

Молекулярная биология, 2020, T. 54, № 3, стр. 469-473
Влияние SGI-1027 на формирование и элиминацию PrPSc в инфицированных прионом клетках
J. J. Li a, C. S. Ryou b, D.-H. Kim c, *
a Department of Chemistry, Gwangju Institute of Science and Technology
61005 Gwangju, Republic of Korea
b Department of Pharmacy, Hanyang University
15588 Ansan, Republic of Korea
c School of Undergraduate Studies, College of Transdisciplinary Studies, Daegu Gyeongbuk Institute of Science and Technology
42988 Daegu, Republic of Korea
* E-mail: dhkim1@dgist.ac.kr
Поступила в редакцию 28.08.2019
После доработки 07.11.2019
Принята к публикации 13.11.2019
Аннотация
Недавно показано, что SGI-1027, хорошо известный ингибитор ДНК-метилтрансферазы (DNMT), эффективно снижает образования патогенного прионного белка PrPSc в инфицированных прионом клетках. Теперь мы подтвердили, что в нейронах, инфицированных прионом болезни хронического истощения (CWD), под действием SGI-1027 происходит элиминация PrPSc, и точно определили область связывания прионного белка человека с SGI-1027. SGI-1027 широко используется против различных типов прионных заболеваний, включая вызванные прионами человека. Нами показано, что SGI-1027 предотвращает распространение PrPSc, в то время как ни обработка энхансером DNMTs S-аденозил-L-метионином (SAM), ни их ингибитором 5-азацитидином не влияют на формирование патогена. Установлено, что антиприонная активность SGI-1027 есть результат его прямого взаимодействия с PrPC, которое и предотвращает конформационные изменения PrPC, приводящие к формированию патогенного PrPSc. На основании полученных результатов сделан вывод, что элиминация патогенного PrPSc под действием SGI-1027 не зависит от DNMT.
ВВЕДЕНИЕ
Прионная болезнь связана с тем, что нормальный прионный белок PrPC, который локализован на клеточной мембране, меняет свою конформацию на патологическую патогенную форму PrPSc, а накопление PrPSc в результате приводит к гибели нейронов [1]. Образование гетеродимеров PrPC/PrPSc индуцирует дальнейшее конформационное изменение PrPC в PrPSc и с увеличением количества патогенных частиц в мозге развивается прионная болезнь [2]. В отличие от размножения бактериальных и вирусных патогенов механизм репликации патогенной формы приона заключается в изменении структуры белка [3].
Прионные заболевания поражают как людей, так и животных. Болезнь Крейтцфельда–Якоба (CJD), болезнь Герстманна–Штрюсслера–Шейнкера (GSS), семейная фатальная бессонница (FFI), куру и вариант CJD (vCJD) ‒ это примеры прионных заболеваний человека, тогда как скрепи овец, коз и муфлонов, трансмиссивная энцефалопатия норок (TME), хроническая болезнь истощения (CWD) оленей и губчатая энцефалопатия крупного рогатого скота (BSE) поражают животных [4].
Обогащенный β-структурами инфекционный PrPSc образуется вследствие изменения вторичной структуры богатого α-спиралями белка PrPC [5]. Стабилизация структуры α-спирали в PrPC ‒ важнейший этап в предотвращении образования PrPSc и, следовательно, рассматривается как одна из терапевтических стратегий против прионных заболеваний. Недавно сообщалось, что различные соединения, такие как конго красный, хинакрин, голубой Чикаго 6B (Chicago Sky Blue 6B), GJP14 и GJP49, полученный из GN8 (2-пирролидин-1-ил-N-[{4-[4-(2-пирролидин-1-ил-ацетиламино)-бензил]-фенил}-ацетамид; 2-pyrrolidin-1-yl-N-{4-[4-(2-pyrrolidin-1-yl-acetylamino)-benzyl]-phenyl}-acetamide), эффективно элиминируют PrPSc из клеток с индуцированным прионным заболеванием [6, 7]. Антиприонные соединения NPR-053, NPR-056 и BMD42-29 были идентифицированы при структурном скрининге лекарственных средств, а GJP14 и GJP49 идентифицированы in silico и, как показано, непосредственно взаимодействуют с PrPC [8, 9]. Недавно нами показано [10], что SGI-1027, хорошо известный ингибитор DNMT, напрямую связывается с PrPC, стабилизирует его и препятствует конформационному изменению в патогенный PrPSc. Все это указывает на то, что прямое взаимодействие с PrPC критично для антиприонной активности всех этих химических соединений.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Клеточные культуры и обработка химикатами. Клетки CWD и ScN2a (инфицированные прионом клетки нейробластомы мыши) культивировали на среде Игла в модификации Дульбекко (DMEM), дополненной 10% эмбриональной сыворотки крупного рогатого скота (FBS), 1% пенициллина/стрептомицина и 1% GlutaMax (“Invitrogen”, США). Клетки пассировали каждые 5 дней. Первоначально клетки с 2%-ной конфлюэнтностью адгезировали на поверхности 10-см культуральных чашек, затем в течение 5 суток их инкубировали с SGI-1027, SAM или 5-азацитидином, взятых в различных концентрациях, и анализировали содержание PrPSc.
Детекция PrPSc методом иммуноблотинга. После достижения более чем 95%-ной конфлюэнт-ности клетки собирали для анализа PrPSc, лизировали в 1 мл буфера для лизиса клеток: 20 мМ Трис (рН 8.0), 150 мМ NaCl, 0.5% Nonidet P-40, 0.5% дезоксихолата натрия, ‒ измеряли концентрацию белка и отбирали аликвоту, содержащую 30 мкг белка (без обработки протеиназой K, PK) для анализа общего белка PrP и βIII-тубулина методом иммуноблотинга. Для исследования PK-резистентного PrPSc 2 мг белка инкубировали с 20 мкг/мл PK (“Invitrogen) в течение 1 ч при 37°C, центрифугировали (16 000 × g) при 4°C в течение 1 ч и белковый осадок анализировали электрофорезом в 1%-ном SDS-ПААГ. В иммуноблотинге использовали антитела 6D11 (“Biolegend”, США) для детекции прионного белка и антитела против βIII-тубулина (“Santa Cruz”, США) в качестве маркера контроля загрузки. Для визуализации белковых полос использованы реагенты ECL Prime Detection (“GE Healthcare”, Великобритания) в системе G Box Chemi XR5 (“Syngene”, Великобритания). При обработке результатов иммуноблотинга использована программа ImageJ для денситометрического анализа количества PrPSc в полосе, по которому определяли значение IC50 для SGI-1027.
Поверхностный плазмонный резонанс (SPR). Для SPR-анализа использована система массива белковых взаимодействий ProteOn XPR36 (“Bio-Rad”, США). Девять синтетических PrP-пептидов, конъюгированных биотином (см. рис. S1 Приложения), были получены от компании “Biostem” (Korea), причем их α-аминогруппы были защищены Fmoc и Boc во время синтеза. Пептидную смолу инкубировали с раствором для расщепления в течение 3 ч после синтеза пептида. Для проверки молекулярной массы пептидов использовали масс-спектрометрию, а для выделения нужного пептида ‒ ВЭЖХ. Биотинилированные пептиды PrP помещали на сенсорный чип ProteOn NLC (“Bio-Rad”) через связывание с авидином. Разбавленный в PBS (pH 7.0) SGI-1027 впрыскивали в проточную ячейку со скоростью потока 100 мкл/мин. Вводили 4 различные концентрации SGI-1027 (5, 10, 50, 75 мкМ) и исследовали фазу диссоциации. При анализе результатов использовали программное обеспечение ProteOn Manager 2.0 со стандартными моделями Ленгмюра для соответствия кинетическим данным. Для определения ответа на связывание между PrP и SGI-1027 значение равновесного отклика (Req) или максимального отклика в сенсограмме делили на молекулярную массу. Образование устойчивого комплекса рассчитывали по уравнению: KD = Kd/Ka.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Ранее нашей группой исследователей обнаружено, что SGI-1027 препятствует образованию PrPSc из PrPC в клетках ScN2a [10]. С целью проанализировать эффективность SGI-1027 в удалении PrPSc в прионах различного патогенного происхождения использовали CWD-инфицированные клетки. Выявлено, что уровень PrPSc постепенно снижался с увеличением концентрации SGI-1027 до полного исчезновения при концентрации 1 мкM (рис. 1а). В CWD-инфицированных клетках значение IC50 для SGI-1027 составляло около 100 нM, что вдвое выше, чем в клетках ScN2a, то есть, ингибиторный эффект SGI-1027 на формирование PrPSc в клетках с прионом оленей менее эффективно, чем в инфицированных прионом клетках нейробластомы мыши (рис. 1а). Тем не менее, SGI-1027 эффективно удаляет PrPSc в CWD-инфицированных клетках, как и в клетках ScN2a и SMB [10], и, следовательно, антиприонный эффект SGI-1027 не ограничивается определенным типом прионного заболевания.
Рис. 1.
Элиминация PrPSc под действием SGI-1027 в клетках прионного заболевания. а ‒ Элиминация PrPSc в линии CWD-клеток после обработки SGI-1027. Уровень экспрессии белка PrPSc детектировали методом иммуноблотинга. PK-резистентные виды PrP считали формами PrPSc. Уровень общего PrP и тубулина измеряли, используя лизат обработанных SGI-1027 клеток без PK-расщепления. б ‒ SPR-анализ взаимодействия между 9 пептидами PrP и SGI-1027. Для двух пептидов: аминокислотные остатки (а.к.) 112–146 и 138–172 ‒ детектировали положительный сигнал прямого взаимодействия с SGI-1027.
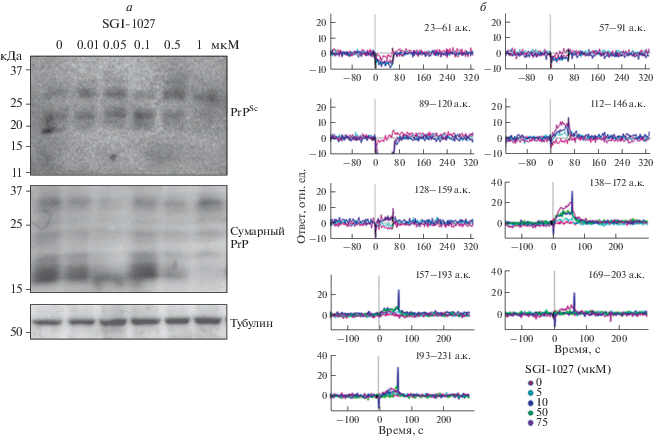
SPR-анализ показал, что SGI-1027 напрямую взаимодействует с PrPC [10]. Для идентификации области связывания PrPC с SGI-1027 провели SPR-анализ для 9 химически синтезированных пептидов PrP. Обнаружены положительные сигналы связывания для 2 пептидов, включающих аминокислотные последовательности 112–146 и 138–172 (рис. 1б). Точные значения KD для каждого пептида не определены из-за слабого взаимодействия между меченными биотином пептидами PrPC и SGI-1027. В отличие от целого белка последовательности, входящие в пептидные фрагменты, не давали сильного связывания с SGI-1027. Несмотря на ограниченные возможности SPR-анализа, выявили, что область SGI-1027-докинга в PrPC включает два остатка в последовательностях 112–146 и 138–172 и эти 2 пептидных фрагмента содержат все важные и критичные остатки, в том числе L130, H155, N159 и Y162, которые предотвращали превращение PrPC в серию β-листов и его трансформацию в PrPSc. Таким образом, SGI-1027 прочно связывается с PrPC и стабилизирует его структуру.
SGI-1027 препятствует переносу метильной группы c S-аденозилметионина (SAM) на цитозин в ДНК. В результате обработка SGI-1027 уменьшает область метилирования во всем геноме и тем самым активирует транскрипцию различных генов без контроля регуляции [11, 12]. Зависимое от SGI-1027 гипометилирование тесно связано с нарушением эпигенетической регуляции, что приводит к нейродегенеративным заболеваниям, включая прионную болезнь [13]. С целью оценить корреляцию между активностью DNMT и распространением PrPSc мы исследовали изменение уровня PrPSc в клетках ScN2a после обработки или кофактором, или ингибитором DNMT. После обработки кофактором DNMT, SAM, интенсивность полосы белка PrPSc в клетках ScN2a оставалась без каких-либо изменений относительно контроля, даже при повышении концентрации SAM до 100 мкМ (рис. 2, слева). Таким образом, усиление активности DNMT не ингибирует конформационные изменения в PrPC и не снижает уровень патогенного белка PrPSc в клетках ScN2a.
Рис. 2.
Исследование уровня PrPSc в клеточной линии ScN2a, обработанной SAM или 5-азацитидином. Детектируемый иммуноблотингом уровень PrPSc не изменялся при увеличении концентрации SAM и 5-азацитидина. PK-резистентные виды PrP считались формами PrPSc. Тубулин, использованный в качестве контроля нанесения, не обрабатывали PK.
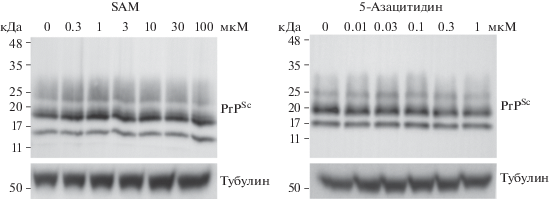
Хорошо известный ингибитор DNMT ‒ 5-азацитидин ‒ предотвращает метилирование ДНК и индуцирует эпигенетический сайленсинг ключевых регуляторных генов, тем самым напрямую влияя на активацию транскрипции определенных генов [14]. Сообщалось, что значение IC50 для ингибирования активности DNMT 5-азацитидином составляет 3 мкM, что в 2 раза лучше, чем для SGI-1027 [15]. Учитывая это, мы выбрали 5-азацитидин, как более сильный ингибитор DNMT, и проверили его антиприонный эффект в клетках ScN2a. Уровень PrPSc в клетках ScN2a не снижался при увеличении концентрации 5-азацитидина. При обработке 1-мкМ 5-азацитидином обнаружено небольшое уменьшение содержания PrPSc по сравнению с контролем (рис. 2, справа). Однако нами обнаружено, что обработка 5-азацитидином в концентрации 1 мкМ и выше индуцирует сильный апоптоз клеток (данные не представлены), по-видимому, из-за цитотоксического действия этого препарата, обычно используемого при химиотерапии миелоидного лейкоза [14]. Таким образом, ослабление полосы PrPSc, видимо, связано с уменьшением числа клеток, то есть токсическим действием 5-азацитидина. Хотя 5-азацитидин гораздо более сильный ингибитор DNMT, чем SGI-1027, он не снижал уровень PrPSc в клетках ScN2a. В анализе вестерн-блот ни кофактор DNMT, ни ингибитор DNMT не оказывали влияния на уровень белков PrPSc, что свидетельствует об отсутствии антиприонного действия. Следовательно, можно сделать вывод, что элиминация PrPSc в модельных клеточных линиях прионного заболевания не связана с контролем регуляции активности DNMT.
Хотя SGI-1027 ингибирует функцию DNMT и, таким образом, полностью изменяет паттерн генной экспрессии под эпигенетическим контролем, его антиприонная активность не связана с эпигенетическими изменениями в клетках прионной болезни. На основании результатов проведенного исследования можно предположить, что антиприонная активность SGI-1027 обусловлена его прямым, высокоаффинным взаимодействием с PrPC, что предотвращает конформационную конверсию PrPC в PrPSc в клетках прионного заболевания. Дальнейшее повышение мощности SGI-1027 как ингибитора конверсии PrPC и антиприонного агента может быть достигнуто введением модификаций в его структуру. Надеемся, что этот путь в конечном итоге приведет к разработке лекарственных средств против прионных заболеваний.
Работа поддержана грантами Программы фундаментальных научных исследований Национального исследовательского фонда Кореи (Basic Science Research Program through the National Research Foundation of Korea; NRF-2013R1A1A2011210) и Программы исследований для студентов (URP) Корейского фонда содействия развитию науки и творчества (Undergraduate Research Program through Korea Foundation for the Advancement of Science and Creativity; SBJ000031876). Эта работа также поддержана фондами INGE Института науки и технологии Кванджу (INGE funds of Gwangju Institute of Science and Technology; GK11750).
Авторы заявляют об отсутствии конфликта интересов.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Список литературы
Prusiner S.B., Dearmond S.J. (1994) Prion diseases and neurodegeneration. Annu. Rev. Neurosci. 17, 311‒339.
Soto C., Satani N. (2011) The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 17, 14‒24.
Cobb N.J., Surewicz W.K. (2009) Prion diseases and their biochemical mechanisms. Biochemistry. 48, 2574‒2585.
Collinge J. (2001) Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 24, 519‒550.
Prusiner S.B. (1982) Novel proteinaceous infectious particles cause scrapie. Science. 216, 136‒144.
Barreca M.L., Iraci N., Biggi S., Cecchetti V., Biasini E. (2018) Pharmacological agents targeting the cellular prion protein. Pathogens. 7(1), pii: E27. https://doi.org/10.3390/pathogens7010027
Ferreira N.C., Marques I.A., Conceicao W.A., Macedo B., Machado C.S., Mascarello A., Chiaradia-Delatorre L.D., Yunes R.A., Nunes R.J., Hughson A.G., Raymond L.D., Pascutti P.G., Caughey B., Cordeiro Y. (2014) Anti-prion activity of a panel of aromatic chemical compounds: in vitro and in silico approaches. PLoS One. 9, e84531. https://doi.org/10.1371/journal.pone.0084531
Hosokawa-Muto J., Kamatari Y.O., Nakamura H.K., Kuwata K. (2009) Variety of antiprion compounds discovered through an in silico screen based on cellular-form prion protein structure: correlation between antiprion activity and binding affinity. Antimicrob. Agents Chemother. 53, 765‒771.
Ishibashi D., Nakagaki T., Ishikawa T., Atarashi R., Watanabe K., Cruz F.A., Hamada T., Nishida N. (2016) Structure-based drug discovery for prion disease using a novel binding simulation. Ebiomedicine. 9, 238‒249.
Kim D.H., Ren C., Ryou C., Li J. (2019) Direct interaction of DNMT inhibitors to PrPC suppresses pathogenic process of prion. Acta Pharm. Sin. B. 9, 952‒959.
Datta J., Ghoshal K., Denny W.A., Gamage S.A., Brooke D.G., Phiasivongsa P., Redkar S., Jacob S.T. (2009) A new class of quinoline-based DNA hypomethy-lating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 69, 4277‒4285.
Valente S., Liu Y.W., Schnekenburger M., Zwergel C., Cosconati S., Gros C., Tardugno M., Labella D., Florean C., Minden S., Hashimoto H., Chan Y.Q., Zhang X., Kirsch G., Novellino E., Arimondo P.B., Miele E., Ferretti E., Gulino A., Diederich M., Cheng X.D., Mai A. (2014) Selective non-nucleoside inhibitors of human DNA methyltransferases active in cancer including in cancer stem cells. J. Med. Chem. 57, 701‒713.
Saijo E., Kang H.E., Bian J.F., Bowling K.G., Brow-ning S., Kim S., Hunter N., Telling G.C. (2013) Epige-netic dominance of prion conformers. PLoS Pathog. 9, e1003692.
Christman J.K. (2002) 5-Azacytidine and 5-aza-2'-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 21, 5483‒5495.
Kim H.J., Kim J.H., Chie E.K., Young P.D., Kim I.A., Kim I.H. (2012) DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat. Oncol. 7, 39. https://doi.org/10.1186/1748-717X-7-39
Дополнительные материалы
- скачать ESM_1.docx
- Additional file
Инструменты
Молекулярная биология