

Российские нанотехнологии, 2021, T. 16, № 1, стр. 23-49
НАНОСТРУКТУРИРОВАННЫЕ ПОЛИМЕРСОДЕРЖАЩИЕ КОМПОЗИТЫ КАК ЭФФЕКТИВНЫЙ ИНСТРУМЕНТ МОЛЕКУЛЯРНОЙ ДИАГНОСТИКИ
Д. В. Капустин 1, *, А. И. Простякова 1, Д. И. Зыбин 2, В. П. Зубов 1
1 Институт биооpганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Москва, Россия
2 МИРЭА – Российский технологический университет, ИТХТ
Москва, Россия
* E-mail: kapustin@ibch.ru
Поступила в редакцию 26.06.2020
После доработки 03.12.2020
Принята к публикации 03.12.2020
Аннотация
Рассмотрена роль нанотехнологий в разработке методов пробоподготовки в молекулярной диагностике, в частности способы выделения нуклеиновых кислот (НК) из биологических образцов и физико-химические процессы, положенные в основу этих методов. Показано, что наиболее эффективными являются методы, основанные на обратимой сорбции (т.е. на твердофазной селективной экстракции), обеспечивающие миниатюризацию и автоматизацию процессов. До последнего времени наиболее распространенный подход был основан на связывании с сорбентом и последующем “смывании” НК из биологических проб (“позитивная селекция”). В обзоре обсуждаются возможности и преимущества альтернативного одностадийного подхода к выделению НК, основанного на использовании сорбентов, связывающих белки и другие компоненты биологических проб, одновременно сорбционно инертных по отношению к НК (“негативная селекция”). Рассматриваются методы получения композиционных наноструктурированных сорбентов на основе твердых матриц (пористые кремнеземы, стеклянные мультикапилляры, синтетические мембраны), модифицированных нанотолщинными слоями полимеров, демонстрирующих эффект негативной селекции в отношении НК, в первую очередь фторполимеров и полианилинов, варианты их практического использования, а также альтернативные области применения этих композитов.
ОГЛАВЛЕНИЕ
Введение
1. Методы пробоподготовки биологических образцов для выделения нуклеиновых кислот, основанные на “позитивной селекции”; применяемые носители, полимерные модификаторы и композиционные сорбенты
1.1. Принципы выделения биополимеров из биологических образцов
1.2. Способы лизиса биологических проб
1.3. Решения для автоматизированного выделения биополимеров
1.4. Микрофлюидные устройства для выделения НК
2. Полимерсодержащие сорбенты для выделения и очистки биологически активных соединений. Методы получения, модификаторы и области применения
2.1. Композиты, получаемые в результате физической адсорбции полимеров
2.2. Композиты, получаемые в результате хемосорбции полимеров
2.3. Композиты, получаемые в результате проведения графт-полимеризации
2.4. Фторполимерсодержащие композиционные сорбенты
2.5. ПАНИ-содержащие композиционные сорбенты
3. Фторполимер- и полианилинсодержащие сорбенты для выделения и очистки биополимеров – новые материалы, демонстрирующие эффект “негативной селекции” в отношении НК. Способы получения и области применения
3.1. Методы получения композитов, демонстрирующих эффект “негативной селекции” в отношении НК
3.2. Применение ФП- и ПАНИ-содержащих композитов в молекулярной диагностике (одностадийное выделение НК)
3.3. Применение ФП-и ПАНИ-содержащих композитов в смежных областях бионанотехнолгии и в биоаналитке
Заключение
ВВЕДЕНИЕ
Биотехнология в качестве самостоятельного научного направления оформилась в 20-х годах прошлого века, сначала она развивалась как сельскохозяйственная, а затем как научно-промышленная дисциплина, решая проблемы повышения эффективности обработки биологического сырья, оптимизации условий ферментации и биотрансформации, а также очистки конечного продукта. Спустя примерно полвека благодаря развитию технологии рекомбинантных ДНК сущность биотехнологии радикально изменилась, поскольку появилась возможность создавать новые микроорганизмы и эукариотические клетки, производящие необходимые человеку биологически активные соединения. В результате возникла молекулярная биотехнология – дисциплина, в рамках которой разрабатываются эффективные способы получения биополимеров (нуклеиновых кислот (НК) и белков) с заданными свойствами, вакцины, а также методы молекулярной диагностики инфекционных и генетических заболеваний [1].
Важнейшее направление современной биотехнологии связано с использованием молекулярно-генетических технологий в лабораторной диагностике инфекционных заболеваний, что стало возможным благодаря открытию в середине 80-х годов XX века процесса искусственного многократного копирования ДНК, известного как полимеразная цепная реакция (ПЦР) [2]. ПЦР-анализ стал рутинной процедурой для определения специфических полинуклеотидных последовательностей благодаря своей универсальности и высокой чувствительности. Преимущества, обеспечиваемые методом ПЦР, очевидны. Однако до сих пор серьезной проблемой часто оказывается выбор конкретного способа подготовки образца к исследованию, поскольку до последнего времени не существовало универсального подхода к выделению НК разных видов организмов из различных биологических источников. Сказанное относится и к методам выделения веществ белковой природы, когда присутствие примесей в выделенном образце может существенно снизить эффективность их применения в диагностике или биоинженерии.
В результате совершенствования так называемых омиксных технологий (включающих в себя геномику, протеомику, транскриптомику, метаболомику) к способам выделения и очистки биополимеров (т.е. к методам пробоподготовки) стали предъявлять дополнительные требования. Ранее эти требования относились в основном к степени очистки и количественному выходу биополимера, выделяемого в нативном состоянии. Сегодня также необходимо учитывать сокращение времени, затрачиваемого на процесс выделения, возможность его автоматизации, роботизации и миниатюризации применяемых для этого систем с одновременным максимальным упрощением процедуры выделения. Все это предполагает использование нанотехнологических подходов и методов.
Используемые до сих пор методы выделения биополимеров из биологических смесей основаны на различиях в растворимости (и/или в сродстве к сорбенту) выделяемых соединений и прочих компонентов смеси и включают в себя жидкостную экстракцию, осаждение и адсорбцию с использованием специальных сорбентов. Как правило, эти методы многостадийны, трудоемки и сопровождаются потерями выделяемого компонента. Методы с применением сорбентов основаны на концепции “улавливания” и удерживания целевого биополимера сорбентом на первом этапе разделения (“позитивная селекция”), а вслед за этим необходимо смывать прочие компоненты смеси и элюировать целевой компонент с поверхности (или из объема пор) сорбента. Такой многостадийный механизм широко применяется на практике, несмотря на то, что каждый этап выделения сопровождается потерями выделяемого компонента и не всегда удается количественно воспроизвести результат процедуры выделения.
В предлагаемом обзоре наряду с методами, основанными на “позитивной селекции” в отношении НК, широко применяемыми в клинических и исследовательских лабораториях, рассмотрен альтернативный подход, основанный на использовании сорбентов, связывающих белки и другие компоненты биологических проб, одновременно сорбционно инертных по отношению к НК “негативная селекция”). Рассмотрены физико-химические процессы, лежащие в основе таких методов; представлены различные типы носителей (матриц), которые применяются при получении композиционных сорбентов. Отдельно рассмотрены физические и химические методы синтеза композиционных полимерсодержащих сорбентов, применяемых при разделении компонентов биологических смесей. Отмечена роль нанотехнологий в получении материалов такого рода. Обзор отражает хронологию развития методов синтеза таких материалов, а также дает представление о современных подходах к их синтезу и применению.
1. МЕТОДЫ ПРОБОПОДГОТОВКИ БИОЛОГИЧЕСКИХ ОБРАЗЦОВ ДЛЯ ВЫДЕЛЕНИЯ НУКЛЕИНОВЫХ КИСЛОТ, ОСНОВАННЫЕ НА “ПОЗИТИВНОЙ СЕЛЕКЦИИ”; ПРИМЕНЯЕМЫЕ НОСИТЕЛИ, ПОЛИМЕРНЫЕ МОДИФИКАТОРЫ И КОМПОЗИЦИОННЫЕ СОРБЕНТЫ
С целью выделения чистых препаратов НК (или белков) из многокомпонентных смесей вначале необходимо обеспечить условия, позволяющие изолировать выделяемые макромолекулы из биологического субстрата. Важнее всего обеспечить разрушение клеточных мембран (или вирусных капсидов), а также разрушение нуклеопротеидных комплексов за счет солюбилизации макромолекул НК и белков в результате лизиса. Затем следует очистить выделяемый биополимер от твердых и растворенных примесей и (если необходимо) сконцентрировать выделенный компонент для последующего анализа и использования.
1.1. Принципы выделения биополимеров из биологических образцов
Впервые ДНК выделил в 1869 г. Мишер из ядер лейкоцитов гноя, назвав новое вещество “нуклеином” [3]. И только спустя почти 90 лет была разработана первая рутинная процедура выделения ДНК в градиенте плотности CsCl из бактериального лизата Escherichia coli, полученного под действием додецилсульфата натрия [4]. Сегодня используется множество способов выделения биополимеров из разнообразных биологических источников, и ежегодно предлагаются новые модификации известных протоколов. Тем не менее все эти методы сводятся к использованию одного из трех физико-химических процессов (или их комбинации). Первая группа методов включает в себя экстракцию, т.е. разделение/выделение компонентов смесей или суспензий (биосепарацию) благодаря избирательному растворению индивидуальных компонентов смеси в подходящих растворителях. При этом выделяемое вещество должно лучше растворяться в экстрагенте, нежели в исходной среде (или в случае проведения твердофазной экстракции (ТЭ) проявлять большее сродство к поверхности сорбента, чем к среде). В качестве примеров таких методов следует привести фенол-хлороформную экстракцию геномной ДНК или иРНК [5] и щелочную экстракцию плазмидной ДНК с использованием стеклянных порошков [6]. Для выделения НК (в частности иРНК) из клеток и тканей используют экстракцию из образцов смесью фенол-гуанидинтиоцианат–хлороформ [7] и др. Эти методы отличаются длительностью выделения (не менее 3.5 ч), многостадийностью, необходимостью применения токсичных органических растворителей и низким выходом НК (не более 50% для ДНК, около 7% для РНК). В отдельную подгруппу жидкостных методов можно выделить способы выделения биополимеров (НК и белков), клеток, мембран, клеточных органелл и микроорганизмов в смеси несмешивающихся водных растворов полимеров (т.е. в двухфазной водной системе), например в смеси 5% раствора декстрана и 3.5% полиэтиленгликоля (ПЭГ) [8]. Еще одна группа методов биосепарации включает в себя способы осаждения целевого компонента, основанные на физическом осаждении за счет преципитации, центрифугирования, нагревания, охлаждения, концентрирования или разбавления, что приводит к изменению агрегатного состояния компонентов смеси (раствора), либо на селективной химической сорбции за счет химического или кулоновского взаимодействия молекул сорбата с функциональными группами на поверхности сорбента. В качестве примеров можно упомянуть осаждение ДНК или белков ультрацентрифугированием [9, 10], препаративное ультрацентрифугирование ДНК в градиенте плотности хлорида цезия [5, 11], переосаждение белков в спиртах [5, 12–14], осаждение белков ацетатом аммония или сульфированным декстраном [15], осаждение олигонуклеотидов и ДНК спиртами при пониженной температуре [5], высаливание [5, 15], различные варианты аффинной хроматографии белков [15] и др. В качестве частного случая экстракции допустимо рассматривать упомянутую ТЭ (которую одновременно можно отнести к адсорбционным процессам). Следует учитывать, что при ТЭ концентрирование целевого компонента происходит на поверхности специального сорбента, т.е. на границе раздела сорбент/жидкая среда. К этой группе методов относятся, например, очистка НК в результате адсорбции на поверхности пористых кремнеземов [5, 16], различные способы очистки НК и белков на магнитных частицах [17, 18].
При проведении ТЭ происходит концентрирование отдельных веществ или группы сходных по свойствам веществ в соответствии с растворимостью выделяемых соединений в зависимости от их ионообменных или комплексообразующих свойств и т.д. Важно, чтобы целевой компонент отличался наибольшим (или наименьшим) фактором удерживания, чтобы все остальные компоненты элюировались ранее (или наоборот, позднее) [19]. С этой целью используют немодифицированные кремнеземы (КЗ), химически модифицированные кремнеземы (ХМКЗ), синтетические полимерные мембраны или композиты, содержащие указанные компоненты. ТЭ применяют для концентрирования органических соединений из морской и пресной воды и из почв, для извлечения биологически активных веществ из сыворотки и плазмы крови, мочи, желчи и экстрактов различных тканей, для подготовки проб продовольственного сырья, пищевых продуктов и кормов. ТЭ эффективна при концентрировании, очистке и количественном определении стероидов, пептидов, некоторых витаминов, нуклеотидов, ряда лекарственных препаратов и метаболитов [20], при определении нормируемых токсикантов (например, микотоксинов) в пищевой промышленности [21]. К середине 90-х годов прошлого века было известно около 400 методик подготовки проб с помощью ТЭ, а в настоящее время их уже несколько тысяч. Методы с использованием ХМКЗ обеспечивают возможность селективной десорбции биологически активных веществ с сорбента для дальнейшего анализа. Другие сорбенты (например, активированный уголь) также адсорбируют природные соединения, однако последние чаще всего удерживаются необратимо.
Наконец, в группу хроматографических методов объединены способы разделения смесей веществ или частиц, основанные на различиях в скоростях их перемещения в системе несмешивающихся и движущихся друг относительно друга фаз. Различные варианты хроматографии широко применяются при разделении смесей белков и/или НК [22]. Как правило, при хроматографическом разделении в различной степени реализуются механизмы взаимодействия растворенных и/или адсорбируемых молекул, которые имеют место при экстракции, осаждении и адсорбции (за исключением, пожалуй, методов, основанных на гель-фильтрации, когда сорбционную активность сорбента стремятся свести к минимуму).
1.2. Способы лизиса биологических проб
С целью извлечения НК из клеточных органелл (ядро, митохондрии, пластиды) клетки могут быть физически разрушены различными методами, такими как замораживания–оттаивания [23], суспензионной гомогенизации [24], обработки ультразвуком [25, 26], растирания в жидком азоте [27] или их комбинацией.
Химические методы лизиса основаны на использовании различных солюбилизирующих и дестабилизирующих агентов, таких как ионогенные и неионогенные поверхностно-активные вещества (например, додецилсульфат натрия [5, 28], тритон X-100, твины, лаурилсаркозин [5, 29] и др.), хаотропные агенты (гидрохлорид гуанидинтиоцианата и перхлорат натрия) [5, 16, 30]. Нередко химический лизис включает в себя стадию высокотемпературной инкубации (при 60–95°С) [5]. Часто процедуры лизиса основаны на разрушении клеточных стенок и мембран благодаря использованию ферментов, таких как лизоцим, субтилизин, лизостафин, протеиназа К [5, 31]. Иногда применяют метод химической флокуляции, в частности для извлечения ДНК из почвы [31].
При пробоподготовке образцов, содержащих бактериальные клетки и/или вирусные частицы, наилучшие результаты достигаются благодаря проведению комбинированного химического и энзиматического лизиса (иногда с термической обработкой). В компактных устройствах (например, в спин-колонке) при работе с малыми объ-емами образца (от 25 до 250 мкл) с помощью фильтрации через специальную мембрану удается быстро выделять клетки (вирусы) для их последующего лизиса непосредственно на мембране. Для фильтрации образца используют фильтры из боросиликатного волокна, целлюлозные мембраны, политетрафторэтиленовые (ПТФЭ) фильтры, мембраны из поливинилиденфторида [32], а также композиты на основе мембраны, содержащей “встроенные” частицы объемно-пористого КЗ. После фильтрации эти мембраны, покрытые слоем клеток и/или вирусных частиц, обрабатываются раствором фермента для разрушения клеточных мембран и/или капсидов, затем лизирующим буферным раствором, содержащим хаотропный агент и детергент. Этот этап позволяет исключить стадию экстракции НК и автоматизировать процедуру выделения. Иногда перед проведением лизиса и последующей ТЭ выделяемой НК целесообразно сконцентрировать клетки или вирусы из объема биологических образцов. Для этого используют фильтрацию образца [33], электрокинетическую фокусировку [34], аффинную хроматографию [35, 36] или микродиализ [37].
Классический способ выделения ДНК из биологических образцов основан на использовании немодифицированных КЗ (силикагелей и макропористых стекол) [5, 16]. Принцип “позитивной селекции”, лежащий в основе этого типа разделения компонентов, содержащихся в образце, базируется на специфическом взаимодействии ДНК с поверхностью КЗ. ДНК адсорбируется на поверхности частиц КЗ в присутствии хаотропной соли (гуанидинтиоцианата), взятой в высокой концентрации, при щелочных значениях рН. В физиологических условиях поверхность КЗ представлена в основном группами Si–O, а не Si–OH [38]. В условиях высокой концентрации хаотропной соли присутствующие в растворе катионы образуют более или менее стабильный слой противо-ионов на отрицательно заряженной поверхности КЗ. В результате заряд поверхности КЗ становится положительным. Почти все биомолекулы в водных растворах при физиологическом значении рН заряжены. Присутствие хаотропной соли вызывает денатурацию большинства биомолекул из-за разрушения их гидратных оболочек, что часто приводит к выпадению биомолекул в осадок. Исключением являются молекулы ДНК, которые устойчивы к денатурации и при физиологическом значении рН характеризуются наиболее высокими плотностями отрицательного заряда по сравнению с любыми другими биомолекулами (эффективная плотность заряда составляет один отрицательный заряд на 0.17 нм) [39–41]. Фосфатные группы ДНК в водной среде при физиологическом рН в основном депротонированы. Поэтому в хаотропных условиях молекулы НК эффективно связываются с положительно заряженной поверхностью КЗ, вытесняя другие молекулы с его поверхности и оставляя их в растворенном состоянии. Образовавшийся комплекс “ДНК–КЗ” отмывают солевым раствором или водно-спиртовой смесью от не связавшихся примесей, после чего очищенную ДНК элюируют с сорбента буферным раствором с низким содержанием солей или водой [42] (рис. 1).
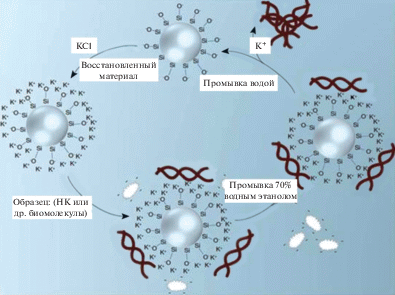
Данный многостадийный механизм выделения НК, основанный на сорбции–десорбции, широко применяется на практике. Однако немодифицированные КЗ характеризуются низкой емкостью (0.5 мкг ДНК/1 мг кремнезема), а при их использовании удается выделять ДНК только длиной более 40 000 п.о. (пар оснований) Таким образом, элюируется не более 50% всей ДНК, так как 50–60% низкомолекулярной ДНК адсорбируется на поверхности КЗ необратимо. Это означает, что выход ДНК составляет всего 40–50% [43]. В этом отношении интересна работа [44], в которой исследовали влияние температуры на выход ДНК микобактерий туберкулезного комплекса при ее выделении с помощью КЗ-содержащих микроколонок из модельных растворов в интервале температур 20–90°С. В [44] показано, что выход ДНК линейно возрастает при повышении температуры вплоть до 70°С, а затем несколько снижается при более высоких температурах. Так, максимальный выход при 70°С составил 73% (на 50% выше, чем при 20°С). По-видимому, описанный эффект связан с изменением вязкости растворов ДНК при росте температуры.
Несмотря на существенные потери при выделении ДНК с помощью немодифицированных (“нативных”) КЗ, последние широко используются в пробоподготовке в самых разнообразных вариантах. Так, выделение НК в результате адсорбции на поверхности КЗ проводят с помощью материалов, поверхностно модифицированных слоями диоксида кремния, в миниатюризированном и автоматизированном формате, например, с использованием микроканалов в составе особых устройств, так называемых “lab-on-a-chip” (или “лаборатории на чипе”), биосенсоров, а также с помощью гибридных наночастиц [45, 46]. В последнем случае биосепарацию НК проводят на поверхности наногибридных частиц с намагничиваемой сердцевиной, покрытой КЗ-оболочкой [47]. В этом случае частицы не упакованы в микроколонку, а используются в виде суспензии (т.е. осуществляется “batch-процесс” [48]). Использование кремнеземных или КЗ-содержащих частиц облегчает работу с вязкими и неоднородными образцами, что сокращает продолжительность процедуры и делает возможным ее автоматизацию [49, 50]. КЗ-матрицы с успехом применяли при выделении высокомолекулярной ДНК из почвы [51] и фитопланктона [52].
При выделении НК из лизатов клеток микроорганизмов и/или вирусных частиц иногда на этапе очистки и/или концентрирования применяют гель-фильтрацию с помощью сшитых смол на основе агарозы (сефароза) или декстрана (сефадекс) [53], а также ионообменные сорбенты [54]. Все же наибольшее распространение получила твердофазная экстракция НК, поскольку принцип “положительной селекции”, положенный в основу этого метода (последовательные этапы удерживания выделяемого компонента смеси, отмывки его от примесей и последующей элюции), легко адаптировать к формату микрофлюидной (МФ) системы. Следующие примеры иллюстрируют многообразие разработанных систем – от простейших (с использованием спин-колонок или суспензий частиц КЗ) до полностью автоматизированных.
В компании Qiagen GmbH (Германия) разработана процедура последовательного выделения ДНК и РНК из одного образца [55]. Образец обрабатывали хаотропным агентом, затем НК осаждали спиртом в присутствии частиц носителя, связывающего НК (использовали КЗ, магнитные частицы с карбоксилированной поверхностью и др.). Затем ДНК и РНК последовательно элюировали с поверхности частиц, изменяя время контакта образца с поверхностью носителя. Поскольку ДНК связывается с носителем значительно быстрее, чем РНК, при малых временах контакта (5–60 с) элюировались образцы, обогащенные ДНК, а при увеличении времени контакта в 10–20 раз – образцы, обогащенные РНК. Очевидно, что для получения препаратов ДНК, очищенных от РНК, требуется дополнительная очистка (либо добавление РНКазы к образцу), и наоборот, для получения чистых препаратов РНК требуется дополнительная очистка от ДНК.
Методики выделения НК, основанные на “позитивной селекции”, как правило, включают в себя ряд последовательно проводимых вручную процедур, включающих в себя подготовку и нанесение образцов на носитель, замену картриджей (спин-колонок) и других расходных элементов, отбор получаемых препаратов. Этапу элюции НК предшествует лизис клеток, за которым следует адсорбция НК, отмывка от растворенных примесей и нерастворимых частиц, что нередко требует термостатирования и/или центрифугирования. С ростом числа этапов выделения повышается риск контаминации получаемых препаратов, а выход НК снижается (вследствие неспецифического и необратимого удерживания определенного количества макромолекул на поверхности носителя, а также из-за возможных ошибок лаборанта). Кроме того, получаемый лизат наряду с целевыми НК содержит множество компонентов, способных ингибировать ПЦР. Отчасти проблема повышения выхода очищенных препаратов НК решается благодаря использованию автоматизированных устройств.
1.3. Решения для автоматизированного выделения биополимеров
С целью обнаружения вирусов гепатитов В, С и ВИЧ в сыворотке и плазме крови компания Roche Diagnostics (США/Швейцария) разработала автоматическую систему выделения ДНК (РНК) COBAS AmpliPrep на магнитных частицах с последующим ПЦР-анализом проб в реальном времени. Для устранения контаминации образцы периодически обрабатываются раствором, содержащим урацил-N-гликозилазу (фермент, разрушающий ранее синтезированные ампликоны) [56].
Компания Qiagen GmbH (Германия) предлагает устройство QIAsymphony SP для одновременного автоматического выделения вирусной или геномной НК из 96 образцов крови. В этом устройстве используются колонки с магнитными частицами, покрытыми никель-нитрилотриацетатной агарозой (Ni-NTA) [57]. Эта же компания разработала автоматизированную станцию BioRobot MDx, содержащую восемь колонок со сменными фильтрами, для выделения геномной ДНК из крови или вирусной НК из сыворотки и плазмы крови. Ввод образца и его перемещение по каналам системы осуществляются с помощью вакуумного насоса [58].
В автоматизированном устройстве Thermo Scientific KingFisher, разработанном в компании Thermo Scientific (США), с помощью магнитных частиц осуществляется одновременное выделение ДНК, РНК и белков из 24 образцов биологических жидкостей (кровь, плазма, моча, слюна) объемом 20–200 мкл [59]. В компании Corbett Technologies (Австралия) разработали настольную роботизированную станцию X-tractor GeneTM System для выделения НК (из образцов объемом до 200 мкл), воспроизводящую ручное выделение НК [60].
Интересное решение, позволяющее снизить риск контаминации, реализовано в роботизированной системе компании Ionis Pharmaceuticals, Inc. (США) для идентификации опасных инфекций [61], включающей в себя два блока, разделенных воздушным шлюзом. В первом блоке происходит выделение и очистка ДНК при давлении выше атмосферного, а во втором осуществляется ПЦР-диагностика при давлении ниже атмосферного.
Автоматизированные устройства для пробоподготовки (в которых вмешательство оператора в ход процесса не предусмотрено), как правило, громоздки и отличаются высокой стоимостью как самого оборудования (более 70 тыс. долл. США в ценах 2020 г.), так и компонентов для проведения анализов. Нередко компании-производители создают условия, когда применение этих систем возможно лишь при использовании реагентов, предназначенных для конкретной модели прибора.
Рядом компаний предложены более компактные частично автоматизированные устройства, используя которые, оператор может заменять или модифицировать их компоненты и регулировать процесс выделения НК, принимая во внимание особенности исследуемого образца. Так, компания Millipore Corporation (США) запатентовала устройство для выделения НК из клеток, вирусов и микоплазм с помощью пористых фильтров (на основе полисахаридного волокна, полиэфирсульфона или стекловолокна), через которые последовательно пропускают образец [62]. На первом фильтре клетки лизируют. Лизат очищают, пропуская его через следующий фильтр, и вслед за этим элюируют очищенную НК. Такое устройство пригодно для одноразового и многоразового использования. Для одновременной обработки нескольких образцов фильтрующие элементы можно объединить в кассету. Реагенты вводят вручную, в процессе выделения требуется неоднократное центрифугирование.
В компании Valentis, Inc. (США) разработан способ выделения плазмидной ДНК в результате пропускания образца через систему статических смесителей (без участия подвижных механических устройств), в которых происходит лизис клеток, осаждение клеточного дебриса и выделение ДНК из лизата. Здесь также необходимы центрифугирование и дополнительная очистка выделенной плазмиды на ионообменной колонке [63].
Компанией Inivitrogen Corporation (США) запатентован картридж для выделения НК [64], включающий в себя фильтрующий элемент с несколькими слоями фильтров и очищающую колонку, содержащую носитель, связывающий НК при определенных значениях рН среды и не удерживающий НК при повышении рН. Это устройство принципиально отличается от автоматизированных систем, поскольку перед нанесением образца на картридж необходима предварительная пробоподготовка.
1.4. Микрофлюидные устройства для выделения НК
В последние годы для обработки биологических образцов и выделения НК нередко используют устройства (разного уровня сложности) на основе микрофлюидных (микрожидкостных) систем. Такие устройства, как правило, содержат сеть микроканалов диаметром до 100 мкм, через которые пропускают малые объемы образцов и растворов реагентов.
Эти системы включают в себя специальные резервуары для проведения лизиса, разделения компонентов биологической смеси, очистки и концентрирования с помощью перемешивающих устройств, микронасосов, микродозаторов, фильтров и т.д. Выделение НК можно осуществлять в полностью автоматическом режиме внутри модуля, изолирующего образец от внешней среды, либо в полуавтоматическом режиме (осуществляя ввод образца и реагентов вручную). Различные типы микрофлюидных устройств (МФУ) для выделения НК и принципы их работы рассмотрены, например, в обзоре [65]. Появление таких систем для выделения НК подтвердило эффективность перехода от жидкостно-жидкостной экстракции к ТЭ с помощью микрочипов, например, с использованием покрытий на основе диоксида кремния, кремниевых элементов и др. [66, 67].
Впервые такое МФУ было описано в 1999 г. [68]. На внутренней поверхности извилистого (с целью максимального увеличения длины на ограниченной площади чипа) канала, предназначенного для ввода и пробоподготовки образца, были размещены микроскопические кварцевые столбики, имеющие площадь поверхности 3.5 нм2. Это обеспечило 10-кратное увеличение поверхностной емкости канала в отношении молекул ДНК по сравнению с гладким каналом. В похожих устройствах [68, 69] в каналах имелись столбики, покрытые слоем диоксида кремния, увеличивающие площадь поверхности не менее чем в 6 раз (рис. 2а). Однако выделить очищенную ДНК с высоким выходом с помощью таких устройств не удалось. Кроме того, требовались относительно большой объем вводимой пробы (до 250 мкл) и высокое давление для продвижения пробы внутрь устройства, что препятствовало интегрированию таких чипов с устройствами, в которых предусмотрено использование меньших объемов жидкости. В обзоре [70] представлен кремниевый чип с микроемкостью эллиптической формы с плоским дном, на котором размещены кремниевые столбики квадратного сечения, боковые поверхности которых имеют по пять вертикальных выемок каждая (по три глубоких и по две менее глубоких) с целью увеличения площади контакта с молекулами ДНК (рис. 2б).
Рис. 2.
РЭМ-изображение кварцевых столбиков. Ширина колонны и расстояние между колоннами – 10 мкм. Глубина каналов и высота столбиков могут варьироваться в диапазоне 20–50 мкм (а). РЭМ-изображение микроструктуры кремниевого чипа, полученного с использованием технологии LIGA. Длина канала – 1 см, ширина – 800 мкм, глубина – 50 мкм; общая площадь поверхности – 1.29 см2. Ширина выемок на поверхности столбиков – 2 мкм, расстояние между ними – 4 мкм (б) [67].
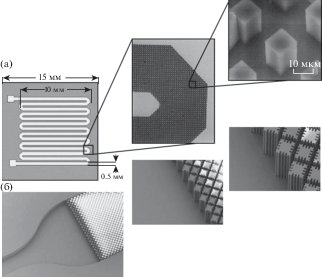
В данном случае речь идет об использовании так называемых LIGA-технологий (т.е. методов получения элементов изделия, основанных на использовании литографии, гальваностегии и формования литьем), позволяющих заменить стеклянные, кремниевые или кварцевые элементы на элементы, изготовленные из подходящих полимеров, что представляется весьма перспективным направлением при разработке МФУ.
В [71] (2000 г.) для выделения ДНК из лейкоцитов использовали микроколонки, содержащие нанограммовые количества КЗ-частиц размером 30–40 мкм. Позже эта методика была воспроизведена в микрочиповом формате [72]. Микроканалы в устройстве заполняли КЗ-частицами или создавали непрерывную КЗ-сетку с помощью золь-гель технологии либо комбинировали эти подходы. Процедура выделения нанограммовых количеств ДНК (отнимающая около 25 мин) включала в себя три этапа: адсорбцию ДНК в присутствии хаотропной соли, удаление примесей водно-спиртовой смесью и элюирование адсорбированной ДНК в небольшом объеме буфера, подходящего для последующего проведения ПЦР. В [73, 74] показано, что с помощью “гибридных” микрочипов с КЗ-частицами, иммобилизованными в сетке геля, удается выделить ДНК из бактериальных лизатов, цельной крови и спермы. Так как эффективность экстракции ДНК на “гибридных” носителях в составе микрочипов снижается из-за возникновения трещин в геле [72], предпринимались попытки усовершенствовать структуру геля с использованием порообразующего агента – ПЭГ [75], что упрочило пористую структуру наполнителя микроканалов в чипе и оптимизировало гидродинамику при пропускании пробы через систему.
В альтернативном решении [76] обеспечена периодическая смена направления потока образца (и рабочих буферных растворов) над иммобилизованным КЗ-покрытием в объеме канала. Это решение, по сути, аналогично разработке “ZipTips” компании Millipore (США), в которой слой частиц гидрофобизованного КЗ иммобилизован в кончике наконечника для ручной или автоматической пипетки [77].
С конца 80-х годов прошлого века независимо в России, США и Японии образован задел для разработки нового поколения устройств, основанных на использовании полимерных монолитов в качестве альтернативы материалам, используемым в колоночной высокоэффективной жидкостной хроматографии (ВЭЖХ). В [78] в качестве такой замены использовали жесткие пористые органические монолиты, размер пор которых контролируемо варьировался от нано- до микроразмеров, причем поры образовывали непрерывную сеть каналов с высокой площадью поверхности. В небольшой колонке (30 × 8 мм) сополимеризовали глицидилметакрилат с этилендиметакрилатом в среде порогенов (смесь циклогексанола и додеканола) в присутствии азо-бис-изобутиронитрила. Затем поверхностные эпоксигруппы гидролизовали с целью получения диольных функций или аминировали. Полученные мини-колонки применяли в различных вариантах хроматографии. В [79] сообщали о получении фотополимеризуемых полимерных монолитов в результате полимеризации 3-метакрилоксипропилтриметоксисилана в присутствии катализатора, порогена и фотоинициатора. Полученный монолитный сорбент эффективен при разделении смеси двух кумариновых красителей. Осознание очевидных преимуществ использования монолитных материалов (контролируемая пористость и возможность реализации интенсивного массопереноса) спустя относительно короткое время привело к созданию устройства на основе полимерных монолитов для очистки ДНК в капиллярном формате [80]. Полимерный монолит формировали в капиллярах в результате полимеризации 3-(метилоксисилил)пропилметакрилата с последующей прививкой на поверхность полученного монолита тетраметилортосиликата, что повышало количество участков, связывающих молекулы ДНК. Объем пробы цельной крови с целью экстракции из нее ДНК был ограничен 100 нл в виду высокой сорбционной активности поверхности материала в отношении белков крови, конкурирующих за сайты связывания с молекулами ДНК. Для решения этой проблемы предложено МФУ для двухстадийной очистки ДНК из крови [81]. На первой стадии до 70% белков удерживалось на микроколонке с сорбентом с фазой С18, а затем на монолите дочищали ДНК. Эффективность очистки ДНК повышалась на 2 порядка.
В [82] описано микроустройство для выделения ДНК, содержащее полимерный монолит с импрегнированными частицами КЗ. В этом устройстве поверхность монолита не удерживала ДНК, а само устройство оказалось одноразовым из-за низкой стабильности материала монолита.
В [83] сообщалось о разработке полимерных монолитных дисков на основе макропористого полиглицидилметакрилата, сшитого этилендиметакрилатом (ГМА-ЭДМА) с различными функциональными поверхностными группами. Эти материалы, демонстрирующие эффект “позитивной селекции” в отношении определяемых соединений, применяли для разделения смесей белков, в иммуноферментном анализе, в качестве ферментного реактора и др. Ряд сополимеров, пригодных для получения монолитных сорбентов, был расширен (в частности, использовали сополимеры глицидилметакрилата, 2-цианоэтилметакрилата и 2-гидроксиэтилметакрила, сшитые глицерин-1,3-диметакрилатом) [84, 85]. Монолитные диски применяли в составе “мембранных колонок”, а стеклянные пластины, на поверхности которых были сформированы монолитные слои указанных сополимеров, использовали в качестве чипов для определения наноколичеств биополимеров.
Отметим работы, в которых при выделении НК исключали обработку биологического образца хаотропным агентом и отмывку органическими растворителями. Так, японскими исследователями в [86] предложено силаминировать поверхность каналов КЗ-чипа с целью получения ионообменной фазы для обратимого рН-зависимого связывания НК, в частности, при выделении ДНК из крови. ДНК удерживалась при pH 6.0 и после отмывки от примесей элюировалась при pH 7.5 и выше. В [87] на поверхность кварцевых микростолбиков в составе МФУ наносили слой оксида алюминия с целью электростатического удерживания молекул НК в кислых условиях с последующей их элюцией в щелочных условиях. В [88] описана рН-чувствительная система для выделения ДНК из крови, основанная на использовании каналов или частиц, покрытых слоем хитозана, на котором молекулы ДНК удерживаются при рН 5.0 и элюируются при рН 9.0.
В качестве альтернативы кремниевым (и кремнеземным) субстратам предложены микрофлюидные (МФ) системы на основе фотоактивируемых поликарбонатов, на поверхности которых в результате УФ-обработки образуются функциональные группы, удерживающие НК при использовании специального связывающего буферного раствора [89]. Такие материалы использовали в формате 96-луночного МФ-чипа для выделения бактериальной ДНК. Однако неравномерность потока элюента при промывке системы не обеспечивала совпадения результатов в разных лунках [90].
В [91] описана сэндвичевая система для удерживания ДНК фага λ, которая является прототипом МФУ на основе пористой мембраны из оксида алюминия, размещенной между двумя слоями полидиметилсилоксана, в каждом из которых создана сеть микроканалов.
Заслуживают внимания результаты разработки микроформатных устройств для экстракции иРНК эукариот, макромолекулы которых имеют поли-А-хвосты, комплементарно связывающиеся с политиминдезоксирибонуклеотидами (поли-dT). В таких устройствах могут использоваться магнитные частицы с привитыми остатками олиго-dT [92], пористые монолиты, полученные на основе замещенных полиметакрилатов, поверхность которых модифицирована олиго-dT [93] и др.
Принципы, положенные в основу создания МФ-чипов, используются технологическими компаниями при разработке устройств, различающихся как своим назначением, так и степенью интегрированности различных подходов в составе одного устройства. Так, компанией Microfluidic systems Inc. (США) предложен МФ-картридж для проведения дифференцированного лизиса различных типов клеток. С помощью этого устройства проводят ультразвуковую обработку образца и химический лизис, получая частично очищенную ДНК [94]. Стадии обработки образцов могут быть автоматизированы.
В компании Bio-Rad Laboratories, Inc. (США) разработано МФУ для очистки ДНК или РНК с помощью частиц диатомита и последующей амплификацией очищенных НК с помощью ПЦР [95]. Запатентовано МФУ для очистки геномной ДНК в результате ее селективного связывания на поверхности канала, модифицированного ДНК-связывающим реагентом, с одновременным определением количества связанной ДНК [96]. В университете Виржинии (США) запатентовано МФУ, в котором очистка ДНК происходит в результате последовательного прохождения через серию микроколонок, селективно сорбирующих примеси при пропускании через систему подвижных фаз специального подобранного состава, причем последняя колонка селективно связывает ДНК [97].
МФУ, разработанное компанией 3M Innovative Properties Company (США) для очистки и концентрирования НК (ДНК, РНК и пептидно-нуклеиновых кислот) из биологических и экспериментальных образцов [98], включает в себя резервуар для загрузки образца и камеры для его обработки и перемешивания, которые соединены каналами. В качестве так называемых “осаждающих агентов” в устройстве используются частицы декстрана или желатина, которые либо вводят в микроканалы устройства, либо предварительно добавляют к образцу с целью получения фракции, обогащенной целевой НК. С помощью этой системы, основанной на многостадийном протоколе выделения, НК очищаются лишь до известной степени в основном от ингибиторов ПЦР.
В Национальном университете Сингапура запатентовано монолитное МФУ для экстракции ДНК и РНК из крови [99], в котором используется кремнеземный носитель.
Для устранения перекрестной контаминации компания Sarnoff Corporation (США) предложила устройство для одновременного автоматического выделения НК из 24 биологических образцов [100]. Устройство включает в себя кассету с 380 стрипами (полосками) для лизиса образцов и съемный герметичный контейнер для хранения и транспортировки стрипов с выделенной НК.
Компанией Microchip biotechnologies, Inc. (США) разработана система, в первом модуле которой происходит связывание и очистка НК на магнитных частицах с аффинным покрытием во вращающемся магнитном блоке. Лизат получают в проточном режиме в результате ультразвуковой обработки образца. Во втором модуле, представляющим собой МФУ, осуществляется анализ выделенной НК [101].
Компания Siemens AG (Германия) разработала одноразовый плоский картридж для автоматического анализа ДНК и белков, представляющий собой систему миниатюрных каналов и емкостей, заполненных сухими реагентами [102]. После ввода образца в картридж и выделения очищенных фракций биополимеров можно получать данные о свойствах выделенных биомолекул, помещая картридж в совместимое считывающее устройство.
Компанией Micronics Inc. (США) разработана одноразовая МФ-карта, на которой одновременно выделяется и амплифицируется ДНК с целью диагностики некоторых бактериальных возбудителей в биологических жидкостях [103]. Клетки концентрируются на специальной мембране, на которой затем проводятся лизис и амплификация ДНК. ПЦР-продукт элюируется с мембраны и поступает на детектор для визуализации.
В Пенсильванском университете (США) разработано устройство, представляющее собой “лабораторию-на-чипе”, предназначенную для ПЦР-диагностики некоторых бактериальных и вирусных патогенов, определяемых в слюне [104]. Система включает в себя коллектор образца слюны, одноразовый МФ-чип для его обработки со встроенной системой для лизиса, экстракции НК, проведения ПЦР и мечения ампликонов. Также в системе имеется платформа-контроллер, управляющая подачей реагентов, температурой, работой клапанов, а также сканер для считывания результатов.
Для анализа геномной ДНК компания Canon U.S. Life Sciences, Inc. (США) разработала устройство, включающее в себя картридж, в котором проводится выделение ДНК (с помощью магнитных частиц либо носителей, изменяющих свои свойства под действием электрического заряда), и инжектор, подающий раствор ДНК на МФ-чип, имеющий зоны для ПЦР-амплификации, детекции и анализа ДНК [105].
Полностью автоматизированная портативная система (применимая в лабораторных и полевых условиях) на основе МФ-чипа для обнаружения патогенных микроорганизмов методом ПЦР в реальном времени разработана компанией Cornell Research Foundation, Inc. (США) [106]. Микрочип содержит модули для очистки ДНК и ПЦР-детекции. После лизиса клеток проводится отмывка НК водно-спиртовым раствором. Система включает в себя микропроцессор, клапаны и насосы, устройства для термоциклирования и флуоресцентной детекции.
МФУ для диагностики вирусов гриппа разработано компанией CombiMatrix Corp. (США). В состав устройства включены ДНК-микрочип (содержащий 12 000 зондов) и МФ-картридж. Непосредственно на чипе происходит синтез олигонуклеотидных зондов. Жидкости перемешиваются за счет пропускания микропузырьков, генерируемых электрохимическими микронасосами, которые также используются для перекачивания жидкостей внутри картриджа. РНК, выделенные из образца, используются в ПЦР и гибридизации на микрочипе. При использовании этого устройства не нужно вносить реагенты, что устраняет контаминацию образцов. С помощью устройства идентифицировали субтипы вируса гриппа А [107], а также исследовали экспрессию генов клеток лейкемии человека [108].
Таким образом, сегодня известно множество способов, обеспечивающих выделение и очистку НК в процессе пробоподготовки биологических образцов. Необходимые для этого манипуляции могут быть частично или полностью автоматизированы. Тем не менее существующие автоматизированные системы дороги и нередко сложны в эксплуатации, а использование более простых систем сопровождается неполной очисткой и недостаточным выходом НК. Часто эти системы предназначены для выделения НК из образца определенного типа (например, только из крови, слюны и др.) либо они пригодны для обнаружения в образце одного конкретного организма (или группы родственных организмов).
Из рассмотренного материала видно, что разработка технологий выделения НК является предметом интенсивных научных и инженерных исследований. Миниатюризация и автоматизация стали результатом широкого использования нанотехнологических методов и подходов. Несмотря на внешнее разнообразие методик и их аппаратурного оформления, все описанные выше системы основаны на реализации эффекта “позитивной селекции” в отношении выделяемой НК, что определяет многостадийность процесса выделения и неизбежность потерь выделяемой НК на каждой стадии. При выделении НК из биологических образцов предпочтение отдается методам, основанным на использовании твердых носителей, способных селективно и обратимо удерживать НК. В качестве таких носителей чаще всего применяют неорганические материалы, такие как КЗ, ХМКЗ, стекла, а также полимерные монолиты или композитные мембраны. В следующем подразделе кратко рассмотрим способы получения (и свойства) композитов, наиболее часто применяемых при выделении НК, и полимерных модификаторов, используемых при получении таких сорбентов, селективно разделяющих компоненты биологических смесей (в частности, биополимеров) в процессах хроматографии и ТЭ.
2. ПОЛИМЕРСОДЕРЖАЩИЕ СОРБЕНТЫ ДЛЯ ВЫДЕЛЕНИЯ И ОЧИСТКИ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ. МЕТОДЫ ПОЛУЧЕНИЯ, МОДИФИКАТОРЫ И ОБЛАСТИ ПРИМЕНЕНИЯ
В качестве сорбентов для выделения биологически активных соединений (БАС) используют как “нативные” носители, так и матрицы, поверхность которых модифицирована низкомолекулярными соединениями, а также материалы, получаемые в результате физической адсорбции или хемосорбции полимеров на поверхности носителя, и, наконец, сорбенты с привитой полимерной фазой. Поскольку обзор посвящен полимерсодержащим нанокомпозитам, подробнее остановимся на методах получения таких материалов и результатах их применения в молекулярной диагностике и биоаналитике.
2.1. Композиты, получаемые в результате физической адсорбции полимеров
Важно отметить, что модельные сорбенты, используемые в исследованиях механизмов адсорбции на твердых поверхностях (проводимых еще в 80-е годы прошлого века), а также сорбенты, получаемые на базе разработанных при этом методик, стали быстро заполнять нишу сорбционных материалов, применяемых для выделения и очистки БАС. Например, сорбенты, полученные в результате физической адсорбции нейтральных полимеров на поверхности КЗ, в ряде случаев оказались пригодны для выделения и очистки биополимеров. В этих материалах полимерная фаза удерживается за счет большого числа слабых взаимодействий сегментов макромолекул с поверхностью носителя. В [109] сорбент получали обработкой водным раствором полиэтиленоксида (ПЭО) макропористого стекла. Этот материал не сорбировал некоторые вирусы, что позволило проводить их очистку от примесей. Аналогичный КЗ, модифицированный ПЭГ [110], оказался эффективен при выделении вируса миелобластоза из плазмы цыпленка в результате эксклюзионной хроматографии. Покрытие поли-N-винилпирролидона [111] на поверхности КЗ применяли для разделения смесей белков.
С целью повышения устойчивости полимерных покрытий исследовали адсорбцию заряженных полимеров на КЗ. В [112] показано, что поли-1,2,4-триазол необратимо сорбируется на поверхности пористых стекол, по-видимому, за счет электростатического взаимодействия и водородных связей. Полученный сорбент оказался пригодным для очистки некоторых вирусов. Устойчивые покрытия получали на основе катионогенных производных декстрана, которые активировали бромцианом для иммобилизации антигена [113] при получении аффинных сорбентов для выделения антител. Исследование свойств силикагелей, модифицированных декстранами, различающимися молекулярной массой (ММ) и содержанием ДЭАЭ-групп [114], показало, что сорбционные свойства таких сорбентов зависят от количества ДЭАЭ-групп. При низком их содержании наблюдали неспецифическую сорбцию белков, а при содержании свыше 10% сорбент приобретал анионообменные свойства.
Одним из приемов, повышающих устойчивость иммобилизованного полимерного покрытия, стала химическая сшивка адсорбированных макромолекул с помощью бифункциональных или олигофункциональных реагентов. В [115] на поверхности силохрома адсорбировали поливиниловый спирт (ПВС), затем сшивали эпоксидной смолой и вводили алкильные радикалы. С помощью этого материала выделяли микробные липазы. В [116] ПВС адсорбировали на цеолите и сшивали эпихлоргидрином. На поверхности полученного материала иммобилизовали группоспецифичный лиганд для выделения киназ и дегидрокиназ. В ряде работ на поверхность пористых силикагелей адсорбировали разветвленный полиэтиленимин [117]. Покрытие сшивали тетраглицидиловым эфиром пентаэритрита, диглицидилэтиленгликолем [118] и глутаровым альдегидом [119]. Аминогруппы в полученных материалах кватернизовали обработкой метилиодидом, получая сорбенты для разделения смеси тРНК или смеси олигонуклеотидов длиной 30–50 оснований [120].
2.2. Композиты, получаемые в результате хемосорбции полимеров
В приведенных примерах использовали композиты с физически иммобилизованными на поверхности неорганических носителей полимерными фазами. С целью повышения стойкости полимерных покрытий и придания им требуемой селективности независимо исследовали свойства материалов, получаемых в результате ковалентной пришивки полимерных молекул к поверхности носителя. Оказалось, что в результате хемосорбции значительно снижается неспецифическая сорбция молекул сорбата. Кроме того, появляется возможность вводить в состав покрытия спейсоры между поверхностными группами носителя и биоспецифичными лигандами с получением аффинных сорбентов. Так, еще полвека назад Вильчек [121] доказал возможность ковалентной пришивки полилизина и поливиниламина к сефарозе. Позже в работах Даванкова и соавт. [122] на поверхность КЗ ковалентно пришивали сополимеры стирола с метилэтилдиэтоксисиланом.
Ряд работ посвящен получению сорбентов, различающихся степенью гидрофобности. Например, в [123] на поверхность силикагеля хемосорбировали полиэтиленимин, обработанный 2-(карбометокси)-этилтрихлорсиланом. Ацилируя остаточные аминогруппы ангидридом дикарбоновой кислоты, получали катионообменники для разделения смесей белков. На поверхности силикагеля с аминопропильными группами Альперт [124] хемосорбировал нейтральный полисукцинимид, подвергая его затем различным полимераналогичным превращениям. Высокую емкость и способность полученных материалов десорбировать белки автор объяснял “пептидностью” иммобилизованных полимерных фаз и их “пушистостью”.
В середине 80-х годов прошлого века много работ было посвящено созданию “мягких” стационарных фаз на основе ПЭО на основе силикагелей, обработанных реакционноспособными силанами. В [125] было показано, что природа соли в составе элюента, структура силана и молекулярная масса ПЭО оказывают различное влияние на эффективность разделения в зависимости от ММ и природы молекул сорбата (т-РНК и белков).
Стойкие сорбенты с низким уровнем неспецифической сорбции биополимеров были получены в результате хемосорбции поли-п-нитрофенилакрилата на аминопропилкремнеземе [126]. Сходные свойства демонстрируют гидрофобные сорбенты, модифицированные бутиламином и фениламином [127], эффективные при ВЭЖХ белков и при выделении некоторых вирусов. Зубов и соавт. в серии работ (например, [128, 129]) “сконструировали” сорбенты с заданными свойствами в результате иммобилизации на поверхность аминопропилкремнеземов водорастворимых сополимеров N-винилпирролидона и акрилоилхлорида. Остаточные хлорангидридные группы преобразовывали в 2-гидроксиэтиламидные, получая гидрофильные и “диффузные” полимерные фазы, 90% сорбционно-активных групп в которых содержались в “петлях” и “хвостах”. Эти материалы оказались эффективны при количественном выделении ряда вирусов, а также РНК из смеси, содержащей бактериальные рибосомальные фрагменты 70S.
2.3. Композиты, получаемые в результате проведения графт-полимеризации
Новые возможности для “конструирования” композиционных сорбентов открыла разработка методов графт-полимеризации, с помощью которых на поверхности твердого (чаще неорганического) носителя можно формировать полимерную фазу, минуя стадию адсорбции предварительно полученного полимера, поскольку полимеризация происходит непосредственно в присутствии носителя. Для осуществления графт-полимеризации необходимо присутствие на модифицируемой поверхности носителя доступных активных участков (функциональных групп). В случае КЗ в качестве таких активных групп могут выступать поверхностные гидроксильные и/или силоксановые группы, а также металлсодержащие группы присутствующих в КЗ примесей.
Реакционноспособные центры можно получать в результате “сухой” активации поверхности носителя механохимическими реакциями в процессе истирания или расщепления носителя [130]. Подобные способы (наряду с активацией поверхности минеральных носителей с помощью плазмы [131]) применяют при получении высоконаполненных композитов. Образование реакционных центров, участвующих в графт-полимеризации, может происходить при радиационном облучении либо под действием инициаторов свободных радикалов [132, 133].
Описаны различные способы проведения радиационно-инициированной полимеризации [134], включающие в себя как прямое облучение системы мономер–носитель (можно использовать жидкий мономер, раствор мономера, мономер в газообразном или в парообразном состоянии), так и прививку мономера на предварительно облученную в инертной атмосфере (или в вакууме) поверхность (так называемая “пост-полимеризация”).
Этот процесс в присутствии твердого носителя наиболее эффективно протекает в слоях мономера, адсорбированных на его поверхности [135]. При облучении КЗ (т.е. при радиолизе) образуются поверхностные радикалы SiO• и Si•, в результате чего на КЗ-поверхности можно полимеризовать, например, стирол, метилметакрилат, акрилонитрил, винилацетат, п-бутилметакриалт и N-винилпирролидон. Присутствие в системе низкомолекулярных радикалов Н• и ОН•, химически не связанных с поверхностью, приводит к образованию большого количества непривитого гомополимера, что типично для прививки полимеров из жидкой фазы. Напротив, при прививке из паровой фазы образуется незначительное количество гомополимера, количество которого прямо зависит от дозы облучения и имеет предельное значение [136].
В [137] показано, что скорость низкотемпературной пост-полимеризации, протекающей на поверхности пористых носителей, определяется концентрацией мономера (тетрафторэтилена, ТФЭ), адсорбированного на поверхности носителя, но не его содержанием в газовой фазе. При этом важно контролировать условия полимеризации во избежание закупорки пор.
Для получения однородных стойких полимерных покрытий на поверхности твердых носителей разработан ряд приемов. Во-первых, используют мономеры, содержащие реакционноспособные группы (например, 2,3-эпоксипропилметакрилат [138]). Во-вторых, поверхность КЗ предварительно активируют, например, органосиланами, способными сополимеризоваться с винилсодержащими мономерами [139]. В-третьих, используют бифункциональные сшивающие агенты (например, азо-бис-изобутиронитрил при полимеризации дивинилбензола). Образующийся в жидкой фазе полимер адсорбируется на поверхности носителя с образованием трехмерной сетки [140].
В [141] получали аффинные сорбенты на основе силикагелей, последовательно проводя их силанизацию органосиланами с галогенсодержащими, алкокси-, алкил- или арильными группами, а затем – поверхностную (со)полимеризацию бифункциональных винильных мономеров и иммобилизацию подходящих биолигандов. Таким способом синтезировали материалы с привитыми сополимерами N-метилолакриламида с N-замещенными акриламидами с активированными сложноэфирными N-оксисукцинимидными группами, а также с аллиламином и N-аллилбромацетамидом, химически модифицированными диольной фазой. В качестве аффинного лиганда к активированным носителям присоединяли гепарин. Полученные сорбенты оказались эффективны, например, при выделении бычьего антитромбина-ΙΙΙ.
В некоторых способах мономер на поверхности носителя адсорбировали из паровой фазы. В [142] на поверхность КЗ в присутствии паров акриловой кислоты и винилацетата воздействовали ионизирующим излучением (40–100 рад/с). Затем активировали функциональные группы полученных привитых полимеров, обрабатывая их бифункциональными реагентами (диамины, диальдегиды, дихлорангидриды) с последующей иммобилизацией биолигандов (ферментов, нуклеотидов, уридина и т.п.). В результате удалось ввести примерно в 8 раз больше функциональных групп, чем после обработки органосиланами.
Методами прививочной полимеризации получают сорбенты с бóльшим количеством привитой органической фазы (и с высокими ММ – до 106 и более) по сравнению с методами адсорбции полимеров [136]. Однако при этом сложнее регулировать как величину ММ, так и количество адсорбированного мономера. В технологическом плане немаловажно, что прививочная полимеризация в присутствии носителя нередко включает в себя большее число стадий, чем способы получения сорбентов в результате адсорбции полимеров на поверхности носителя.
2.4. Фторполимерсодержащие композиционные сорбенты
Потребность в сорбентах, содержащих полимерные фазы, эффективно экранирующие поверхность носителя, стойкие к воздействию агрессивных сред и к частой смене подвижной фазы при хроматографическом анализе стимулировала создание новых типов полимерсодержащих композитов, в первую очередь фторполимерсодержащих сорбентов. В ранних подобных работах исследовали свойства химически модифицированных частиц полихлортрифторэтилена (ПХТФЭ, Kel-F), отличающихся гидрофобностью, высокой хемостойкостью и механической прочностью. Частицы ПХТФЭ модифицировали металлоорганическими соединениями (н-бутиллитий, ариллитий, алкилмагний и арилмагний), вводя в состав получаемых материалов алькильные группы с уменьшенным содержанием хлора [143]. В результате модификации ароматического ядра в цепи ПХТФЭ получали катион- и анионообменники, сочетающие в себе свойства обращенно-фазовых и ионообменных сорбентов [144], с помощью которых разделяли производные витаминов группы В, нитрофенолы и карбоновые кислоты [145].
Первые работы по исследованию свойств фторсодержащих композитов были посвящены взаимодействию фторсодержащих поверхностей с низкомолекулярными соединениями. Берендсен модифицировал силикагель (гептадекафтордецил)диметилсиланом (ГФД) и сравнил свойства полученного материала с кремнеземами, модифицированными пропил- и н-децил-аналогами [146]. Оказалось, что ГФД-фаза удерживает фторсодержащие соединения, благодаря чему удалось разделить бензол и фторбензол, в то время как на аналогичных не содержащих фтор носителях эти вещества не разделялись. Оказалось, что носители с перфторированной полимерной фазой менее полярны, нежели сорбенты с С-18-фазой [147].
Отметим работу, в которой изучали сорбцию предварительно фторированных белков на фторированных полимерных матрицах. В ней предполагалось, что удерживание перфторалкилсодержащего белка фторсодержащей углеродной поверхностью определяется особым “фтор-обусловленным” взаимодействием, механизм которого отличается от механизма обращенно-фазового разделения [148]. В [149] впервые приведен пример успешного разделения белковых смесей на фтормодифицированных носителях в режиме ВЭЖХ.
В [150] описаны синтез и применение бифильного сорбента, полученного в результате модифицирования наружной поверхности частиц КЗ остатками перфторбутилэтилендиметилсилана (для уменьшения удерживания липофильных сорбатов, таких как белки крови) с одновременным модифицированием внутренней поверхности пор октадецильными группами (для удерживания низкомолекулярных веществ). Этот сорбент использовали для определения содержания различных лекарственных соединений в крови.
Заметим, что аналогичный принцип положен в основу создания известных “сорбентов Пинкертона” [151], имеющих гидрофильную внешнюю поверхность (чтобы не удерживать молекулы лекарственных препаратов и пептидов, содержащихся в биологических жидкостях) и гидрофобизованную трипептидом глицил-фенилаланил-фенилаланином внутреннюю поверхность пор. В результате из-за больших размеров молекулы белка не проникают внутрь пор, а низкомолекулярные метаболиты лекарственных веществ хроматографически разделяются во внутреннем объеме пористого сорбента.
Параллельно исследовали полифторполимерсодержащие материалы и их свойства. Поскольку олеофобно-гидрофобная поверхность таких материалов отличается низкой поверхностной энергией, они слабо смачиваются органическими растворителями и водными растворами. В результате в определенных условиях на фторсодержащих поверхностях слабо адсорбируются как гидрофобные, так и гидрофильные молекулы, что позволяет эффективно разделять компоненты смесей биологических молекул, в частности биополимеров, даже незначительно изменяя состав элюента. Еще одно важное свойство фторполимеров заключается в их способности образовывать сплошные привитые однородные нанопокрытия на твердых поверхностях, эффективно экранирующие поверхность носителя от воздействия агрессивных сред и обеспечивающие снижение неспецифической сорбции [152].
Пластичность частиц фторполимеров не позволяет достичь высокой эффективности их применения при повышенных давлениях (например, при проведении ВЭЖХ) и интенсивном перемешивании (при реализации “batch-процессов”). Поэтому предполагалось, что фторполимерсодержащие композиты на основе кремнеземов будут сочетать контролируемую пористость и механическую прочность твердого носителя с хемостойкостью и специфическими сорбционными свойствами иммобилизованной полимерной фазы, экранирующей поверхность носителя.
В литературе обсуждались различные подходы к модифицированию носителей фторполимерами. Так, в качестве инициаторов полимеризации фтормономеров использовали стабильные озониды перфторолефинов, получаемые путем низкотемпературного озонолиза перфторолефинов при отсутствии растворителей [153, 154]. При контакте с мономером озонид инициирует полимеризацию в интервале температур 240–300 К. После удаления избытка озонида в систему вводят мономер (ТФЭ, винилфторид, метилметакрилат, акрилонитрил, ацетилен и др.), а затем систему нагревают до температуры распада озонида, вызывая полимеризацию мономера. Применение озонидов перфторолефинов позволило проводить полимеризацию в более мягких условиях по сравнению с полимеризацией фтормономеров. С помощью полученных сорбентов обессоливали тРНК, очищали плазмидную ДНК от РНК и белков, разделяли смеси пептидов или компоненты смеси жирорастворимых витаминов.
Альтернативные способы синтеза композиционных фторполимерсодержащих сорбентов основаны на проведении γ-инициированной пост-полимеризации виниловых мономеров (в частности, ТФЭ) на поверхности пористых кремнеземов [155]. ТФЭ адсорбировали на поверхности носителя, а затем подвергали воздействию ионизирующего излучения. ПТФЭ-модифицированные КЗ можно получать в различных режимах (“раздельный радиолиз” при комнатной температуре; облучение носителя при комнатной температуре с последующим намораживанием мономера и проведением пост-полимеризации в процессе разогрева; “раздельный радиолиз” при температуре жидкого азота; “совместный радиолиз” при температуре жидкого азота). Оптимальным является совместный радиолиз, поскольку в этом случае ТФЭ сорбируется из газовой фазы равномерно по всей поверхности КЗ. Растущие радикалы на концах цепей прививаемого ПТФЭ на воздухе образуют кислородсодержащие полярные группы, что приводит к неспецифической сорбции. Для решения этой проблемы проводили так называемый “эндкэппинг” концевых полярных групп различными соединениями, например гесафторпропиленом (ГФП). Полученные материалы использовали в обращено-фазовой и эксклюзионной хроматографии биополимеров, а также в качестве гемосорбентов [156].
Сорбционное поведение фторполимерсодержащих материалов существенно отличает их от других полимерсодержащих сорбентов, что впервые продемонстрировано в 1978–80 годах в работах [157, 158], посвященных исследованию сорбционных свойств ПТФЭ в отношении смесей биополимеров. Было показано, что перфторполимеры обратимо сорбируют белки и пептиды, но при этом не удерживают (или слабо удерживают) НК. В [159] с помощью ПТФЭ-КЗ, полученного методом радиационной пост-полимеризации, очищали плазмидную ДНК. Через колонку с сорбентом пропускали раствор плазмиды рВR322, содержащий также РНК и белки, в нейтральном буфере. РНК и белки оставались на колонке, а плазмида выходила в составе исключенного объема. РНК элюировали с колонки 10%-ным изопропанолом (либо метанолом или ацетонитрилом). При повышении доли органического растворителя в составе элюента до 50% удавалось полностью элюировать белковую фракцию.
В [160] описаны альтернативные способы получения фторполимерсодержащих сорбентов, основанные на использовании сополимера трифторстирола (ТФС) с метилвинилдиэтоксисиланом, полученного радикальной сополимеризацией. По первому способу частицы силикагеля обрабатывали раствором сополимера в абсолютном толуоле, а остаточные силанольные группы – гексаметилдисиланом. По второму способу ампулу с ПТФЭ-силикагелем вакуумировали, охлаждали, а затем облучали с помощью γ-источника дозой 3–5 Мрад. Затем ампулу медленно нагревали, вводили пары ТФС и выдерживали 18 ч. Оба полученных сорбента по своим характеристикам занимали промежуточное положение между КЗ, модифицированными ПТФЭ и полистирольными сорбентами. На полученных сорбентах в обращено-фазовом режиме разделяли компоненты смесей белков, пептидов, антибиотиков и др.
Широкое применение перфторполимерсодержащих сорбентов ограничивается сложностью и трудоемкостью технологий их получения. Поэтому очевидна необходимость разработки новых способов иммобилизации на поверхности твердых носителей фторполимерных покрытий, а также альтернативных не содержащих фтор полимерных модификаторов. Одним из таких модификаторов мог бы стать открытый в 1947 г. поли-п-ксилилен, известный также как парилен N (незамещенный поли-п-ксилилен). Поли-п-ксилиленсодержащий КЗ-сорбент, описанный в [161], служит примером композита с нанопокрытием на основе не содержащего фтор полимерного модификатора. Материал устойчив в условиях щелочного гидролиза, пористость исходного носителя в значительной мере сохраняется. В ряде работ доказаны био- и гемосовместимость медицинских изделий, покрытых слоями хлорзамещенного поли-п-ксилилена [162].
2.5. ПАНИ-содержащие композиционные сорбенты
Данную главу обзора нельзя считать завершенной, не рассмотрев возможности, которые дает использование полианилина (ПАНИ) – полимерного модификатора, весьма перспективного для синтеза полимерсодержащих композитов. ПАНИ относится к классу особых макромолекул с системой полисопряжения, обладающих высокой химической стойкостью и уникальными сорбционными свойствами, которые можно обратимо изменять в результате допирования–раздопирования. Благодаря рН-чувствительности ПАНИ относят к smart-полимерам (“стимул-чувствительным” полимерам), что определяет широкий спектр его применений в различных областях науки и техники.
Макромолекула ПАНИ включает в себя фрагменты, относительное число которых определяется уровнем окисления цепи, поскольку ПАНИ может обратимо окисляться и восстанавливаться. В зависимости от уровня окисления различают три основные формы ПАНИ: пернигранилин – полностью окисленная форма; лейкоэмеральдин – полностью восстановленная форма; эмеральдин – форма с соотношением окисленных и восстановленных фрагментов, равным 1:1 [163]. Каждая форма ПАНИ может вступать в кислотно-основные реакции и существовать как в виде соли, так и в виде основания. Протонированный эмеральдин, в цепи которого содержится равное количество окисленных и восстановленных звеньев, является наиболее устойчивой формой ПАНИ [164].
Чаще всего ПАНИ получают в результате химической или электрохимической окислительной полимеризации анилина [165]. Технологически проще осуществлять и масштабировать процессы химического синтеза ПАНИ. На структуру и свойства конечного продукта влияют природа окислителя и кислоты-допанта, соотношение мономер: допант: окислитель, а также рН реакционной среды [166].
В реакции синтеза ПАНИ можно выделить по крайней мере две стадии: индукционный период (когда не наблюдается окрашивания реакционной смеси и образования твердой фазы полимера) и стадию роста цепи, во время которой скорость образования полимера растет, а затем снижается до нуля, в результате чего кинетическая кривая реакции окислительной полимеризации анилина приобретает S-образный вид [167].
Сложность механизма полимеризации анилина и условия проведения реакции определяют образование разнообразных по химической структуре и морфологии полимерных продуктов. Можно ожидать, что сорбционные свойства материалов, модифицированных ПАНИ, также будут в значительной степени определяться условиями их получения. Крайне важно отметить, что все типы надмолекулярных структур ПАНИ могут формироваться не только в объеме реакционной среды, но и на разнообразных поверхностях раздела фаз [168–170]. ПАНИ-покрытия, как правило, однородны по толщине и прочно удерживаются носителем.
Не рассматривая варианты проведения реакции полимеризации анилина с целью получения ПАНИ-структур на границе раздела жидких фаз, кратко остановимся на способах получения ПАНИ-покрытий на твердых носителях. Описанные в литературе методы получения композиционных ПАНИ-содержащих материалов можно отнести к четырем основным типам: методы химического поверхностного и химического диффузионного осаждения [171]; образование пленок на поверхности носителя с использованием дисперсий ПАНИ-частиц [172, 173]; получение покрытий электрохимическим осаждением [166]; метод механического перемешивания [174]. Когда речь идет о получении ПАНИ-содержащих композитов, наиболее технологичными представляются методы, отнесенные к первой группе. Исследованию процессов образования ПАНИ-покрытий в ходе окислительной полимеризации анилина in situ в присутствии внесенного субстрата посвящен ряд работ. ПАНИ можно легко высадить из смеси водных растворов анилина в присутствии допанта (кислоты) и окислителя в виде хорошо удерживающейся пленки на поверхности различных материалов в результате так называемой адсорбционной полимеризации [171, 173]. С этой целью носитель погружают в указанную смесь. Скорость образования покрытия и его характеристики зависят от ряда факторов: соотношения реагентов, концентрации, температуры и др.
Цзоу и Грегори [175] одними из первых показали, что скорость окислительной полимеризации анилина, как правило, увеличивается в присутствии твердого субстрата, однако при этом важно учитывать природу носителя. Так, по мнению авторов, на инертной поверхности рост ПАНИ-покрытия обусловлен осаждением частиц ПАНИ, образующихся в объеме реакционной смеси. В случае носителей, активных в реакции окисления, наблюдали заметное сокращение индукционного периода.
В [176] сообщалось о возможности нанесения покрытий ПАНИ на поверхность пленок из пористого кремния n+-типа. Структура полимера внутри пор не отличалась от структуры полимера на поверхности кремния. Оказалось, что полимерное нанопокрытие, состоящее из пяти монослоев ПАНИ, покрывает всю поверхность пор, в то время как нанопокрытие на внешней поверхности носителя состоит из двух монослоев.
Орлов и соавт. [177, 178] показали, что по мере увеличения концентрации исходных реагентов, взятых в реакцию, в водных средах происходят повышение скорости полимеризации анилина и сокращение индукционного периода. Авторы отмечали сложный характер зависимости выхода ПАНИ, образующегося на поверхности субстрата, от времени.
Технологическое преимущество, обеспечиваемое синтезом ПАНИ-содержащих сорбентов по сравнению, например, с процессами получения фторполимерсодержащих материалов радиационными методами, определяется мягкими условиями полимеризации (комнатная температура, водная среда, отсутствие сложного оборудования).
Отдельного внимания заслуживают методы на основе “матричной полимеризации” анилина в присутствии растворимых поликислот. Это в первую очередь связано с возможностью получения новых материалов, сочетающих особые свойства ПАНИ с механическими свойствами полимерных материалов – носителей [179]. В матричном синтезе ПАНИ использовали различные поликислоты, например полиакриловую кислоту, полиметакриловую кислоту, полистиролсульфокислоту и другие [180–182], отличающиеся регулярным расположением остатков кислотных групп вдоль полимерной цепи. Также было показано, что эффективный синтез ПАНИ можно осуществить в присутствии поликислот, в макромолекулах которых кислотные группы распределены нерегулярно. Такие поликислоты являются ароматическими полиамидами [183, 184], которые получают в результате поликонденсации хлорангидридов изо- и терефталевой кислот и 4,4´-диаминофенил-2,2´-дисульфоната натрия (а именно, изо-ПСК из изофталевой кислоты и тере-ПСК из терефталевой кислоты) [184]. При полимеризации анилина в присутствии указанных полисульфокислот наблюдали рост индукционного периода полимеризации. Автокаталитический характер матричной полимеризации анилина подтверждается результатами полимеризации анилина в присутствии “затравки” (предварительно синтезированные частицы ПАНИ).
Интерполимерные комплексы ПАНИ с поликислотами можно рассматривать в качестве эффективных полимерных модификаторов поверхности твердых носителей при разработке технологичных способов синтеза ПАНИ-содержащих композитов для разделения компонентов биологических смесей.
Таким образом, в распоряжении исследователей и диагностов имеется широкий арсенал полимерсодежащих композитов, получаемых разнообразными физико-химическими и химическими методами. В этих материалах полимерная фаза особым образом структурирована на поверхности носителя и включает в себя наноразмерные элементы. Однако следует еще раз отметить, что большинство известных методов выделения с применением сорбентов основаны на удерживании целевого биополимера сорбентом на первом этапе выделения (“позитивная селекция”), после чего необходимо отмыть удерживаемый компонент от примесей и элюировать его с поверхности сорбента. Многостадийность процедуры не только приводит к потерям выделяемого из биологической смеси компонента, но и ухудшает воспроизводимость по его выходу и степени очистки.
3. ФТОРПОЛИМЕР- И ПОЛИАНИЛИНСОДЕРЖАЩИЕ СОРБЕНТЫ ДЛЯ ВЫДЕЛЕНИЯ И ОЧИСТКИ БИОПОЛИМЕРОВ – НОВЫЕ МАТЕРИАЛЫ, ДЕМОНСТРИРУЮЩИЕ ЭФФЕКТ “НЕГАТИВНОЙ СЕЛЕКЦИИ” В ОТНОШЕНИИ НУКЛЕИНОВЫХ КИСЛОТ. СПОСОБЫ ПОЛУЧЕНИЯ И ОБЛАСТИ ПРИМЕНЕНИЯ
Обзор литературы позволяет выявить круг проблем, связанных с необходимостью разработки новых технологий, позволяющих проводить выделение биополимеров по принципиально более простой одностадийной схеме, основанной на эффекте “негативной селекции” в отношении НК. Известны работы, в которых эти проблемы решаются с использованием междисциплинарного подхода, предусматривающего комплексное применение химико-технологических, физико-химических и молекулярно-биологических методов. С целью получения материалов, демонстрирующих эффект “негативной селекции” в отношении НК, разработан ряд масштабируемых технологий, обеспечивающих формирование нанотолщинных покрытий фтор- и анилинсодержащих полимеров (проявляющих такие свойства) на поверхности твердых носителей. Кроме того, разработаны специальные биосепарирующие элементы (БЭ), в состав которых включены полученные сорбенты, а также оптимальные протоколы пробоподготовки, позволяющие выделять биополимеры из различных источников в соответствии с заданными параметрами. Наиболее интересные особенности таких материалов и методик рассмотрены ниже.
3.1. Методы получения композитов, демонстрирующих эффект “негативной селекции” в отношении НК
Чтобы установить, пригодны ли ПАНИ-покрытия для селективного разделения смесей биополимеров, Капустиным и соавт. проведено сравнительное исследование сорбционных свойств ряда полимеров в отношении НК и белков. Авторы полагали, что эффект “негативной селекции” в отношении НК может проявляться при использовании не только перфторированных, но и частично фторированных или не содержащих фтор полимеров, таких как ПАНИ, а возможно, и других синтетических полимеров.
Эффект “негативной селекции” в отношении НК проиллюстрирован результатами использования ВЭЖХ-колонок, упакованных ПТФЭ- и ПАНИ-содержащими КЗ-сорбентами для разделения смеси плазмиды pBR 322 из E. coli, РНК и сопутствующих белков [159, 167]. Было известно, что ДНК с ПТФЭ-колонки выходит в первой фракции в исключенном объеме, в то время как РНК слабо удерживается, но также выходит в изократическом режиме в составе второй фракции. Как оказалось, аналогичный результат достигается при использовании ПАНИ-модифицированного КЗ. Различия в сорбционном поведении ПТФЭ- и ПАНИ-содержащих сорбентов выявляются при десорбции удерживаемых сорбентами белков. Суммарная белковая фракция полностью десорбируется с ПТФЭ-колонки в обращенно-фазовом режиме в условиях возрастающего градиента концентрации органического растворителя (как и при использовании С18-фаз), когда скорость элюирования белков (пептидов) тем выше, чем ниже их гидрофобность. Напротив, удержанные рН-чувствительным ПАНИ-покрытием молекулы белков выходят с колонки в условиях понижающегося градиента рН в зависимости от значения их изоэлектрической точки (pI), но не ММ или гидрофобности.
С целью оценки влияния морфологии, химического состава и заряда поверхностного слоя полимерных покрытий на их сорбционные свойства в [185–188] исследовали свойства кремнеземов, модифицированных полиарамидами, содержащими набор таких “ключевых” элементов, как ароматический азот, фтор, а также донорные и акцепторные фрагменты. При исследовании динамической сорбции биополимеров (ДНК, РНК и белков, различающихся ММ и значениями pI) в режиме реального времени методом спектрально-корреляционной интерферометрии аналогичные полимерные покрытия наносили методом spin-coating на плоские стеклянные подложки. Свойства всех полученных покрытий сравнивали со свойствами фторполимер- и ПАНИ-содержащих сорбентов. Было установлено, что в отличие от сорбционных материалов, в определенных условиях удерживающих НК (таких как, например, полиэтиленимин или кремнеземы) и не связывающих белки (например, полипропилен или полиакриламид), композиты, модифицированные фторполимерами (ФП), ПАНИ и некоторыми полиарамидами, ведут себя противоположным образом. Подбирая соответствующий полимерный модификатор, можно решать конкретные задачи по селективному выделению биополимеров из сложных смесей, используя “нулевую” сорбционную способность к удерживанию ДНК и способность к обратимой сорбции белков в зависимости от значения их pI. Количественные параметры сорбции при этом зависят как от химической структуры полимерного покрытия в составе сорбента, так и от природы молекул сорбата, и в значительно меньшей степени – от морфологии поверхности сорбента.
В упомянутых работах показано, что сорбционные свойства исследованных материалов определяются суммарным вкладом различных механизмов сорбции, происходящей в результате гидрофобных, диполь-дипольных взаимодействий и образования водородных связей. Зная химическую структуру полимерного покрытия композита, можно уверенно предсказывать его сорбционные свойства в условиях разделения смесей НК и белков. Это также означает, что для получения одинакового хроматографического эффекта при разделении смесей биополимеров можно использовать сорбенты, обработанные различными по химической структуре полимерными модификаторами, демонстрирующими эффект “негативной селекции” в отношении НК.
В литературе представлен ряд технологичных способов синтеза фторполимер- и ПАНИ-содержащих композитов для выделения биополимеров. Поскольку ни ФП, ни ПАНИ не обладают удовлетворительными механическими и технологическими свойствами, позволяющими изготовить из них качественные пористые сорбенты, созданы композиционные материалы на основе твердых носителей, модифицированных нанослоями указанных полимеров.
Поверхность таких композитов ведет себя как соответствующий полимер, а жесткость и морфология (пористость и удельный объем носителя) определяются свойствами исходной матрицы (рис. 3). При проведении прямого синтеза таких композитов полагали, что формирование макромолекул будет сопровождаться образованием однородных полимерных нанопокрытий, прочно связанных с поверхностью носителя, в том числе с внутренней поверхностью пор.
Рис. 3.
Морфология композиционных полимерсодержащих сорбентов. Полимерное покрытие должно быть тонким и равномерно распределенным по поверхности носителя, обеспечивая снижение неспецифической сорбции и повышение химической стабильности композиционного сорбента. При этом значения внутреннего диаметра и объема пор носителя не должны значительно уменьшаться.
Разработанные подходы к получению композитов удобно формально разделить на две группы. К первой группе относятся способы, в которых композит с полимерным нанопокрытием получали в результате полимеризации мономера в присутствии неактивированного носителя. К этой группе также следует отнести способы, в которых слой готового прекурсора (олигомера или полимера) предварительно наносили на поверхность носителя методом “кастинга”, а затем иммобилизовали на поверхности носителя за счет химического отверждения (в том числе, в результате полимераналогичных превращений).
Так, ПАНИ-покрытия могут быть получены на поверхности твердых носителей различной природы методом окислительной осадительной полимеризации анилина. При этом важно получить первый слой ПАНИ, а образование последующих слоев будет катализироваться предыдущими слоями. Очевидно, что природа поверхности носителя может влиять на характер полимеризации анилина. Например, ПАНИ-покрытия на поверхности частиц катионообменной смолы формировались без добавления допанта, а S-образная форма кинтетической кривой в целом сохранялась. Присутствие частиц КЗ, не оказывающего каталитического воздействия на полимеризацию анилина в кислой среде, практически не изменяет вид кинетической кривой, получаемой в отсутствие носителя. Однако при этом полимеризация параллельно протекает на поверхности и в объеме реакционной смеси, и до 60% мономера расходуется на образование взвеси полимерных частиц в объеме реакционной смеси. Эти частицы оседают на поверхность ПАНИ-модифицированного носителя, слабо удерживаясь на ней и загрязняя сорбент (особенно поры), что весьма существенно усложняет процедуру отмывки материала. Напротив, при использовании поверхностей, представляющих собой поликислоты, мономер расходуется преимущественно на образование ПАНИ-покрытия, а не на образование взвеси полимерных частиц в объеме реакционной среды. Таким образом, существует по крайней мере один путь для локализации процесса формирования ПАНИ-покрытия на поверхности носителя – использование носителей с кислотными поверхностными группами [189].
В [190, 191] описан способ получения полифторбутадиенсодержащих сорбентов на основе кремнезема с олигобутадиеновым покрытием, который затем обрабатывали in situ парами XeF2 в атмосфере аргона в “режиме псевдоожижения”. В результате фторирования происходило химическое структурирование за счет рекомбинации макрорадикалов и взаимодействия радикалов с кратными связями с образованием поперечно-сшитого фторированного полимерного покрытия. Благодаря замещению атомов водорода атомами фтора модифицируются поверхностные свойства полимерного покрытия композита (без изменения его объемных свойств). Использование XeF2 в качестве фторирующего агента упростило технологию фторирования по сравнению с обработкой смесью фтора с азотом.
Вторая группа разработанных методов включает в себя способы прямого синтеза композитов, основанные на локализации процесса полимеризации на активированной поверхности носителя, т.е. когда полимеризация инициируется поверхностными функциональными группами носителя и протекает на его поверхности. С целью локализации полимеризации анилина на поверхности носителя разработан ряд способов, основанных на предварительной иммобилизации на поверхности носителя полимерных нанослоев, содержащих функциональные группы – источники протонов.
При окислительной полимеризации анилина вначале образуется катион-радикал анилиния. В [192, 193] предположили, что матрицами, одновременно обеспечивающими протонирование анилина и локализацию процесса полимеризации на поверхности носителя, могут служить твердые нерастворимые поликислоты (кислые катионообменники). Для получения таких матриц методом пост-радиационной прививочной полимеризации получали ПТФЭ-кремнезем, на поверхность которого методом пост-радиационной блоксополимеризации прививали сополимер стирола с дивинилбензолом (ПС-ДВБ). Полученный ПС-ДВБ-кремнезем сульфировали разбавленной серной кислотой, варьируя продолжительность обработки и получая материалы с различным содержанием поверхностных сульфогрупп. При полимеризации анилина на поверхности таких носителей практически отсутствовал индукционный период и не образовывались частицы ПАНИ в объеме реакционной среды. Конверсия мономера повышалась пропорционально содержанию поверхностных сульфогрупп [192].
Локализовать процесс формирования ПАНИ-покрытия на поверхности носителя также удалось в результате проведения матричной полимеризации, протекающей с образованием полимерных комплексов, состоящих из растворенных макромолекул “полимера-матрицы” и синтезируемого на них ПАНИ. В отличие от осадительной полимеризации анилина в присутствии низкомолекулярной кислоты в процессе матричной полимеризации система остается фазово-гомогенной. Поликомплексы проявляют свойства, отличные от свойств образующих их полимеров. Этот принцип положен в основу получения новых композитов, в которых предварительно силаминированный 3‑аминопропилтриэтоксисиланом КЗ модифицировали продуктами матричной окислительной полимеризации анилина на ароматических полисульфокислотах (ПСК). Описаны варианты матричного синтеза ПАНИ в присутствии поли(п,п'-(2,2'-дисульфокислота)дифениленизофталамида (изо-ПСК): модификация силаминированного стекла готовыми комплексами изо-ПСК-ПАНИ и полимеризации анилина в присутствии силаминированного стекла, предварительно покрытого изо-ПСК. Образованию устойчивых покрытий на основе поликомплексов изо‑ПСК–ПАНИ способствовало повышение содержания протонированных сульфогрупп в ПСК [194].
Полагали, что придание поверхности сорбента дополнительной функциональности за счет введения в состав полимерного модификатора определенных функциональных групп позволит расширить область применения получаемых сорбентов. Поскольку химическая модификация готового ПАНИ в силу его химической стойкости неэффективна, поверхность КЗ модифицировали предварительно синтезированными сополимерами анилина с 3-аминобензойной кислотой (3-АБК), п-нитроанилином или о-толуидином [194].
Для локализации полимеризации мономеров, полимеризующихся по различным механизмам (фтормономеры, анилин и др.), был разработан способ активации КЗ-матрицы озоном, что позволило получить гетерофазный инициатор полимеризации, поверхность которого одновременно служит подложкой при синтезе композита [194, 195].
Показано, что поверхность КЗ-носителя может быть активирована в результате обработки озоном с образованием активных центров различной природы. Кремнезем (диоксид кремния) не реагирует с озоном, но, как правило, содержит примеси оксидов железа или алюминия, которые под действием озона образуют ион-радикальные и/или пероксидные группы, способные инициировать полимеризацию различных мономеров. Установлено, что активированный озоном КЗ можно использовать в качестве гетерогенного инициатора полимеризации мономеров, полимеризующихся по различным механизмам (фтормономеры, анилин, стирол и др.). Таким способом получены сорбенты, модифицированные ПТФЭ (с использованием образующихся поверхностных радикалов без добавления инициатора) и ПАНИ (с использованием поверхностных пероксидов без добавления окислителя). В систему дополнительно вводили ГФП во избежание образования концевых заряженных кислородсодержащих групп с целью уменьшения неспецифической сорбции биополимеров либо аллиламин или аллиловый спирт с целью введения дополнительных функциональных групп для повышения гидрофильности поверхности и селективности получаемых материалов в процессах разделения смесей одно- и двухнитевых НК. Сомономеры вводили одновременно с ТФЭ в режиме пост-полимеризации либо после исчерпания ТФЭ на первом этапе, обеспечивая повышенное содержание поверхностных амино- или гидроксигрупп соответственно [196].
На стадии индукционного периода при полимеризации анилина образуются гидрофобные феназинсодержащие олигомерные продукты. В [195, 197] показано, что синтез ПАНИ можно локализовать на поверхности гидрофобизованной матрицы, например покрытого фторполимером КЗ. Кремнезем обрабатывали раствором фторопласта-42Л (ФП) в ацетоне, после удаления растворителя на поверхности носителя получали нанотолщинное гидрофобное ФП-покрытие. При полимеризации анилина в присутствии полученного носителя, низкомолекулярной кислоты и окислителя образующиеся гидрофобные олигомеры прочно сорбировались на гидрофобизованной поверхности, служащей в данном случае “матрицей”, удерживающей растущие макромолекулы ПАНИ. Расход мономера на образование полимерных частиц в объеме смеси понижался по сравнению с синтезом ПАНИ в присутствии необработанного кремнезема в 6–8 раз. Полученный технологический эффект определяется тем, что локализация полимеризации анилина на поверхности носителя достигается в отсутствие сомодификаторов ПАНИ и без предварительной химической активации поверхности носителя.
В рассмотренных выше работах показано, что в результате полимеризации анилина в присутствии твердых носителей различной природы (с нейтральной, кислой, гидрофобизованной поверхностью) образуется устойчивое ПАНИ-покрытие, что позволяет в качестве носителей использовать не только объемно-пористые частицы, но и синтетические мембраны, капилляры и др. В [197] представлена линейка материалов, из которых легко выбрать наиболее подходящий вариант для эффективного выделения биополимеров из конкретного биологического образца (рис. 4).
Рис. 4.
Примеры БЭ: 1 – спин-картриджи с ПАНИ сорбентом, 2 – двухслойная концентрирующая спин-колонка (верхний очищающий слой – ФП-сорбент, нижний концентрирующий слой – гранулы сшитого полиакриламида), 3 – стеклянные мультикапилляры, покрытые нанослоем ПАНИ, впаянные в наконечник для механического дозатора, 4 – ПАНИ-модифицированная мембрана ММК-2, размещенная в картридже, 5 – кремниевые пластины с 3-АБК-ПАНИ-покрытием для масс-спектрометрии.

Так, использование стеклянных мультикапилляров (МК) в качестве носителей при получении ПАНИ-модифицированных сорбентов обеспечивает возможность контролировать точный объем анализируемой пробы (что особенно важно при количественном выделении). Промодифицировать поверхность МК анилинсодержащими покрытиями можно двумя способами. Во-первых, заполняя объем МК реакционной смесью и проводя полимеризацию анилина, протекающую как на внутренней поверхности стенок капилляров, так и в их объеме. Частицы полимера, слабо связанные с покрытием, легко удаляются промывкой. Во-вторых, в качестве полимерного модификатора можно использовать растворы сополимеров анилина с замещенными анилинами подходящего состава. Модифицированные МК могут быть впаяны в стандартные пластиковые наконечники для ручных или автоматических дозаторов. Для работы с наконечниками не требуется дополнительного оборудования (центрифуги, насосы и пр.). Эти наконечники также можно присоединять к шприцам, обеспечивая работу с бóльшими объемами образца [198].
В [199] в качестве подложек при получении ПАНИ-модифицированных сорбентов использовали синтетические мембраны МФФК (фторопластовая гидрофобная), МФФК-Г (фторопластовая гидрофилизованная), ММК (микрофильтрационная полиамидная), МКМ (капроновая), ММПА+ (нейлоновая с положительным поверхностным зарядом), ПА-66 (полигексаметиленадипинамидная) и МПС (полиэфирсульфоновая). Важно подчеркнуть, что модифицированную ПАНИ-покрытием мембрану в составе БЭ использовали не в качестве фильтра, а в качестве селективного сорбента (в скрученном особым образом виде), благодаря чему не происходила фильтрация образца через мембрану, но обеспечивался максимальный контакт образца с поверхностью мембраны.
3.2. Применение ФП- и ПАНИ-содержащих композитов в молекулярной диагностике (одностадийное выделение НК)
Способы получения ФП- и ПАНИ-содержащих композитов разрабатывались в первую очередь с целью реализации эффекта “негативной селекции” в отношении НК, демонстрируемого включенными в их состав полимерами. За счет высокой сорбционной емкости и селективности ФП- и ПАНИ-содержащих сорбентов удалось разработать схему одностадийного разделения смесей НК и белков без изменения состава элюента или даже в отсутствие такового (рис. 5). С помощью одноразовых картриджей с ПТФЭ-, ПФБД-, ПАНИ- или ФП-ПАНИ-сорбентами обеспечено эффективное одностадийное выделение ДНК из лизатов грамположительных и грам-отрицательных бактериальных культур, пищевых продуктов, косметических препаратов [194, 200].
Рис. 5.
Схема одностадийного выделения НК и последующего выделения белков (пептидов) с помощью разработанных сорбентов.
При использовании ФП-содержащих сорбентов с дополнительно введенными функциональными группами, полученных на основе озонированного кремнезема, удалось разделить смеси НК, различающихся вторичной структурой, в частности двунитевую ДНК (днДНК) и РНК из лизатов A. tumifaciens C58, а также разделить компоненты смеси синтетических олигонуклеотидов и фрагментов днДНК [194, 197].
В клинической ПЦР-диагностике весьма эффективной оказалась пробоподготовка с использованием ПАНИ-сорбентов. В [196] выделяли ДНК двух групп микроорганизмов, включающих в себя Gardnerella vaginalis и Candida albicans, а также Chlamydia trachomatis, Ureaplasma urealyticum, Mycoplasma hominis и Mycoplasma genitalium, сравнивая эффективность одностадийной пробоподготовки с помощью ПАНИ-сорбента и стандартной многостадийной методики. Выделенную ДНК использовали в ПЦР-анализе. Чистота ДНК, выделенной с помощью быстрого одностадийного протокола, оказалась значительно выше, чем по стандартному многостадийному протоколу. Высокая эффективность применения ФП-ПАНИ-сорбентов в диагностике патогенов человека подтверждена результатами выделения ДНК из Mycobacterium tuberculosis complex. В [201] сравнивали количество ДНК, выделенной из образцов мокроты больных туберкулезом на сорбентах и с помощью роботизированной станции для выделения и амплификации Tecan Freedom EVO® PCR (Швейцария). Количество копий ДНК при выделении с помощью ФП-ПАНИ-сорбента во всех случаях значительно превышало количество копий при автоматическом выделении (максимально в 33 раза).
Важным объектом исследования является облигатный аэроб – палочковидная агробактерия Agrobacterium tumefaciens C58, инфицирующая клетки растений (а в ряде случаев опасная для ВИЧ-инфицированных людей). Установлено [195, 197], что одностадийная процедура выделения ДНК из агробактерии с использованием ФП-ПАНИ-сорбента обеспечивает значительно более высокий выход ДНК с одновременным отсутствием РНК в получаемых элюатах по сравнению с многостадийным протоколом.
Разработанные сорбенты также эффективны при выделении вирусной ДНК. Так, с помощью ФП-ПАНИ-сорбента выделяли днДНК вируса гепатита В человека из плазмы крови пяти групп пациентов, различающихся концентрацией вируса. Выделенную ДНК использовали в ПЦР. Показано, что материал одинаково эффективен при выделении ДНК возбудителя из образцов с высоким и низким содержанием вирусной ДНК. Для оценки эффективности применения ФП-ПАНИ-сорбента при выделении однонитевой ДНК (онДНК) выделяли преимущественно циклическую онДНК из вируса гепатита TTV, содержащегося в образцах плазмы крови [202]. Таким образом, с помощью ФП-ПАНИ-сорбентов удается эффективно выделять вирусную ДНК с различной третичной структурой.
В ряде работ показано, что поверхность ПАНИ-содержащих сорбентов эффективно связывает металлсодержащие соединения (ингибиторы ПЦР), такие как гем и хлорофиллы. Поэтому при их использовании не требуется предварительно удалять гемоглобин и его фрагменты (при выделении ДНК из лизатов крови) или хлорофиллы (при выделении ДНК из лизатов тканей растений). Образцы ДНК, полученные при пропускании лизатов цельной крови животных (коров и свиней) и лизатов листьев табака Nicotiana Tabacum L. через картриджи с ПАНИ-содержащими сорбентами, не содержали гема или хлорофиллов [200]. Высокую связывающую способность в отношении хлорофиллов демонстрируют изо-ПСК-ПАНИ-сорбенты [193, 200]. ФП-ПАНИ-сорбент эффективен при одновременном выделении ДНК из тканей растений и грибов, что доказано результатами одностадийного выделения ДНК фитопатогенного гриба (Fusarium graminearum) как из зараженных семян пшеницы, так и непосредственно из мицелия гриба [195].
Кроме того, ФП-ПАНИ-сорбент эффективен при очистке ПЦР-фрагментов (ампликонов) от примесей, присутствующих в реакционной смеси после проведения ПРЦ, таких как Taq-полимераза (сорбентом удерживается свыше 95%), дНТФ (до 84%), олигонуклеотиды (до 100%) и флуорофоры (до 100%) [197]. Очищенные ампликоны можно использовать для клонирования генов, секвенирования НК и т.д.
3.3. Применение ФП- и ПАНИ-содержащих композитов в смежных областях бионанотехнологии и в биоаналитике
Использование частично фторированных полимеров с активными функциональными группами в качестве модификаторов поверхности позволило проводить иммобилизацию биолигандов. Разработан носитель для твердофазного синтеза олигонуклеотидов на основе пористого кремнезема, предварительно покрытого нанотолщинным слоем сополимера этилена с метил-3-(1-(дифтор-((трифторэтенил)-окси)-метил)-1,2,2,2-тетрафторэтокси)-2,2,3,3-тетрафторпропанатом (EVE; Du Pont, США). Этот сополимер содержит активные эфирные группы, что позволяет использовать их в качестве “якоря” для иммобилизации биолигандов. Наличие фторированных групп снижает неспецифическую сорбцию синтезируемых олигонуклеотидов на поверхности носителя (благодаря эффекту “негативной селекции”). На поверхности EVE-модифицированного КЗ иммобилизовали остатки нуклеозида DMT-dT-SNPE за счет взаимодействия с якорными группами. Поверхностная концентрация нуклеозидных остатков составила ~40 мкмоль/г сорбента. Материал сохранял свои свойства при длительном хранении при комнатной температуре [194, 195].
КЗ-носители, поверхностно модифицированные ФП-42Л, проявляют себя как мягкие обращено-фазные сорбенты (проявляющие как олео-, так и гидрофобные свойства в зависимости от состава используемого элюента). Таким образом, их можно использовать не только для выделения биополимеров, но и для выделения низкомолекулярных биологически активных веществ, различающихся гидрофильно-липофильным балансом. Разработан воспроизводимый метод одновременного выделения производных пяти водорастворимых (С, В1, В5, В6, В12) и четырех жирорастворимых (А, D3, E, K) витаминов (витамеров) из одной пробы крови (не более 10 мл). Из образца получали и концентрировали сыворотку, затем проводили жидкофазную экстракцию неполярным растворителем. Органическую фазу упаривали, а сухой остаток растворяли в метаноле. Полученный раствор, как и водную фазу, независимо пропускали через картриджи с ФП-сорбентом, получая элюаты, содержащие соответственно смесь жиро- и смесь водорастворимых витамеров. Элюаты анализировали с помощью ВЭЖХ [203].
Выше представлены примеры использования полимерсодержащих композитов, полученных на основе пористых неорганических и органических носителей. В биоаналитике широко используются также плоские матрицы, которые служат типичными “рабочими телами” биочипов, биосенсоров и ряда других диагностических устройств.
Введение дополнительных функциональных групп в ПАНИ-покрытия открывает новые возможности для их использования в биоаналитике. В [204] показано, что разделение смесей белков (пептидов) можно проводить непосредственно на кремниевой пластине (чипе), покрытой нанослоем сополимера 3-АБК-ПАНИ. Белки с различными рI предварительно метили люминесцентными полупроводниковыми нанокристаллами (CdSe)ZnS. Смесь меченых белков наносили на поверхность модифицированного чипа, систему промывали раствором с определенным рН, удерживание белков на пластине визуализировали в УФ-свете. На пластине удерживались белки с pI < pH отмывочного раствора. Эти пластины использовали в качестве подложек для нанесения образца и его обогащения аналитом при проведении масс-спектрометрии. Было обнаружено, что анилинсодержащее покрытие обратимо абсорбирует лазерную энергию, что позволяет использовать чипы с таким покрытием в масс-спектрометрии в формате SELDI-TOF-MS без добавления к пробе “веществ-матриц” (ароматических кислот), поглощающих энергию лазера и способствующих переводу аналита в газовую фазу. Присутствие “вещества-матрицы” приводит к появлению значительного фонового сигнала в низкомолекулярной части масс-спектра. Напротив, при использовании чипов, покрытых сополимером анилина с 3-АБК для анализа пептидов (брадикинина и др.), фоновые пики в масс-спектре не наблюдали (рис. 6).
Рис. 6.
Определение брадикинина методом SELDI-TOF-MS на кремниевых чипах, модифицированных сополимером анилина с 3-АБК.
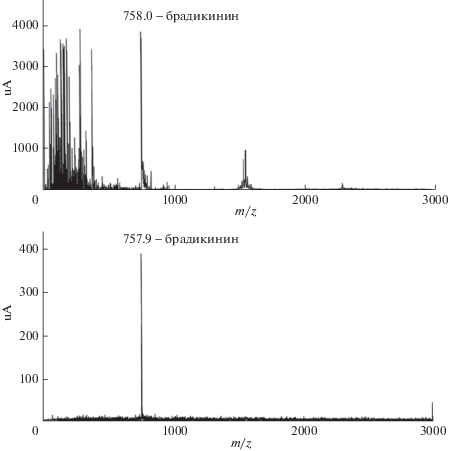
Было показано, что покрытие на основе сополимера анилина с 3-АБК обладает высокой “матричной” активностью и обеспечивает образование протонированных молекул ([M+H]+). Таким образом, один и тот же композитный материал обеспечивает удаление примесей из пробы и одновременно повышает информативность анализа за счет существенного упрощения структуры масс-спектра.
ЗАКЛЮЧЕНИЕ
Стремительное развитие молекулярной диагностики обусловлено широким применением методов современной нанобиотехнологии, а также разработкой новейших сорбционных композитных материалов. Можно утверждать, что переход от использования немодифицированных неорганических носителей к полимерсодержащим композитам (через использование матриц, модифицированных низкомолекулярными соединениями) позволил не только повысить эффективность диагностических методов, но и расширить область применения таких материалов. Использование нетрадиционных полимерных модификаторов с комплексом особых свойств позволило изменить сам принцип выделения биополимеров из биологических смесей сложного состава. Иными словами, стал возможен переход от использования материалов, демонстрирующих эффект “позитивной селекции” в отношении НК, к применению материалов и методов, основанных на использовании эффекта “негативной селекции”. В результате стало возможным осуществлять одностадийные процедуры выделения биополимеров (в частности, НК) из биологических смесей (в отличие от традиционных многостадийных методов).
Использование ФП- и ПАНИ-содержащих сорбентов не только обеспечивает одностадийное выделение НК из разнообразных биологических объектов в процессе пробоподготовки в молекулярной диагностике, но и позволяет решать особые задачи, такие как иммобилизация биолигандов, выделение низкомолекулярных биологически активных соединений, разделение смеси НК, различающихся третичной структурой и др. Наконец, композиты с нанопокрытиями на основе сополимеров анилина с замещенными анилинами эффективны в качестве рабочих тел в масс-спектрометрии.
Таким образом, полимерсодержащие композиты являются полезным и эффективным инструментом молекулярной биотехнологии, а область их применения постоянно расширяется.
Список литературы
Glick B.R., Pasternak J.J. Molecular Biotechnology Principles and Applications of Recombinant DNA. 3rd ed. USA: ASM Press, 2005. 584 p.
Ребриков Д.В., Саматов Г.А., Трофимов Д.Ю. и др. ПЦР в реальном времени / Под ред. Ребрикова Д.В. М.: БИНОМ, 2009. 223 с.
Miescher F. // Medicinisch-chemische Untersuchungen. 1871. V. 4. P. 441.
Meselson M., Stahl F.W. // Proc. Natl. Acad. Sci. USA. 1958. V. 44 (7). P. 671.
Sambrook J., Russel D.W. Molecular Cloning: A Laboratory Manual. 3rd ed. NY: Cold Spring Harbor Laboratory Press, 2001. 2100 p.
Marko M.A., Chipperfield R., Birnboim H.C. // Anal. Biochem. 1982. № 121. P. 382.
Chomczynski P., Sacchi N. // Nature Protocol. 2006. № 1 (2). P. 581.
Albertson P.-A. Partion of cell and macromoleculs. Separation and purification of biomolecules, cell organelles, membranes and cell in aqueous polymer two-phase systems and their use in biochemical analysis and biotechnology. 3rd Ed. NY: J. Wiley & Sons. 1986. 346 p.
Carr S.M., Griffiths O.M. // Biochem. Genet. 1987. № 25. P. 385.
Perez-Ramirez B., Steckert J.J. // Therapeutic Proteins: Methods and Protocols. 2005. NJ: Humana Press, V. 308. P. 301.
Noles S.R. // Thermo Scientific, application note AN-LECF-PLASDNA-0408. 2008. P. 1.
Crouse J., Amorese D. // Focus. 1987. № 9 (2). P. 3.
Zeugin J.A., Hartley J.L. // Focus. 1985. № 7 (4). P. 1.
Paithankar K.R., Prasad K.S. // Nucl. Acids Res. 1991. № 19 (6). P. 1346.
Janson J.-Ch. Protein Purification: Principles, High Resolution Methods, and Applications. 3rd edition. NJ: John Wiley & Sons, Inc. 2011. 548 p.
Rother D., Sen T., East D., Bruce I.J. // Nanomedicine. 2011. № 6 (2). P. 281.
Tan S.C., Yiap B.C. // J. Biomed. Biotechnol. 2009. № 574398. P. 1.
Berensmeier S. // Appl. Microbiol. Biotechnol. 2006. V. 73 (3). P. 495.
Антонова О.С., Корнева Н.А., Белов Ю.В., Курочкин В.Е. // Научное приборостроение. 2010. Т. 20. № 1. С. 3.
Kong M.Li, Laurent P.R., Stephen J.C. // Curr. Pharm. Analysis. 2006. V. 2 (2). P. 95.
Durhan E.J., Lukasewycz M.T., Amato J.R. // Environ. Toxicol. Chem. 1990. V. 9 (4). P. 463.
Остерман Л.А. Хроматография белков и нуклеиновых кислот. М.: Наука, 1985. 536 с.
Kuske C.R., Banton K.L., Adorada D.L. et al. // Appl. Environ. Microbiol. 1998. V. 64 (7). P. 2463.
Yamamoto N., Matsuzaka Y., Kimura M. et al. // J. Biosci. Bioeng. 2009. V. 107 (4). P. 464.
Heller M.J., Robinson R.A., Burgart L.J. et al. // Modern Pathology. 1992. V. 5 (2). P. 203.
Marentis T.C., Kusler B., Yaralioglu G.G. et al. // Ultrasound Med. Biol. 2005. V. 31. P. 1265.
Abdel-Latif A., Osman G. // Plant Methods. 2017. № 13:1. https://plantmethods.biomedcentral.com/track/pdf/10.1186/s13007-016-0152-4
Zhou J., Bruns M.A., Tiedje J.M. // Appl. Environ. Microbiol. 1996. V. 62. P. 316.
Sekikawa T., Kawasaki Yu., Katayama Y. // New Biotechnol. 2011. V. 29 (1). P. 139.
Chacon-Cortes D., Griffiths L.R. // J. Biorepos. Sci. Appl. Med. 2014. V. 2. P. 1.
Braid M.D., Daniels L.M., Kitts C.L. // J. Microbiol. Methods. 2003. V. 52 (3). P. 389.
Elkins K.M. DNA Extraction. Forensic DNA Biology: A Laboratory Manual. USA: Academic Press, 224 p.
Ikuta K., Maruo S., Fujisawa T., Yamada A. MEMS’99: Twelfth IEEE international conference on micro electro mechanical systems, technical digest. Orlando. 1999. P. 376.
Cabrera C.R., Yager P. // Electrophoresis. 2001. V. 22 (2). P. 355.
Chockalingam P.S., Jurado L.A., Jarrett H.W. // Mol. Biotech. 2001. V. 19 (2). P. 189.
Smith J.S., Johnson F.B. // Methods Mol. Biol. V. 608. P. 207.
Xiang F., Lin Y.H., Wen J. // Anal. Chem. 1999. V. 71. P. 1485.
Goloub T.P., Koopal L.K., Bijsterbosch B.H., Sido-rova M.P. // Langmuir. 1996. V. 12 (13). P. 3188.
Nelson D.L., Cox M.M. Lehninger Principles of Biochemistry. USA: W.H. Freeman and Company. 2008. 1130 p.
Gelbart W.M., Bruinsma R.F., Pincus P.A., Parsegian V.A. // Phys. Today. 2000. V. 53 (9). P. 38.
Fogolari F., Brigo A., Molinari H. // J. Mol. Recognit. 2002. V. 15 (6). P. 377.
Melzak K.A., Sherwood C.S., Turner R.F.B., Hay-nes C.A. // J. Colloid Interface Sci. 1996. V. 181 (2). P. 635.
Beld M., Sol C., Goudsmit J., Boom R. // Nucl. Acids Res. 1996. V. 24 (13). P. 2618.
Петров Д.Г., Макарова Е.Д., Корнева Н.А. др. // Научное приборостроение. 2015. Т. 25. № 2. С. 91.
Magnani M., Galuzzi L., Bruce I.J. // J. Nanosci. Nanotech. 2006. V. 6 (8). P. 2302.
Sen T., Bruce I.J. // Microporous Macroporous Mater. 2009. V. 120 (3). P. 246.
Taylor J.I., Hurst C.D., Davies M.J. et al // J. Chromatogr. A. 2000. V. 890 (1). P. 159.
Odenbach S. Colloidal Magnetic Fluids: Basics, Development and Application of Ferrofluids. Germany: Springer, 2009.
Bruce I.J., Taylor J., Todd M. et al. // J. Magn. Magn. Mater. 2004. V. 284. P. 145.
Amagliani G., Brandi G., Omiccioli E. et al. // Food Microbiol. 2004. V. 21 (5). P. 597.
Sebastianelly A., Sen T., Bruce I.J. // Lett. Appl. Microbiol. 2008. V. 46 (4). P. 488.
Bertozzini E., Penna A., Pierboni E. et al. // J. Appl. Phycol. 2005. V. 17. P. 223.
Henco K., Stichel A., Colpan M. Patent US 5057426 A. 1991.
Smith C.E., Holmes D.L., Simpson D.J. et al. Patent US 6310199 B1. 2001.
Reitan E.H., Deggerdal A., Skagestad V. Patent WO2004/108925. 2004.
http://www.rosmed.ru/scatalog/show/7860/PCR_analizator_Cobas_AmpliPrep
https://www.qiagen.com/us/resources/resourcedetail?id=15606127-cc31-4681-a27a-bcec58502f72&lang=en
Kruhøffer M., Dyrskjøt L., Voss T. // J. Mol. Diagn. 2007. V. 9 (4). P. 452.
https://www.thermofisher.com/ru/ru/home/life-science/dna-rna-purification-analysis/automated-purification-extraction/kingfisher-instruments.html
https://www.labortechnik.com/en/pipetting-robot/x-tractor-gene
McNeil J., Ecker D., Hofstadler S.A. et al. Patent WO/2005/009202. 2005.
Baggio R.F., Gagne Jr. G.A. Patent EP 1873242A3. 2008.
Bridenbaugh R., Dang W., Bussey L. Patent WO/2000/005358. 2000.
Stevenson A., Potts A., Donovan D., Baker M. Patent WO/2005/012521. 2005.
Wen J., Legendre L.A., Bienvenue J.M., Landers J.P. // Anal. Chem. 2008. V. 80. P. 6472.
Boom R., Sol C.J., Salimans et al. // J. Clin. Microbiol. 1990. V. 28 (3). P. 495.
McCormick R.M. // Anal. Biochem. 1989. V. 181. P. 66.
Christel L.A., Petersen K., McMillan W., Northrup M.A. // J. Biomech. Eng-T. ASME. 1999. V. 12. P. 22.
Cady N.C., Stelick S., Batt C.A. // Biosens. Bioelectron. 2003. V. 19 (1). P. 59.
Hindson B., Gutierrez D.M., Ness K.D. et al. // Analyst. 2008. V. 133 (2). P. 248.
Tian H.J., Huhmer A.F.R., Landers J.P. // Anal. Bioch. 2000. V. 283 (2). P. 175.
Wolfe K.A., Breadmore M.C., Ferrance J.P. et al. // Electrophoresis. 2002. V. 23 (5). P. 727.
Breadmore M.C., Wolfe K.A., Arciba I.G. et al. // Anal. Chem. 2003. V. 75 (8). P. 1880.
Bienvenue J.M., Duncalf N., Marchiarullo D. et al. // J. Forensic Sci. 2006. V. 51 (2). P. 266.
Wu Q., Bienvenue J.M., Hassan B.J. et al. // Anal. Chem. 2006. V. 78 (16). P. 5704.
Chung Y.C., Jan M.S., Lin Y.C. et al. // Lab. Chip. 2004. V. 4 (2). P. 141.
https://www.sigmaaldrich.com/catalog/product/mm/ZTC18S?lang=en®ion=RU
Svec F., Frechet J.M.J. // Anal. Chem. 1992. V. 64 (7). P. 820.
Moshimara K., Bennett B.D., Dulay M.T. et al. // J. Sep. Sci. 2002. V. 25 (15–17). P. 1226.
Wen J., Guillo C., Ferrance J.P., Landers J.P. // Anal. Chem. 2006. V. 78 (5). P. 1673.
Wen J., Guillo C., Ferrance J.P., Landers J.P. // Anal. Chem. 2007. V. 79 (16). P. 6135.
Bhattacharyya A., Klapperich C.M. // Anal. Chem. 2006. V. 78 (3). P. 788.
Тенникова Т.Б. Дис. “Высокоэффективная мембранная хроматография: формирование структуры порового пространства полимерных носителей для межфазового распределения белков”… д-ра хим. наук. М.: МГУ, 1998.
Синицына Е.С., Власова Е.Н., Влах Е.Г., Тенникова Т.Б. // Журн. прикл. химии. 2008. Т. 81. № 8. С. 1326.
Синицина Е.С. Дис. “Полимерные монолитные материалы для биочипов с контролируемой пористостью и различными реакционноспособными группами”… канд. хим. наук. С.-Петербург: Институт высокомолекулярных соединений РАН, 2014.
Nakagawa T., Tanaka T., Niwa D. et al. // J. Biotechnol. 2005. V. 116 (2). P. 105.
Ogawa R., Kaji N., Hashioka S. et al. // Jpn J. Appl. Phys. Pt 1. 2007. V. 46 (4B). P. 2771.
Cao W.D., Christopher C.J., Ferrance J.P., Landers J.P. // Anal. Chem. 2006. V. 78 (20). P. 7222.
Witek M.A., Lopis S.D., Shawn D. et al. // Nucl. Acids Res. 2006. V. 34 (10). Article number: e74.
Witek M.A., Hupert M.L., Park D. S.-W. et al. // Anal. Chem. 2008. V. 80 (9). P. 3483.
Kim J., Voelkerding K.V., Gale B.K. // J. Micromech. Microeng. 2006. V. 16 (1). P. 33.
Jiang G.F., Harrison D.J. // Analyst. 2000. V. 125 (12). P. 2176.
Satterfield B.C., Stern S., Caplan M.R. et al. // Anal. Chem. 2007. V. 79 (16). P. 6230.
Belgrader P., Yuan B., Aflatooni N. Patent WO/2005/028635. 2005.
Ugozzoli L. Patent US 20080003585A1. 2008.
Weigl B.H., Bardell R.L. Patent WO/2006/081324. 2006.
Landers J.P., Ferrance J.P., Wen J., Guillo C. Patent WO/2008/058204. 2008.
Partha Sarathy R.V., Ericson K.K., Bedingham W. Patent WO/2005/068626. 2005.
Samper V., Hongmiao J.I., Chen Yu. et al. Patent WO/2005/066343. 2005.
Southgate P.D., Loewy Z.G. Patent WO/1997/047967. 1997.
Jovanovich S.B., Blaga I.I., Rank D. Patent WO/2008/030631. 2008.
Barlag H., Birkle S., Gumbrecht W. et al. Patent WO/2006/042838. 2006.
Battrell C.F., Weigl B.H., Shen M. et al. Patent WO/2004/065010. 2004.
Chen Z., Mauk M.G., Wang J. et al. // Ann. N.Y. Acad. Sci. 2007. V. 1098. P. 429.
Inoue H., Knight I.T., Dale G.A., Colwell R.R. Patent WO/2007/028084. 2007.
Cady N.C., Batt C.A., Stelick S.J. et al. Patent WO/2006/085948. 2006.
Liu R.H., Lodes M.J., Nguyen T. et al. // Anal. Chem. 2006. V. 78 (12). P. 4184.
Liu R.H., Nguyen T., Schwarzkopf K. et al. // Anal. Chem. 2006. V. 78 (6). P. 1980.
Hiatt C.W., Shelokov A., Rosental E.J., Galimore J.M. // J. Chromatogr. 1971. V. 56. P. 362.
Darling T., Albert J., Russel P. et al // J. Chromatogr. 1977. V. 131. P. 383.
Letot L., Lesec J., Quivoron C. // J. Liq. Chromatogr. 1981. V. 4. P. 1311.
Копьев В.П., Жигис Л.С., Решетов П.Д. и др. // Вопр. вирусологии. 1989. Т. 34. № 6. С. 760.
Tardy M., Tayot J.L., Roumyantseff M. et al. Chromatography of Synthetic and Biological Polymers. V. 2. Chichester: Ellis Horwood, 1978. 236 p.
Santarelli X., Muller D., Jozefonvicz J. // J. Chromatogr. 1988. V. 443. P. 55.
Букбарде В.У., Арен А.К., Грузинь И.В. и др. Авт. свид. СССР № 935121. 1980.
Rosevear A., Mattock P. Patent US 1602432. 1981.
Alpert A., Regnier F.E. // J. Chromatogr. 1979. V. 185. P. 375.
Lawson T.G., Regnier F.E., Weith H.L. // Anal. Biochem. 1983. V. 133 (1). P. 85.
Watanabe K., Chow W.S., Royer G.P. // Anal. Biochem. 1982. V. 127 (1). P. 155.
Drager R.R., Regnier F.E. // Anal. Biochem. 1985. V. 145 (1). P. 47.
Wilchek M. // FEBS Lett. 1973. V. 33 (1). P. 70.
Kurganov A., Kuzmenko O., Davankov V.A. et al. // J. Chromatogr. 1990. V. 506. P. 391.
Gupta S., Pfannkoch E., Regnier F.E. // Anal. Biochem. 1983. V. 128 (1). P. 196.
Alpert A. // J. Chromatogr. 1983. V. 266. P. 23.
Elrassi Z., Horvath C. // J. Liq. Chromatogr. 1986. V. 9 (15). P. 3245.
Papirer E., Nguyen V.T. // J. Polym. Sci. B: Polym. Lett. 1972. V. 10. P. 167.
Иванов А.Е., Верховская Л.В., Хилько С.Н., Зубов В.П. // Биоорган. хим. 1990. Т. 16. № 8. С. 1028.
Иванов А.Е., Жигис Л.С., Чеховских Е.Ю. и др. // Биоорган. химия. 1985. Т. 11. № 11. С. 1527.
Иванов А.Е., Жигис Л.С., Турчинский М.Ф. и др. // Мол. ген., микробиол. и вирусол. 1987. Т. 11. С. 39.
Таубман А.Б. В кн.: Матер. V Всесоюз. симп. по механоэмиссии и механохимии твердых тел. Окт. 1975. Т. 1. С. 79.
Berg D., Tiller H.J.G., Kopka J., Lanyguth B. // Z. Chem. 1978. V. 18 (6). P. 219.
Иванчев С.С. Радикальная полимеризация. Л.: Химия, 1985. 280 с.
Брык М.Т. Полимеризация на твердой поверхности неорганических веществ. Киев: Наукова думка, 1981. 288 с.
Цетлин Б.Л., Власов А.В., Бабкин И.Ю. Радиационная химия полимеров. М.: Наука, 1973. С. 108.
Брук М.А. // Успехи химии. 1987. Т. 56. № 1. С. 148.
Оленин А.В., Кристюк А.Л., Голубев В.Б. и др. // Высокомол. соед. А. 1983. Т. 25. С. 423.
Брук М.А., Абкин А.Д., Демидович В.В. и др. // Высокомол. соед. А 1975. Т. 17. С. 3.
Bleha M., Votavova E., Tlustakova M. et al. // Angew. Makromol. Chem. 1982. V. 107. P. 25.
Hayakawa K., Kawase K., Yamakita H. // J. Appl. Polym. Sci. 1977. V. 21 (11). P. 2921.
Abuelafiya R., Pesek J. // J. Liq. Chrom. 1989. V. 12 (9). P. 1571.
Schutijser J. US Patent № 4415631. 1983.
Варламов В.П., Власов А.В., Банникова Т.Е. и др. Авт. свид. СССР № 689200. 1980.
Huth J., Danielson N. // Anal. Chem. 1982. V. 54 (6). P. 930.
Siergiej R.W., Danielson N.D. // J. Chromatogr. Sci. 1983. V. 21 (8). P. 362.
Kruepelman M., Danielson N.D. // Anal. Chem. 1985. V. 57 (1). P. 340.
Berendsen G.E., Pikaart K.A., de Galan L., Olieman C. // Anal. Chem. 1980. V. 52 (12). P. 1990.
Billiet H.A.H., De Galan L. // J. Chromatogr. A. 1981. V. 218. P. 443.
Kobos R.K., Eveleigh J.W., Arentzen R. // Trends Biotechnol. 1989. V. 7 (4). P. 101.
Xindu G., Carr P.W. // J. Chromatogr. 1983. V. 269 (2). P. 96.
Williams D.E., Kabra P.M. // Anal. Chem. 1990. V. 62 (8). P. 807.
Pinkerton T.C. // J. Chromatogr. 1991. V. 544 (1–2). P. 13.
Сабуров В.В., Муйдинов М.Р., Гурьянов С.А. и др. // Журн. физ. химии. 1991. Т. 65. № 10. С. 2692.
Муйдинов М.Р. // Рос. хим. журн. (Журн. Рос. хим. о-ва им. Д.И. Менделеева). 2002. Т. XLVI. № 3. С. 72.
Муйдинов М.Р. // Рос. хим. журн. (Журн. Рос. хим. о-ва им. Д.И. Менделеева). 2008. Т. LII. № 3. С. 81.
Муйдинов М.Р. Дис. “Синтез и исследование композиционных материалов, модифицированных поверхностно привитым политетрафторэтиленом”… докт. хим. наук. М.: МИТХТ, 2006.
Муйдинов М.Р. Патент РФ 2104695. 1998.
Hjerten S. // J. Chromatogr. A. 1978. V. 159. P. 47.
Hjerten S., Hellman U. // J. Chromatogr. A. 1980. V. 202 (3). P. 391.
Ivanov A.E., Saburov V.V., Zubov V.P. // Adv. Polym. Sci. 1992. V. 104. P. 135.
Катаев А.Д. Дис. “Синтез композиционных политрифторстиролсодержащих сорбентов и их использование в хроматографии биополимеров”… канд. хим. наук. М.: МИТХТ, 1993.
Gorham W.F. // J. Polym. Sci. Pt 1 A.: Polym. Chem. 1966. V. 4 (12). P. 3027.
Park S.Y., Blackwell J., Chvalun S.N. et al. // Macromolecules. 1999. V. 32 (23). P. 7845.
Кобрянский В.М., Арнаутов С.А., Мотякин М.В. // Высокомол. соединения. А. 1995. Т. 37 (1). С. 35.
Сапурина И.Ю. Дис. “Наноструктурированный полианилин и композиционные материалы на его основе”… докт. хим. наук. С.-Петербург, 2015.
Омельченко О.Д. Дис. “Полимеризация анилина в присутствии полимерных сульфокислот: влияние конформации поликислоты на свойства комплексов полианилина”… канд. хим. наук. М., 2014.
Милакин К.А. Дис. “Структура и свойства поли-анилина, полученного в присутствии углеродных матриц”… канд. хим. наук. М., 2015.
Ягудаева Е.Ю. Дис. “Синтез полианилинсодержащих содержащих сорбентов для выделения и очистки нуклеиновых кислот и белков”… канд. хим. наук. М.: МИТХТ, 2013.
Riede A., Helmstedt M., Sapurina I., Stejskal J. // J. Colloid Interface Sci. 2002. V. 248 (2). P. 413.
Stejskal J., Sapurina I. // Materials Syntheses, A Practical Guide. Wien: Springer-Verlag, 2008. P. 199.
Trchova M., Sapurina I., Prokes J., Stejskal J. // Synth. Met. 2003. V. 135 (1–3). P. 305.
Ruckenstein E., Sun Y. // Synth. Met. 1995. V. 74 (2). P. 107.
Zhu C., Wang C., Yang L. et al. // Appl. Phys. A. 1999 V. 68 (4). P. 435.
Gregory R.V., Kimbrell W.C., Kuhn H.H. // Synth. Met. 1989. V. 28 (1–2). P. 823.
Megha R., Ravikiran Y.T., Chethan B. et al. // J. Mater. Sci: Mater. Electron. 2018. V. 29 (9). P. 7253.
Tzou K., Gregory R.V. // Synth. Met. 1992. V. 47 (3). P. 267.
Nicolau Y.F., Ermolieff A. // Synth. Met. 1995. V. 71 (1–3). P. 2073.
Орлов А.В., Киселева С.Г., Юрченко О.Ю., Карпачева Г.П. // Высокомол. соединения. А. 2000. Т. 42. № 12. С. 2089.
Киселева С.Г. Дис. “Полимеризация анилина в гетерофазной системе”… канд. хим. наук. М.: ИНХС, 2003.
Гусева М.А., Исакова А.А., Грибкова О.Л. и др. // Высокомол. соединения. А. 2007. Т. 49. № 1. С. 9.
Sun L.F., Liu H.B., Clark R., Yang S.C. // Synth. Met. 1997. V. 84 (1–3). P. 67
Liu W., Anagnostopoulos A., Bruno F.F. et al. // Synth. Met. 1999. V. 101. P. 738
Caramyshev A.V., Evtushenko E.G., Ivanov V.F. et al. // Biomacromolecules. 2005. V. 6 (3). P. 1360.
Кирш Ю.Э., Федотов Ю.А., Иудина Н.Н., Каталевский Е.Е. // Высокомол. соединения. Б. 1990. Т. 32. № 6. С. 403.
Кирш Ю.Э., Федотов Ю.А., Иудина Н.И. и др. // Высокомол. соединения. А. 1991. Т. 33. № 5. С. 1127.
Yagudaeva E.Yu., Liaw D.-J., Ischenko A.A. et al. // J. Mater. Sci. 2014. V. 49. P. 3491.
Liaw Der-Jang, Yagudaeva E., Prostyakova A. et al. // Colloids Surf. B. 2016. V. 145. P. 912.
Yagudaeva E., Zybin D., Vikhrov A. et al. // Colloids Surf. B. 2018. V. 163. P. 83.
Льяо Д.-Дж., Зыбин Д.И., Простякова А.И. и др. // Изв. вузов. Химия и химическая технология. 2018. Т. 61. № 1. С. 422.
Zubov V.P., Kapustin D.V., Generalova A.N. et al. // Polym. Sci. A. 2007. V. 49 (12). P. 1247.
Капустин Д.В., Сабуров В.В., Завада Л.Л. и др. // Биоорган. химия. 1998. Т. 24. № 11. С. 868.
Капустин Д.В. Дис. “Синтез композиционных сорбентов на основе фторированных полибутадиенов для выделения нуклеиновых кислот”… канд. хим. наук. М., 2000.
Yagudaeva E.Yu., Muydinov M.R., Kapustin D.V., Zubov V.P. // Rus. Chem. Bull. Int. Ed. 2007. V. 56 (6). P. 1166.
Yagudaeva E.Yu., Bukina Ya.A., Prostyakova A.I. et al. // Polym. Sci. A. 2009. V. 51 (6). P. 675.
Kapustin D., Prostyakova A., Bryk Ya. et al. // Nanocomposites and polymers with analytical methods. Croatia: Intech, 2011. P. 83.
Простякова А.И. Дис. “Синтез полимерсодержащих сорбентов и их использование для одностадийного выделения ДНК”… канд. хим. наук. М., 2013.
Kapustin D.V., Prostyakova A.I., Ryazantcev D.Yu., Zubov V.P. // Nanomedicine. 2011. V. 6 (2). P. 241.
Капустин Д.В. Дис. “Фторполимер- и полианилинсодержащие композиты как эффективный инструмент молекулярной биотехнологии”… докт. хим. наук. М., 2020.
Скибина Ю.С., Белоглазов В.И., Тучин В.В. и др. Патент РФ № 2547597. 2015.
Капустин Д.В., Тарасов А.В., Простякова А.И. и др. Патент РФ № 2631934 C1. 2017.
Kapustin D.V., Yagudaeva E.Y., Zubov V.P. et al. // Frontiers in DNA Research. USA: Nova Science Publishers, 2006. P. 113.
Kapustin D.V., Prostyakova A.I., Alexeev Ya.I. et al. // Acta Naturae. 2014. V. 6 № 2 (21). P. 6.
Kapustin D.V., Prostyakova A.I., Zubov V.P. // Bioanalysis. 2014. V. 6 (7). P. 957.
Зайцева И.П., Серебрянский Е.П., Скальная М.Г., Капустин Д.В. // Вестник восстановительной медицины. 2014. Т. 2 (60). С. 62.
Vaczine-Shlosser G., Ribbing C., Bachman P.K. et al. Patent WO 2011004308 (A1). 2011.
Дополнительные материалы отсутствуют.
Инструменты
Российские нанотехнологии