеренных рефлексов | 50 592 | 18 927 |
| Число независимых рефлексов | 7812 (Rint = 0.0360) | 3889 (Rint = 0.0611) |
| Полнота сбора данных по θ = 25.00°, % | 99.9 | 99.8 |
| S-фактор по F 2 | 1.048 | 1.068 |
| R1, wR2 (I > 2σI) | 0.0250, 0.0726 | 0.0546, 0.1414 |
| R1, wR2 (все отражения) | 0.0306, 0.0754 | 0.0729, 0.1526 |
| Остаточная электронная плотность, min/max, e/Å3 | 0.323/–0.546 | 1.809/–0.630 |
Дополнительные структурные данные для соединений I и II депонированы в Кембриджском банке структурных данных (ССDС № 2118498, 2118499;; deposit@ccdc.cam.ac.uk или http://www.ccdc.cam. ac.uk).
Спектры фотолюминесценции и возбуждения комплексов в твердом состоянии регистрирова-ли с помощью спектрофлуориметра Horiba Fluorolog 3, оснащенного непрерывными 450 Вт и импульсными ксеноновыми лампами в качестве источников света, охлаждаемым детектором и двойными решетчатыми монохроматорами возбуждения и излучения. Для измерения квантового выхода использовали спектрофлуориметр Fluorolog 3, снабженный квантовой сферой (Quanta-ϕ). Спектры фотолюминесценции и возбуждения корректировали с учетом интенсивности источника (лампа и решетка) и спектрального отклика излучения (детектор и решетка) с помощью стандартных кривых коррекции. Температурно-зависимые измерения проводили с помощью Optistat DN в диапазоне 77–300 К.
Кинетику затухания фотолюминесценции в наносекундном временном диапазоне регистрировали методом коррелированного по времени подсчета одиночных фотонов с использованием импульсного источника света NanoLED и контроллера NanoLED-C2. Для комплекса I для возбуждения использовали длину волны 350 нм, регистрацию проводили в максимуме излучения 530 нм, в случае II для возбуждения использовали длину волны 300 нм, регистрацию проводили в максимуме излучения 387 нм (Т = 300 К). Кинетику затухания люминесценции в миллисекундном диапазоне времени записывали с помощью импульсной ксеноновой лампы (λex = 320 нм, λem = 500 нм).
Исследование фотостабильности комплекса [AgL2]PF6 проводили по двум методикам.
Методика № 1. Свежую порцию поликристаллического образца комплекса I помещали между двумя кварцевыми стеклами, для нее записывали спектр ФЛ при возбуждении 360 нм. Далее образец облучали последовательно набором длин волн в диапазоне от 300 до 500 нм с шагом 10 нм (при таком подходе суммарное время облучения составляло ~10 мин) и повторно записывали спектр эмиссии при первоначальных параметрах, после сравнивали два спектра эмиссии: до интенсивного облучения и после интенсивного облучения.
Методика № 2. Для такого же свежеприготовленного образца записывали подряд 10 спектров ФЛ при возбуждении 320 нм. В этом случае общее время облучения образца составляло от 8 до 10 мин в зависимости от диапазона записи спектра эмиссии. По изменению интенсивности спектра ФЛ как в первой методике, так и во второй делали выводы о фотостабильности образца. Отметим, что для веществ, не деградирующих при воздействии электромагнитного облучения, спектр эмиссии не изменяется при записи в одинаковых условиях.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Реакция между лигандом L и гексафторофосфатом серебра(I) в смеси CH2Cl2−MeCN (1 : 3) приводит к образованию белого поликристаллического осадка (I, cхема 2).
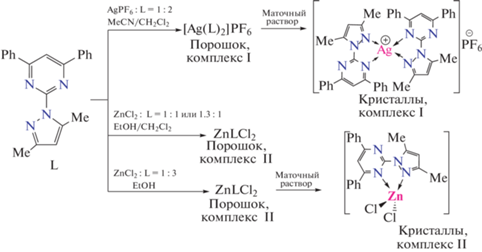
Схема 2 .
По данным элементного анализа, полученный продукт имеет состав AgL2PF6. В маточном растворе в течение недели появляются прозрачные бесцветные крупные кристаллы, пригодные для РСА (рис. S1 ). Полученная расчетная дифрактограмма из данных монокристального РСА хорошо согласуется с экспериментальной дифрактограммой полученной с порошка комплекса I (рис. S2 ).
Комплекс II получен при взаимодействии хлорида цинка(II) и лиганда L в этанольном растворе (или в смеси EtOH–CH2Cl2) в виде белого порошка состава ZnLCl2 согласно данным элементного анализа (см. схему 2 ). Эту поликристаллическую фазу можно получить при использовании и других мольных соотношений Zn : L (1 : 1, 1.3 : 1 или 1 : 3), что подтверждено данными РФА (рис. S3 ). Наиболее близкий к теоретическому CHN-анализ получен в реакции с небольшим избытком соли цинка(II) по отношению к соединению L (Zn : L = 1.3 : 1). Это связано с отсутствием примесей L в образце комплекса. В дальнейшем именно этот образец использовали для фотофизических исследований комплекса II. Монокристаллы II для проведения РСА были получены из маточного раствора синтеза, проведeнного в соотношении Zn : L = 1 : 3. Расчетная порошкограмма, полученная по данным монокристалльного РСА, совпадает с экспериментальными порошкограммами для всех выделенных поликристаллических образцов ZnLCl2 (рис. S3 ). На основании этого сравнения cделан вывод об образовании одной и той же фазы во всех случаях.
Кристаллическая структура комплекса I образована ионами [AgL2]+ и [PF6]–. Строение комплексного катиона показано на рис. 1, кристаллографические данные приведены в табл. 1. Две молекулы L координируются к атому серебра бидентатно-циклическим способом через атомы азота N(2) пиразольного и N(3) пиримидинового циклов. Координационный полиэдр атома серебра – искаженный тетраэдр. Длины связей в молекуле комплекса I представлены в табл. 2. Пиразольное, пиримидиновое и фенильное кольца плоские с точностью до 0.01 Å. Плоскости пиримидиновых колец повернуты относительно плоскостей пиразольных колец на 10.4° и 9.2°. Углы между плоскостями пиримидинового и фенильного колец составляют 21.4°, 37.1° и 26.9°, 34.8° соответственно. В структуре I обнаружены межмолекулярные взаимодействия между атомами фтора гексафторофосфат-анионов и атомами водорода фенильных колец комплексных катионов [AgL2]+, расстояния F…Н и F…C составляют около 2.3–2.5 и 3.1–3.3 Å соответственно.
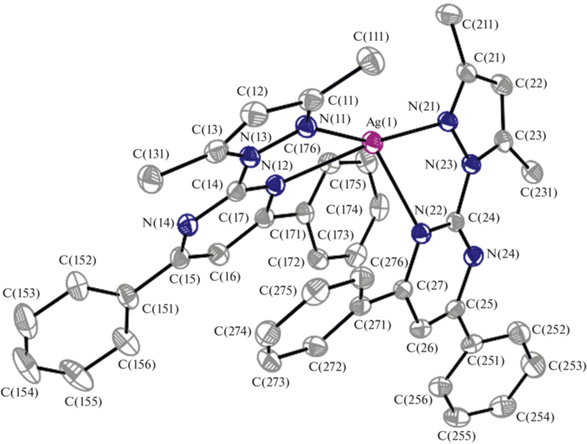
Таблица 2.
Избранные межатомные расстояния (Å) в структурах I, II
| Связь d, Å | Связь d, Å | ||
|---|---|---|---|
| I | II | ||
| Координационный узел | |||
| Ag(1)–N(11) | 2.180(1) | Zn(1)–N(1) | 2.038(4) |
| Ag(1)–N(21) | 2.181(1) | Zn(1)–N(3) | 2.113(3) |
| Ag(1)–N(12) | 2.478(1) | Zn(1)–Cl(1) | 2.217(1) |
| Ag(1)–N(22) | 2.477(1) | Zn(1)–Cl(2) | 2.204(1) |
| Пиразольные кольца | |||
| N–N | 1.373(2)–1.378(2) | N–N | 1.379(4) |
| N–C | 1.323(2)–1.380(2) | N–C | 1.330(5), 1.374(5) |
| C–C | 1.363(2)–1.409(2) | C–C | 1.364(6), 1.403(6) |
| C-CМе | 1.487(2)–1.492(2) | C-CМе | 1.483(6), 1.485(6) |
| Пиримидиновое кольцо | |||
| N–C | 1.318(2)–1.350(2) | N–C | 1.315(5)–1.366(5) |
| C–C | 1.386(2)–1.391(2) | C–C | 1.381(6), 1.387(6) |
| C–CPh | 1.478(2)–1.484(2) | C–CPh | 1.478(5), 1.466(5) |
| Фенильные кольца | |||
| 1.374(3)–1.401(2) | 1.368(7)–1.416(6) Å | ||
| Межмолекулярные взаимодействия | |||
| F(5)…Н(172)' 2.518 | F(5)…C(172)' 3.301 | Cl(1)…Н(56)' 2.804 | Cl(1)…C(56)' 3.485 |
| F(5)…Н(256)' 2.427 | F(5)…C(256)' 3.256 | Cl(1)…Н(2)' 2.773 | Cl(1)…C(2)' 3.565 |
| F(1)…Н(272)' 2.368 | F(1)…C(272)' 3.165 | Cl(2)…Н(73)' 2.872 | Cl(2)…C(73)' 3.606 |
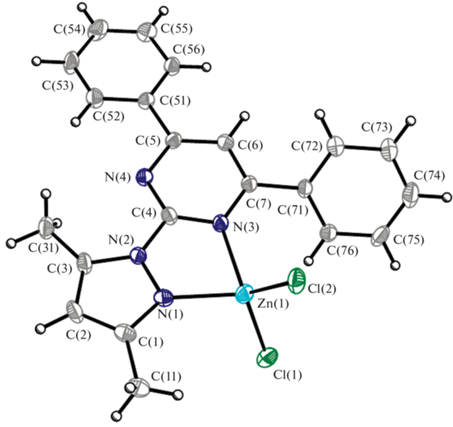
Кристаллическая структура комплекса II построена из молекул ZnLCl2 (рис. 2, табл. 1). Способ координации лиганда L такой же, как в комплексе I. К атому цинка координирована одна молекула L, координационный полиэдр атома цинка дополняется атомами хлора до искаженного тетраэдра. Координация лиганда L приводит к замыканию почти плоского пятичленного хелатного кольца ZnN3C, атом Zn(1) отклоняется от средней хелатной плоскости кольца на –0.0092 Å. Длины связей в молекуле комплекса II представлены в табл. 2. Пиразольное, пиримидиновое и фенильное кольца плоские с точностью до 0.01 Å. Углы между плоскостями пиримидинового и фенильного колец составляют 7.6° и 46.5°, плоскость пиримидинового кольца повернута относительно плоскости пиразольного кольца на 4.1°. В структуре комплекса II обнаружены межмолекулярные взаимодействия между атомами хлора и атомами водорода пиразольных и фенильных ко-лец соседних молекул, расстояния составляют около Cl…Н 2.7–2.8 Å и Cl…C 3.4–3.6 Å. Необходимо отметить π-стэкинг-взаимодействия между центрами пиразольного и фенильного колец соседних молекул, расположенными параллельно друг другу в кристаллической структуре с расстоянием между плоскостями около 3.490 Å. Расстояние между центроидами этих колец составляет 3.682 Å. (рис. S4 ). Длины связей и углы в комплексах хорошо согласуются с известными литературными данными [42].
Для комплексов I и II получены и проанализированы ИК-спектры в диапазоне от 4000 до 100 см–1 (рис. S5 и S6 ). Основные колебания комплексов и лиганда L представлены в табл. 3. Колебания CH-, CH3-групп и валентно-деформационные колебания колец в ИК-спектрах комплексов I и II незначительно сдвинуты относительно соответствующих колебаний в спектре лиганда L. В спектре комплекса I найдена полоса колебания при 840 см–1, соответствующая колебанию некоординированного иона ${\text{PF}}_{6}^{ - }$. В низкочастотной области в спектрах комплексов по сравнению со спектром L появляются новые полосы, некоторые полосы исчезают. В ИК-спектре комплекса II колебания Zn–N и Zn–Cl находятся в одной области – 345–310 см–1, что усложняет их отнесение. Можно предположить, что полосы при 343, 336, 315 см–1, отсутствующие в спектре лиганда L, относятся к валентным колебаниям в координационном узле ZnN2Cl2. В ИК-спектре комплекса I полоса колебаний Ag–N более слабая, чем в ИК-спектре комплексa II и находится при 336 см–1.
Таблица 3.
Основные колебательные частоты в ИК-спектрах лиганда L, комплексов I и II
| Соединение | ν(CH), cм–1 | ν(CH3), cм–1 | (ν + δ)колец | ν(P–F) | ν(M–N) | ν(M–Cl) |
|---|---|---|---|---|---|---|
| L | 3080 3045 | 2981, 2924 | 1604, 1592, 1578, 1570, 1531, 1499 | |||
| I | 3142 3110 3096 3060 | 2971, 2922 | 1602, 1594, 1577, 1567, 1527, 1498 | 840 | 336 | |
| II | 3146 3117 3090 3063 | 2986, 2926, 2849 | 1602, 1594, 1577, 1570, 1531, 1523 | 343, 336, 315 (смешаны с Zn–Cl) |
343, 336, 315 (смешаны с Zn–N) |
Фотолюминесцентные свойства комплексов I и II исследовали в твeрдом состоянии при комнатной температуре, для комплекса I также провeдeны исследования при температурах в диапазоне от 77 до 300 K. Отметим, что для лиганда L и комплексов были записаны спектры поглощения в хлористом метилене (рис. S7 ). Оба комплекса имеют практически идентичный спектр поглощения со спектром L, полосы переноса заряда металл–лиганд выше 350 нм отсутствуют, свидетельствуя о том, что комплексы практически полностью диссоциируют в растворе. Для лиганда L были получены фотофизические характеристики в растворе. Лиганд L обладает одной полосой эмиссии при 380 нм с временем жизни возбужденного состояния 6.8 нс (рис. S8 ) и квантовым выходом 0.4% (λвозб = 320 нм).
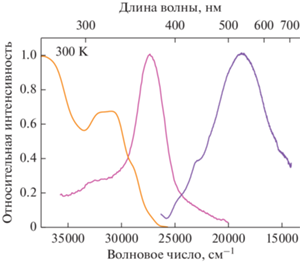
Соединение II обладает типичной для комплексов цинка(II) голубой эмиссией [14–18], полоса ФЛ в спектре комплекса II (λэм = 387 нм) батохромно смещена по сравнению с полосой ФЛ в спектре лиганда L (λэм = 368 нм) на 19 нм (рис. 3). В целом спектр ФЛ комплекса II похож на спектр ФЛ лиганда L (λэм = 368 нм). Полоса эмиссии при 387 нм имеет два времени жизни возбужденного состояния (t1 = 1.6 нс, t2 = 11 нс, табл. 4, рис. S9 ), при этом вклад первого времени менее 0.01%, что может быть вызвано релаксацией возбужденных поверхностных молекул. Близкое положение максимумов в спектрах ФЛ комплекса II и L (387 и 368 нм) и близкие наносекундные времена жизни (11 и 9.1 нс), по-видимому, свидетельствуют о том, что природа эмиссии комплекса II – это внутрилигандные переходы координированной молекулы L. При этом квантовый выход ФЛ комплекса II больше на порядок (табл. 4), что связано с образованием более жeсткой и плоской молекулы комплекса [ZnLCl2].
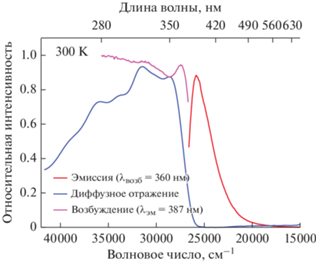
Таблица 4.
Фотофизические данные для лиганда L, комплексов I и II в твердом состоянии
| Соединение | Максимум в спектре ФЛ | Квантовый выход | Время жизни возбужденного состояния |
|---|---|---|---|
| L | 368 нм (λвозб = 340 нм, 300 K) |
0.5% (λвозб = 340 нм, 300 K) |
t = 9.1 нс (λвозб = 300 нм, λэм = 380 нм, 300 K) |
| I | 408, 435, 530 нм (λвозб = 360 нм, 300 K) |
Не менее 5.5% (λвозб = 360 нм, 300 K) |
t1 = 2.8 нс, t2 = 12 нс (λвозб = 350 нм, λэм = 530 нм, 300 K) |
| 455, 485, 520 нм (λвозб = 360 нм, 77 K) |
t1 = 0.16 мс, t2 = 4.8 мс (λвозб = 320 нм, λэм = 500 нм, 77 K) |
||
| II | 387 нм (λвозб = 340 нм, 300 K) |
7.8% (λвозб = 320 нм, 300 K) |
t1 = 1.3 нс, t2 = 11 нс (λвозб = 300 нм, λэм = 387 нм, 300 K) |
Комплекс I при фотовозбуждении различными длинами волн (λвозб = 320, 340, 360, 390 нм) демонстрирует практически белую люминесценцию (рис. 4, рис. S10 , табл. S1 ). Например, при возбуждении длиной волны 390 нм цветность излучаемого света соответствует точке 0.30; 0.37 в координатах CIE. В спектре ФЛ комплекса I наблюдается одна широкая полоса с максимумом при 500–540 нм с колебательной структурой, в спектре возбуждения также проявляется колебательная структура. Полоса эмиссии комплекса I имеет два наносекундных времени жизни (табл. 4, рис. S11 ), которые близки к временам жизни комплекса II и лиганда L. По-видимому, эмиссия комплекса II происходит за счет внутрилигандных переходов, сильно возмущенных координированным атомом серебра.
Рис. 4.
Спектры ФЛ (λвозб = 360 нм), возбуждения (λэм = 530 нм) и диффузного отражения комплекса I при 300 K.
Известно, что соединения серебра(I) могут быть неустойчивыми под действием света, поэтому одной из задач исследования было изучение стабильности полученного комплекса I при действии электромагнитного излучения с различной энергией. Исследована фотостабильность комплекса I при 300 и 77 К при облучении светом с различными длинами волн (рис. S12–S16 ). При комнатной температуре комплекс достаточно быстро разлагается под действием света (рис. S12, S13 ). За 10 мин интенсивность основной полосы ФЛ уменьшается примерно на 15% (рис. S12 ), но при постепенном снижении интенсивности ФЛ форма полосы эмиссии сохраняется (рис. S13 ). Через 2 ч облучения спектр ФЛ практически исчезает (рис. S14 ). Возможно, деструкция комплекса связана с разрушением координационной связи и с восстановлением Ag+ до Ag0. На основании этого можно сделать вывод, что при облучении разрушается более интенсивно люминесцирующий комплекс серебра(I) (φ = 5.5%) в сравнении с менее интенсивно люминесцирующим лигандом L (φ = 0.5%); это приводит к уменьшению интенсивности полосы ФЛ в спектре. Мы попробовали зафиксировать этот процесс методом ИК-спектроскопии. Для исследования фотостабильности комплекса I была выбрана область 600–300 см–1, так как в ней наблюдается наибольшее количество отличий спектра комплекса I от спектра лигандa L (рис. S6 ). Таблетку комплекса I в KBr облучали в течение различных интервалов времени лазером с λ = 405 нм и регистрировали ИК-спектр. При этом не наблюдалось никаких изменений в ИК-спектре комплекса I (рис. S17 ), что свидетельствует о том, что изменения с молекулами комплекса I происходят только на поверхности образца. Oтметим, что оба комплекса являются термически устойчивыми до 300°С (рис. S18 и S19 ).
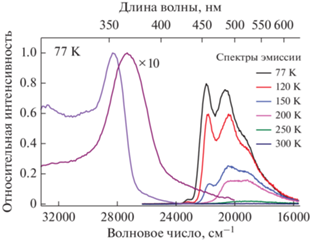
При температуре 77 K комплекс I фотостабилeн. На рис. S15 представлены кривые зависимости интенсивности люминесценции от времени (λэм = 540 нм) при возбуждении 360 нм, показывающие, что интенсивность остаeтся постоянной; на рис. S16 приведены спектры сравнения до облучения и после облучения. Получены спектры ФЛ и возбуждения и измерены времена жизни при разных температурах для комплекса I (рис. 5 и рис. S20 ). При понижении температуры в спектре ФЛ появляется новая линия со своей колебательной структурой. Судя по временам жизни, увеличившимся на шесть порядков (0.16 мс, 4.8 мс, табл. 4), появляется новый механизм эмиссии, связанный с релаксацией триплетных состояний. Такой механизм может быть связан с возбужденными состояниями с переносом заряда (MLCT).
Рис. 5.
Спектры ФЛ при разных температурах (λвозб = = 360 нм) и спектры возбуждения при 77 (λэм = 455 и 530 нм) для комплекса I.
Синтезированы и структурно охарактеризованы хелатные комплексы серебра(I) и цинка(II) с производным 2-(1H-пиразол-1-ил)пиримидина. В обоих комплексах 2-(1H-пиразол-1-ил)пиримидиновый остов координируется бидентатно, что приводит к образованию моноядерных комплексов [AgL2]PF6 и [ZnLCl2].
Как правило, органические молекулы, содержащие ароматические карбоциклические и гетероциклические фрагменты, люминесцируют за счет n–π*- и π–π*-переходов, что закономерно приводит к голубой люминесценции. Вполне ожидаемо, что использованный лиганд L обладает голубой эмиссией. Рассматривая образование комплексов цинка(II) и серебра(I) с L, мы сравнили влияние координации использованных металлов на свойства ФЛ молекулы L. При комнатной температуре оба комплекса обладают флуоресценцией, координация иона цинка(II) не вносит сильное возмущение в уровни лиганда, и комплекс [ZnLCl2] демонстрирует схожую с лигандом L голубую эмиссию, но с большим квантовым выходом. В то же время координация иона серебра(I), предположительно, вносит гораздо большее возмущение в орбитали координированного лиганда, что и приводит к сильному смещению и уширению полосы ФЛ. Полученная широкая полоса перекрывает весь видимый диапазон, что приводит к белому свечению.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Gaffuri P., Stolyarova E., Llerena D. et al. // Renewable and Sustainable Energy Rev. 2021. V. 143. P. 110869. https://doi.org/10.1016/j.rser.2021.110869
Chen S.-A., Chang E.-C. // Semiconducting Polymers / Eds. Hsieh B.R., Wei Y. Washington (DC, USA): American Chemical Society, 1999. Ch. 11. P. 163. https://doi.org/10.1021/bk-1999-0735.ch011
Kim H.U., Jang J.-H., Park H. J. et al. // J. Nanosci. Nanotechnol. 2017. V. 17. P. 5587. https://doi.org/10.1166/jnn.2017.14155
Hao Z., Jiang H., Liu Y. et al. // Tetrahedron. 2016. V. 72. P. 8542. https://doi.org/10.1016/j.tet.2016.11.008
Zeng Q., Li F., Chen Z. et al. // ACS Appl. Mater. Interfaces. 2020. V. 12. P. 4649. https://doi.org/10.1021/acsami.9b18162
Su H.-C., Chen Y.-R., Wong K.-T. // Adv. Funct. Mater., 2020. V. 30. 1906898. https://doi.org/10.1002/adfm.201906898
Tang Y., Wu H., Cao W. // Adv. Opt. Mater. 2020. P. 2001817. https://doi.org/10.1002/adom.202001817
Zhang N., Guan Q.-L., Liu C.-H. et al. // Appl. Organomet. Chem. 2020. P. e5506. https://doi.org/10.1002/aoc.5506
SeethaLekshmi S., Ramya A.R., Reddy M.L.P., Varughese S. // J. Photochemistry Photobiology. C. 2017. V. 33. P. 109.
Fleetham T., Li J. // J. Photon. Energy. 2014. V. 4. 040991-1. https://doi.org/10.1117/1.JPE.4.040991
Chen Z., Ho C.-L., Wang L., Wong W.-Y. // Adv. Mater. 2020. V. 32. P. 1903269. https://doi.org/10.1002/adma.201903269
Kim D.-E., Shin H.-K., Kim N.-K. et al. // J. Nanosci. Nanotechnol. 2014. V. 14. P. 1019. https://doi.org/10.1166/jnn.2014.9140
Hao Y., Meng W., Xu H. et al. // Org. Electronics. 2011. V. 12. P. 136. https://doi.org/10.1016/j.orgel.2010.10.019
Wang S. // Coord. Chem. Rev. 2001. V. 215. P. 79. https://doi.org/10.1016/S0010-8545(00)00403-3
Erxleben A. // Coord. Chem. Rev. 2003. V. 246. P. 203. https://doi.org/10.1016/S0010-8545(03)00117-6
Zheng S.-L., Chen X.-M. // Aust. J. Chem. 2004. V. 57. P. 703. https://doi.org/10.1071/CH04008
Yu C., Wang X., Wu T. et al. // Dalton Trans. 2020. V. 49. P. 12082. https://doi.org/10.1039/D0DT02033H
Miao J., Nie Y., Li Y. et al. // J. Mater. Chem. C. 2019. V. 7. P. 13454. https://doi.org/10.1039/C9TC04033A
Zhu L., Xie W., Qian C. et al. // Adv. Opt. Mater. 2020. V. 8. 2000406. https://doi.org/10.1002/adom.202000406
Jaime S., Arnal L., Sicilia V., Fuertes S. // Organometallics. 2020. V. 39. P. 3695. https://doi.org/10.1021/acs.organomet.0c00510
Sukhikh T.S., Khisamov R.M., Bashirov D.A. et al. // Cryst. Growth Des. 2020. V. 20. P. 5796. https://doi.org/10.1021/acs.cgd.0c00406
Wu J., Ameri L., Cao L., Li J. // Appl. Phys. Lett. 2021. V. 118. P. 073301. https://doi.org/10.1063/5.0043955
Boddula R., Tagare J., Singh K., Vaidyanathan S. // Mater. Chem. Front. 2021. V. 5. P. 159. https://doi.org/10.1039/D1QM00083G
Ilmi R., Khan M.S., Sun W. et al. // J. Mater. Chem. C. 2019. V. 7. P. 13966. https://doi.org/10.1039/C9TC04653D
Ma X., Jia L., Yang B. et al. // J. Mater. Chem. C. 2021. V. 9. P. 727. https://doi.org/10.1039/D0TC04234J
Liu X., Wang Y.-F., Li M. et al. // Organic Electronics. 2021. V. 88. P. 106017. https://doi.org/10.1016/j.orgel.2020.106017
Khammultri P., Kitisriworaphan W., Chasing P. et al. // Polym. Chem. 2021. V. 12. P. 1030. https://doi.org/10.1039/D0PY01541E
Lian L., Zhang P., Liang G. et al. // ACS Appl. Mater. Interfaces. 2021. V. 13. P. 22749. https://doi.org/10.1021/acsami.1c03881
Wu T.-C., Zhao F.-Z., Hu Q.-L. et al. // Appl. Organomet. Chem. 2020. V. 34. P. e5691. https://doi.org/10.1002/aoc.5691
Wang X.-Y., Hu Y.-X., Yang X.-F. et al. // Org. Lett. 2019. V. 21. P. 9945. https://doi.org/10.1021/acs.orglett.9b03875
Xue Z.-Z., Meng X.-D., Li X.-Y. et al. // Inorg. Chem. 2021.V. 60. P. 4375. https://doi.org/10.1021/acs.inorgchem.1c00280
Kaeser A., Moudam O., Accorsi G. et al. // Eur. J. Inorg. Chem. 2014. V. 2014. P. 1345. https://doi.org/10.1002/ejic.201301349
Sun C., Guo Y.-H., Yuan Y. et al. // Inorg. Chem. 2020. V. 59. P. 4311. https://doi.org/10.1021/acs.inorgchem.9b03139
Shekhovtsov N.A., Vinogradova K.A., Berezin A.S. et al. // Inorgan. Chem. Front. 2020. V. 7. P. 2212. https://doi.org/10.1039/D0QI00254B
Bushuev M.B., Krivopalov V.P., Semikolenova N.V. et al. // Russ. J. Coord. Chem. 2006. V. 32. P. 199. https://doi.org/10.1134/s1070328406030067
Sedova V.F., Shkurko O.P., Nekhoroshev S.A. et al. // Heterocycl. Compounds. 2003. V. 34. https://doi.org/10.1002/CHIN.200304150
Fadeeva V.P., Tikhova V.D., Nikulicheva O.N. et al. // J. Analyt. Chem. 2008. V. 63. P. 1197. https://doi.org/10.1134/S1061934808110142
Nakamoto K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, Applications in Coordination, Organometallic, and Bioinorganic Chemistry. New Jersey: John Wiley, 2014. P. 408.
Vinogradova K.A., Plyusnin V.F., Kupryakov A.S. et al. // Dalton Trans. 2014. V. 43. P. 2953. https://doi.org/10.1039/C3DT53040J
Bruker Apex3 software suite. Apex3, SADABS-2016/2 and SAINT. Version 2019.1-0. Madison (WI, USA): Bruker AXS Inc., 2017.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3. https://doi.org/10.1107/S2053273314026370
Allen F.H., Kennard O., Watson D.G. // Perkin Trans. 1987. № 12. S1. https://doi.org/10.1039/p298700000s1
Дополнительные материалы
- скачать ESM.docx
- Рис. S1 - Рис. S20
Таблица S1.
Инструменты
Координационная химия