

Нефтехимия, 2020, T. 60, № 1, стр. 89-100
Дезактивация промышленного Cо–Al2O3/SiO2-катализатора при высоком давлении и циркуляции газа в синтезе Фишера–Тропша
А. П. Савостьянов 1, Р. Е. Яковенко 1, *, Г. Б. Нарочный 1, И. Н. Зубков 1, С. И. Сулима 1, В. Н. Соромотин 1, С. А. Митченко 1, 2
1 Южно-Российский государственный политехнический университет (НПИ) им. М.И. Платова
346428 Ростовской обл., Новочеркасск, Россия
2 Институт физико-органической химии и углехимии им. Л.М. Литвиненко
83114 Донецк, Украина
* E-mail: jakovenko@lenta.ru
Поступила в редакцию 05.07.2019
После доработки 04.09.2019
Принята к публикации 09.09.2019
Аннотация
Представлены результаты исследования процесса синтеза углеводородов (УВ) из СО и Н2 на промышленном Co–Al2O3/SiO2-катализаторе в течение 1000 ч при 6.0 МПа и кратностях циркуляции реакционного газа в диапазоне 2.2–6.0. Скорость дезактивации катализатора в этих условиях зависит от селективности по синтетическим длинноцепочечным УВ (СДУ) С35+. Повышение содержания СДУ в продуктах от 27.6 до 39.8% увеличивает скорость дезактивации катализатора в три раза. С ростом кратности циркуляции от 2.2 до 6.0 при давлении 6.0 МПа увеличивается содержание олефинов в продуктах синтеза. При повышении температуры наблюдается изменение селективности в сторону УВ с более короткой цепью. Вязкость длинноцепочечных углеводородов, конденсирующихся в порах, уменьшается за счет разбавления их более легкими УВ, что способствует увеличению количества доступных активных центров катализатора и его регенерации.
ВВЕДЕНИЕ
Синтез Фишера–Тропша (СФТ) является перспективным методом переработки углеродсодержащего сырья в ценные продукты, среди которых моторное топливо, масла, синтетические воски, сырье для нефтехимии [1, 2]. В качестве катализаторов для этого процесса используются контактные массы преимущественно на основе кобальта и железа [3, 4]. Кобальт считается лучшим активным компонентом для синтеза СДУ не только из-за его высокой активности и селективности по линейным парафинам С35+, но и за счет низкой активности в реакции водяного газа.
Современная промышленная технология СФТ, реализованная компаниями Shell, Sasol, направлена на преобразование синтез-газа (СО и Н2) преимущественно в СДУ (FT-waxes), так как это способствует его более полному использованию [6]. Для получения моторных топлив и базовых масел СДУ подвергают гидрооблагораживанию – гидрокрекингу и гидроизомеризации. СДУ также востребованы на рынке как отдельные продукты – их коммерческая стоимость значительно превышает стоимость жидких (С5–С18) дистиллятов [7].
Синтетические УВ с числом атомов углерода С35+ имеют температуру каплепадения более 105°С, в них отсутствуют ароматические и серосодержащие соединения, поэтому их используют в пищевой промышленности (водостойкая упаковка из бумаги, картона и ткани, одноразовая посуда, декоративные свечи, парафиновая оболочка для пищевых продуктов), в производстве косметических средств (кремы, губная помада, лосьоны), при получении термоплавких чернил, клеев, красок [8].
Кобальтовый катализатор представляет собой диспергированные частицы металлического Co на оксидной подложке из SiO2, Al2O3, TiO2 [9]. Дезактивация ведет к потере активных участков в дорогостоящем кобальтовом катализаторе, уменьшению производительности процесса и снижению эффективности технологии СФТ. Причины дезактивации кобальтовых катализаторов в течение многих лет являются предметом промышленного и научного интереса [10, 11].
Ранее, в работах [12–14], были представлены результаты исследований промышленного нанесенного кобальтсиликагелевого катализатора, промотированного оксидом алюминия, для получения УВ С35+. Комплексное изучение влияния разных технологических факторов (соотношения Н2/СО, давления, температуры, кратности циркуляции (Кц) реакционного газа) на получение УВ С35+ позволило определить оптимальные области проведения процесса СФТ. Одним из способов повышения селективности и производительности процесса по УВ С35+ является увеличение давления до 6.0 МПа и нагрузки по синтез-газу до 1000 ч–1 в условиях циркуляционного режима. Производительность процесса СФТ по УВ С35+ в этих условиях возросла почти в 13 раз (от 3.6 до 46.4 ${{{\text{кг}}} \mathord{\left/ {\vphantom {{{\text{кг}}} {{\text{(м}}_{{{\text{кат}}}}^{3}\,\,{\text{ч)}}}}} \right. \kern-0em} {{\text{(м}}_{{{\text{кат}}}}^{3}\,\,{\text{ч)}}}}$) в сравнении с ранее предложенным проточным режимом при давлении 2.0 МПа и нагрузке по синтез-газу 60–300 ч–1 [15, 16].
Однако при давлении 6.0 МПа катализатор оказался более склонен к дезактивации по сравнению с работой при умеренном давлении (2.0 МПа). Вероятной причиной быстрой дезактивации катализатора может быть блокировка активных центров высокомолекулярными углеводородами, которые затрудняют массоперенос реагентов к активной поверхности контакта [17]. В этой связи остались невыясненными временные характеристики дезактивации Co−Al2O3/SiO2-катализатора при высоком давлении (6.0 МПа) в течение длительного периода его эксплуатации. Такие исследования представляют значительный интерес с точки зрения оптимизации технико-экономических показателей промышленного производства СДУ и определения жизненного цикла катализатора.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Выбор катализатора
Для исследований был выбран Co−Al2O3/SiO2-катализатор синтеза СДУ, разработанный для опытно-промышленной установки СФТ получения церезина (УВ С35+) [12]. Катализатор содержит 20 мас. % Co и 1 мас. % промотирующей добавки оксида алюминия. Получают его методом пропитки силикагелевого носителя марки КСКГ (ГОСТ 3956-76, ООО “Салаватский катализаторный завод”, Россия) растворами солей нитратов Co и Al. Технология этого катализатора отработана в промышленных условиях на оборудовании катализаторной фабрики.
Методики исследований катализатора
Исследования каталитических свойств были выполнены на лабораторной проточно-циркуляционной установке (рис. 1а), которая позволяет: обеспечивать давление в реакционной зоне до 10.0 МПа; создавать электромагнитным насосом 19 циркуляцию реакционного газа с кратностью от 1 до 100; моделировать различные составы синтез-газа.
Рис. 1.
Схема лабораторной проточно-циркуляционной установки (а) и эскиз лабораторного реактора (б): 1, 2, 3, 21, 22 – регуляторы давления; 16 – редукционный клапан регулировки давления водяного пара; 4, 5, 6, 7, 20 – регуляторы массового расхода (MFC) “Bronkhorst”; 8, 15 – манометры; 9 – водяная рубашка; 10 – реактор; 11 – термопары; 12 – регулятор температуры; 13 – электрическая печь; 14 – паросборник; 17 – обогреваемый сепаратор парафинов; 18 – сепаратор легких углеводородов; 19 – электромагнитный циркуляционный насос; 22 – газовые часы. б – эскиз лабораторного реактора.
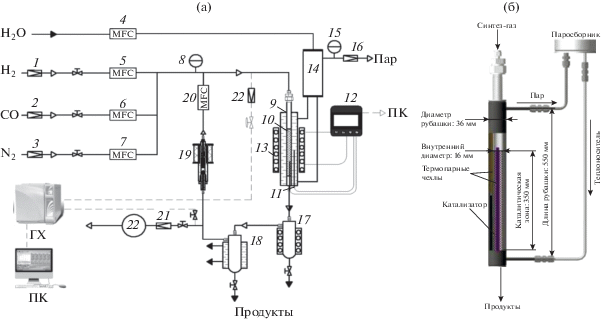
Катализатор объемом 15 см3 и фракцией 2–3 мм загружали в реактор 10, эскиз которого представлен на рис. 1б. Реактор оснащен водяной рубашкой 9 и коаксиально расположенным термопарным карманом. Измерение температуры в каталитическом слое осуществляли с помощью хромель-алюмелевой термопары. Регулирование температуры в реакционной зоне производили электропечью 13, регулятором температуры 12 и отведением избыточного реакционного тепла за счет фазового перехода воды в пар в рубашке 9. Параметры (Т, Р) кипения воды в рубашке 9 задавались с помощью редукционного клапана 16. Изотермичность в каталитическом слое достигалась разбавлением катализатора кварцевой крошкой в соотношении 1 : 2.
Исследование скорости дезактивации проводили на предварительно восстановленном и активированном катализаторе. Восстановление совершали в токе водорода в течение 1 ч при температуре 400°С и объемной скорости газа (ОСГ) 3000 ч–1, при этом степень восстановления кобальта, определенная волюмометрически по методике [18], составила ~50%. Активацию катализатора осуществляли в токе синтез-газа (Н2/СО = 1.85, Р = 0.1 МПа, ОСГ = 100 ч–1) до достижения степени конверсии СО, близкой к 50%. Затем, катализатор разрабатывали последовательно при 2.0 и 4.0 МПа (Кц = 2.2, ОСГ = 1000 ч–1) и конечных значениях конверсии СО 50–60% в течение 90–120 ч. После этого, из сборников 17, 18 удаляли продукты синтеза и, проводили испытания катализатора в непрерывном режиме при давлении 6.0 МПа в течение 1000 ч. Для сравнения физико-химических характеристик был испытан катализатор с продолжительностью работы 100 ч.
Испытания катализатора CДУ выполнены в трех режимах: 1-й – продолжительностью 500 ч (ОСГ = 1000 ч–1, Кц = 2.2, Н2/СО = 1.85); 2-й – продолжительностью 300 ч (ОСГ = 600 ч–1, Кц = 2.2, Н2/СО = 1.85); 3-й – продолжительностью 200 ч (ОСГ = 1000 ч–1, Кц = 6.0, Н2/СО = 1.85). После каждого режима производился слив продуктов. Степень конверсии СО в процессе синтеза УВ за весь период испытаний поддерживали в диапазоне значений 80–82%, а ее падение компенсировали повышением температуры.
Анализ состава газообразных продуктов синтеза выполняли методом газо-адсорбционной хроматографии на хроматографе марки Кристалл 5000, оснащенного детектором по теплопроводности и колонками Haysep R и молекулярные сита NaX. Первую колонку использовали для анализа УВ С1–С5 и СО2 (газ-носитель – гелий, расход – 15 мл/мин), вторую – для анализа СО, Н2, N2 (газ-носитель – аргон, расход – 15 мл/мин). Режим – температурно-программированный, 80–240°С, скорость нагрева 8°С/мин.
Фракционный состав жидких УВ определяли методом фракционирования, выделяя фракции углеводородов в зависимости от температуры кипения, в том числе с температурой кипения выше 450°С (церезин); групповой и индивидуальный состав – методом капиллярной газожидкостной хромато-масс-спектрометрии на газовом хроматографе Agilent GC 7890 с масс-селективным детектором MSD 5975С и капиллярной колонкой HP-5MS.
Для рентгенофазового анализа (РФА) катализаторов использовали специализированный источник синхротронного излучения “КИСИ-Курчатов” из Института им. Курчатова с длинной волны 0.793508, стандарт LaB6. Определение качественного фазового состава осуществляли с помощью PDF-2 [19] в программном комплексе Crystallographica.
Рентгеновские фотоэлектронные спектры (РФ-ЭС) катализаторов были получены с использованием рентгеновского фотоэлектронного микрозонда ESCA-LAB 250. Спектры возбуждались монохроматизированным излучением линии AlKα. Абсолютный разрешенный энергетический интервал соответствовал 0.6 эВ, что было определено по профилю линии Ag3d5/2, диаметр рентгеновского пятна на образце составлял 500 µм. Воздействие медленных электронов использовалось для удаления положительного заряда на образце. Линию C1s применяли для калибровки шкалы энергии для всех уровней всех элементов, используемых в этом исследовании.
Определение удельной поверхности проводили методом БЭТ с использованием анализатора Micromeritics ChemiSorb 2750. Предварительно образцы выдерживали в токе гелия в течение 1 ч при 200°С.
Исследование катализатора методом просвечивающей электронной микроскопии (ПЭМ) осуществляли на электронном микроскопе Hitachi HT7700. Съемку изображений вели в режиме регистрации прошедших электронов (режим светлого поля) при ускоряющем напряжении 100 кВ.
Микроструктуру поверхности образцов изучали методом сканирующей электронной микроскопии (СЭМ-ЭДС) на микроскопе Hitachi SU8000. Съемку изображений вели в режимах регистрации вторичных и отраженных электронов при ускоряющем напряжении до 15 кВ [20]. Оптимизация аналитических измерений проведена в рамках описанного ранее подхода [21].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Каталитические испытания
В процессе работы катализатора наблюдается плавное снижение активности во времени. Поэтому в промышленном синтезе падение степени конверсии СО обычно компенсируют повышением температуры. Мы использовали такой технологический прием в настоящей работе для поддержания конверсии СО на уровне ~80% (рис. 2). Повышение температуры производилось после падения степени конверсии СО ниже этого значения. Каталитические характеристики катализатора СДУ в разных технологических режимах приведены в табл. 1.
Рис. 2.
Временная диаграмма Хсо–T–τ испытаний Co–Al2O3/SiO2-катализатора за 1000 ч непрерывной работы для режимов: I – ОСГ = 1000 ч–1, Кц = 2.2; II – ОСГ = 600 ч–1, Кц = 2.2; III – ОСГ = 1000 ч–1, Кц = 6.0. 1 – конверсия СО; 2 – изменение температуры.
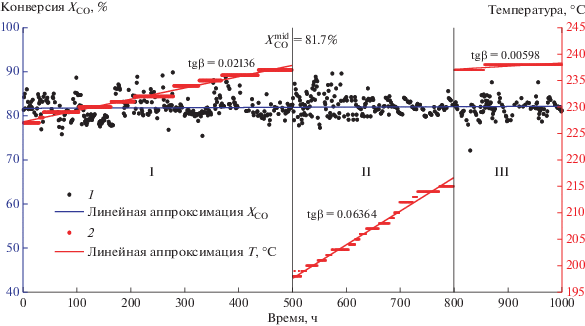
Таблица 1.
Каталитические характеристики Co−Al2O3/SiO2-катализатора при 6.0 МПа и различных условиях работы
| Режим | ОСГ, ч–1 | Интервал температур | Кц | τ, ч | Отношение H2/CO в реакционной циркулирующей смеси | XCO*, % | Селективность*, % | Производительность* по С5+, ${{{\text{кг}}} \mathord{\left/ {\vphantom {{{\text{кг}}} {\left( {{\text{м}}_{{{\text{кат}}}}^{3}\,\,{\text{ч}}} \right)}}} \right. \kern-0em} {\left( {{\text{м}}_{{{\text{кат}}}}^{3}\,\,{\text{ч}}} \right)}}$ | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | С2–С4 | С5+ | CO2 | ||||||||
| I | 1000 | 228–236 | 2.2 | 500 | 1.2 | 81.7 | 12.1 | 7.0 | 79.7 | 1.2 | 147.0 |
| II | 600 | 198–215 | 2.2 | 300 | 1.45 | 82.4 | 5.6 | 4.0 | 90.2 | 0.1 | 100.0 |
| III | 1000 | 236–238 | 6.0 | 200 | 0.84 | 81.7 | 10.0 | 5.7 | 81.7 | 2.6 | 154.0 |
С увеличением кратности циркуляции Кц от 2.2 до 6.0 и ОСГ от 600 до 1000 ч–1 растет производительность катализатора по УВ C5+. Селективность по газообразным углеводородам С1–С4 снижается, а по CO2 повышается.
Как видно из рис. 2, ступенчатое повышение температуры можно аппроксимировать линейной зависимостью (1):
Наклон линии аппроксимации (табл. 2) косвенно характеризует скорость дезактивации катализатора. Такая форма оценки скорости дезактивации катализатора позволяет определить время работы катализатора до его регенерации из уравнения прямой (1). Задаваясь предельной конечной температурой синтеза и зная начальную температуру процесса, можно оценить для выбранного технологического режима временной отрезок жизненного цикла катализатора при условии постоянства конверсии СО на этом временном отрезке. В табл. 2 приведены расчетные значения времени работы катализатора для начальных температур 228, 198 и 236°С и конечной температуры синтеза 250°С для I, II и III технологических режимов.
Таблица 2.
Расчетные значения рабочего времени катализатора синтеза СДУ
| Режим | Рабочий интервал температур ΔТ = (Ткон – Тнач), °С |
Значение a в уравнении (1) | Ожидаемое время работы катализатора, ч |
|---|---|---|---|
| I | 22 | 0.02136 | 1030 |
| II | 52 | 0.06364 | 817 |
| III | 14 | 0.00598 | 2341 |
Максимальное время работы (2341 ч) катализатора при давлении 6.0 МПа обеспечивают параметры процесса в условиях режима III, для которого зафиксирована и наибольшая производительность по УВ C5+ (табл. 1). Наименьшее значение производительности по УВ C5+ (100.0 ${{{\text{кг}}} \mathord{\left/ {\vphantom {{{\text{кг}}} {{\text{(м}}_{{{\text{кат}}}}^{3}\,\,{\text{ч)}}}}} \right. \kern-0em} {{\text{(м}}_{{{\text{кат}}}}^{3}\,\,{\text{ч)}}}}$) отмечено для режима II, где расчетное время эксплуатации катализатора составило 817 ч. Ожидаемое время работы катализатора в условиях I режима испытаний (ОСГ = 1000 ч–1, Кц = 2.2) равно 1030 ч, что соответствует почти половине времени эксплуатации (2341 ч) в режиме III. Для I и III технологических режимов значения производительности и селективности очень близки (147 и 154 ${{{\text{кг}}} \mathord{\left/ {\vphantom {{{\text{кг}}} {{\text{(м}}_{{{\text{кат}}}}^{3}\,\,{\text{ч)}}}}} \right. \kern-0em} {{\text{(м}}_{{{\text{кат}}}}^{3}\,\,{\text{ч)}}}}$ по УВ C5+; 27.6 и 29.1% по УВ C35+) соответственно для режимов I и III (табл. 1). Фракционный и групповой состав УВ, синтезированных в разных технологических режимах, приведен в табл. 3.
Таблица 3.
Фракционный и групповой состав углеводородов
| Режим | ОСГ, ч–1 | Кц | Группа углеводородов | Фракционный состав углеводородов, мас. % | α | о/п* | |||
|---|---|---|---|---|---|---|---|---|---|
| С5–С10 | С11–С18 | С19–С34 | С35+ | ||||||
| I | 1000 | 2.2 | н-Парафины | 7.4 | 30.6 | 23.3 | 27.6 | 0.929 | 0.12 |
| изо-Парафины | 0.5 | 0.00 | 0.00 | 0.00 | |||||
| Олефины | 3.4 | 6.9 | 0.3 | 0.0 | |||||
| II | 600 | 2.2 | н-Парафины | 21.87 | 21.26 | 2.73 | 39.80 | 0.934 | 0.12 |
| изо-Парафины | 1.94 | 0.00 | 0.00 | 0.00 | |||||
| Олефины | 11.99 | 0.32 | 0.09 | 0.00 | |||||
| III | 1000 | 6.0 | н-Парафины | 5.2 | 27.3 | 17.9 | 29.1 | 0.932 | 0.24 |
| изо-Парафины | 0.5 | 0.5 | 0.00 | 0.00 | |||||
| Олефины | 6.9 | 12.4 | 0.2 | 0.00 | |||||
Составы продуктов, полученных в технологических режимах I и III, близки с той лишь разницей, что при более высокой кратности циркуляции Кц = 6.0 в режиме III в продуктах синтеза зафиксировано большее количество олефинов и изо-парафинов, среди которых обнаружены и высокомолекулярные изо-парафины С11–С18. Показатель, характеризующий отношение содержания олефиновых УВ к парафиновым, вырос при этом с 0.12 до 0.24. Также при снижении нагрузки на катализатор по синтез-газу до ОСГ = 600 ч–1 (II режим, табл. 3) количество олефинов фракции С5–С10 возрастает. Для этих условий зафиксирована наивысшая селективность по УВ C35+ 39.8% (табл. 3), что отражается на величине α, характеризующей вероятность роста углеводородной цепи в уравнении Андерсона–Шульца–Флори, описывающего распределение продуктов в СФТ [22]. В целом, для всех исследованных режимов эта величина высокая и превышает значение 0.9.
Образцы катализатора, отработавшего 100 и 1000 ч, были охарактеризованы методами РФА, БЭТ, СЭМ-ЭДС, ПЭМ, РФЭС.
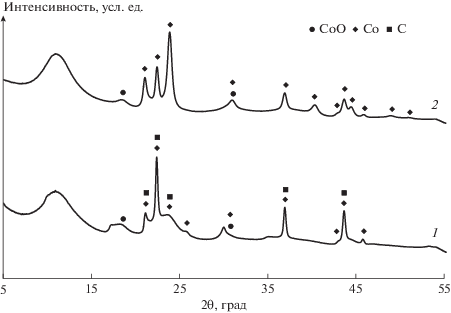
На рентгенограммах (рис. 3) исследованных образцов катализатора идентифицирован кобальт в виде кристаллической фазы Co0 с характерными максимумами дифракции в области углов 2θ = 20°–55°, а также кобальт в оксидной форме CoO при 2θ ≈ 18.6°; 30.9°. Для образца катализатора, отработавшего 1000 ч, характерно наличие кристаллической фазы углерода, возникшей, вероятно, в результате процессов диспропорционирования СО или термического распада углеводородов. Оксид кремния рентгеноаморфный, об этом свидетельствует широкое диффузное гало в области малых углов.
Спектры Co2p РФЭС дополнили информацию о степени окисления кобальта на поверхности катализатора (рис. 4). Для образцов кобальтсодержащего катализатора, отработавших 100 и 1000 ч, в спектрах наблюдается компонент A с энергией связи Eb = 777.5 эВ, которую можно отнести к металлическому кобальту. Заметим, что энергия связи этого компонента несколько ниже эталонного значения 778.1 эВ [23]. Разница, вероятно, возникла из-за неоднозначности в коррекции зарядов спектров или из-за дифференциальной зарядки частиц металлического кобальта на непроводящей подложке, в нашем случае на SiO2. Другие компоненты в спектрах соответствуют частицам Co2+, на что указывает присутствие интенсивного так называемого спутника с переносом заряда (компонент C около 785.0 эВ) на стороне с высокой энергией основного пика окисленного кобальта (компонент B около 780.5 эВ). Это окисленное состояние кобальта может быть отнесено к CoO или Co(OH)2 [23].
Рис. 4
. Co2p рентгеновские фотоэлектронные спектры образцов катализатора: а – отработавший в течение 1000 ч; б – отработавший 100 ч.
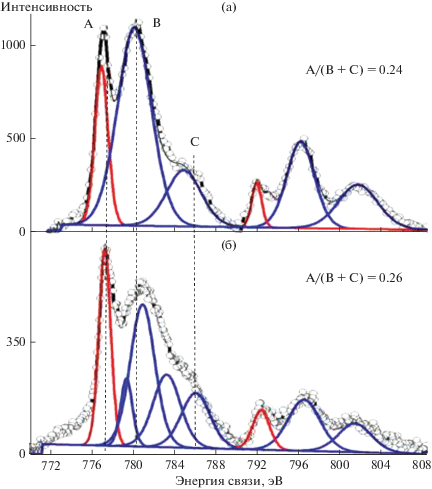
Вклад восстановленного кобальта (компонент А) близок для обоих образцов катализатора, но немного ниже (24% против 26%) для образца (А) (рис. 4). Это может быть связано с медленным окислением металлического кобальта при длительном тестировании в СФТ. Известно, что мелкие частицы кобальта менее устойчивы к окислению, чем крупные [11]. Трехвалентный кобальт имеет сателлит очень низкой интенсивности и его в приведенных спектрах не обнаружено.
Изображения частиц кобальтсодержащего катализатора и диаграммы распределения кристаллитов кобальта по размерам, полученные методом ПЭМ, приведены на рис. 5.
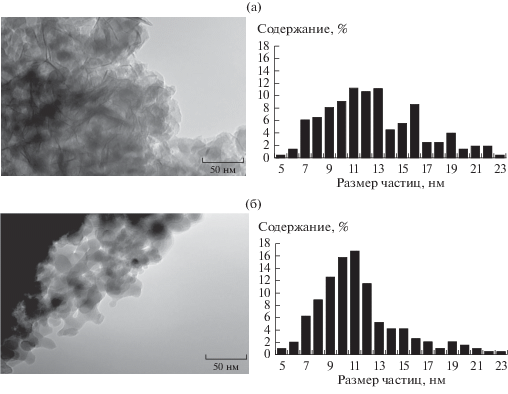
Средние значения величины кристаллитов, рассчитанные по результатам ПЭМ, и удельная поверхность отработавших 1000 ч (образец А) и 100 ч (образец Б) катализатора СДУ представлены в табл. 4. В исходном восстановленном образце катализатора средний размер частиц кобальта согласно данным ПЭМ равен 8 ± 2 нм [13]. В отработанном катализаторе в течение 100 ч (образец Б) размер частиц кобальта составил 13 ± 4 нм. Более длительная работа (1000 ч) в условиях СФТ способствует дальнейшему укрупнению частиц 16 ± 5 нм, в основном за счет снижения доли частиц с размером 9–12 нм.
Таблица 4.
Удельная поверхность и размер кристаллитов кобальта
| Образец | А | Б |
|---|---|---|
| Размер кристаллитов кобальта (ПЭМ), dср, нм | 16 ± 5 | 13 ± 4 |
| Удельная поверхность, Sуд, м2/г | 89 | 230 |
Значения удельной поверхности образцов сильно разнятся по своей величине в зависимости от условий эксплуатации. Для исходного катализатора в восстановленной форме удельная поверхность равна 274 м2/г. После работы катализатора в условиях высокого давления (6.0 МПа) поверхность уменьшается: в течение 100 ч до 230 м2/г (образец Б) и в течение 1000 ч до 89 м2/г (образец А). По-видимому, в процессе СФТ в порах катализатора накапливаются продукты синтеза, что приводит к их закупорке, в первую очередь, мелких пор, и, соответственно, к снижению удельной поверхности катализатора.
СЭM-изображения поверхности образцов кобальтсодержащего катализатора, с увеличением 50 000 и 5000, представлены на рис. 6.

Поверхность отработавшего 100 ч (образец (б)) катализатора выглядит однотипно, в виде плотной, гладкой структуры. На поверхности образца (а), отработавшего 1000 ч, наблюдается дополнительная микроструктура, более развитая, чем у образца (б). При 5000-кратном увеличении на снимках отчетливо видны агломераты кобальта размером 2–3 мкм, как правило, шарообразной формы. Образование подобных скоплений частиц на поверхности отработанных катализаторов описано в работе [24], где их наблюдали после эксплуатации катализатора при низком соотношении Н2/СО = 1.
СЭМ-ЭДС изображения образцов позволяют представить локализацию элементов на поверхности катализатора (рис. 7).
Рис. 7.
Элементное картографирование катализаторов (СЭМ-ЭДС): а – отработавший в течение 1000 ч; б – отработавший 100 ч.

Распределение элементов по поверхности имеет различный вид: у кобальта это фрагментарный, а у кремния и кислорода более равномерный. Углерод после 100 ч работы катализатора начинает понемногу накапливаться на поверхности кобальта, а после 1000 ч – его локализация на частицах кобальта становится весьма заметной.
Таким образом, каталитические испытания катализатора Co–Al2O3/SiO2 в течение 100 и 1000 ч показали, что по мере увеличения длительности эксплуатации его активность, выраженная через степень конверсии СО, плавно снижается. Авторы [10] считают, что основными причинами потери активности (дезактивации) кобальтовых катализаторов СФТ могут быть: отравление, повторное окисление активных участков кобальта парами воды, образование различных поверхностных видов углерода (зауглероживание), реструктуризация поверхности, агломерация кристаллитов кобальта, а также диффузионные ограничения.
В нашем случае, отравление катализатора, например, соединениями серы, маловероятно, так как исходные вещества для приготовления катализатора и реакционные газы не содержали соединений серы и других контактных ядов. Окисление кобальта водой или другими реагентами возможно лишь в минимальной степени, поскольку степень конверсии в условиях циркуляционного режима за один проход невелика (менее 40%). После реактора продукты СФТ охлаждали, тем самым большая часть паров воды удалялась из циркуляционного газа. Количество металлического кобальта, по данным РФЭС, в образцах после 100 и 1000 ч работы характеризуется довольно высокими значениями.
Наиболее существенными фактороми дезактивации, действующими в течение всего периода непрерывной работы катализатора, по мнению многих авторов, считается зауглероживание его поверхности и спекание (термическая агломерация) [25–27]. В нашем случае при переходе от режима I к режиму II наблюдается увеличение скорости дезактивации, что вероятно можно отнести к зауглероживанию, которое обусловлено резким увеличением селективности по углеводородам С35+ и, как следствие, блокировкой активных центров этими углеводородами. Следует отметить, что такая дезактивация является обратимой, так как при переходе от режима II к режиму III наблюдается полное восстановление активности катализатора и замедление скорости дезактивации. По-видимому, данный эффект вызван освобождением активных центров катализатора от тяжелых восков за счет растворения последних короткоцепочечными УВ, синтезируемыми при более высокой температуре в режиме III. Углерод может воздействовать на активный компонент катализатора в различных формах – от атомного до полимерного и от аморфного до кристаллического [25]. В этом случае блокировка активного металла происходит в значительной степени. Это подтверждается результатами исследований СЭМ-ЭДС и РФА, из которых видно, что на поверхности катализатора, отработавшего 1000 ч, область локализации углерода весьма заметно совпадает с расположением частиц кобальта (рис. 7). В меньшей степени это наблюдается и для образца, отработавшего 100 ч. Блокировка пор катализатора углеродсодержащими соединениями, по-видимому, является и основной причиной снижения удельной поверхности.
С ростом длины углеводородной цепи повышается вязкость продуктов СФТ, которые конденсируются в порах. В этом случае неизбежно происходит нарастание внутридиффузионных ограничений, препятствующих подводу реагентов и отводу продуктов синтеза от активных центров катализатора. При повышении температуры синтеза наблюдается сдвиг селективности в сторону УВ с короткой цепью, которые растворяют более тяжелые воски. Вязкость восков, конденсирующихся в порах, уменьшается за счет их разбавления, что способствует увеличению количества доступных активных центров катализатора и его регенерации. Снижение вязкости продуктов должно приводить и к частичному снятию внутридиффузионных ограничений.
Снижение производительности катализатора может возникать и в результате наступления внешнедиффузионного торможения [28]. При значениях ХСО > 80%, большая часть реакционного газа превращается в парожидкостное состояние и, как результат, снижается степень турбулентности реакционного потока. За счет циркуляции реакционного газа и повышения ОСГ внешнедиффузионные ограничения можно устранить. На это указывает тот факт, что при увеличении кратности циркуляции (Кц) от 2.2 до 6.0 в III этапе исследований (табл. 2), селективность по УВ С35+ несколько подросла (с 27.6 до 29.1%) в сравнении с аналогичным технологическим режимом I.
Укрупнение частиц кобальта, особенно для образца, отработанного в течение 1000 ч, также может оказывать в определенной степени влияние на активность катализатора. По данным [26], основное увеличение размера частиц происходит в течение начальных 10–15 суток эксплуатации катализатора, после чего оно становится незначительным. За указанный промежуток времени размер частиц увеличился с 9 до 14 нм, а в последующие 85–90 суток – до 15 нм [26]. Регенерация катализатора путем окислительно-восстановительной обработки обычно позволяет восстановить размеры кристаллитов кобальта, близкие к исходным значениям.
Проследить причину потери активности/селективности катализатора во многих случаях сложно, особенно когда несколько процессов дезактивации влияют на эти каталитические характеристики. СФТ охватывает большое количество взаимодействующих факторов, отрицательно влияющих на каталитическую активность. Изменение условий эксперимента, микро- и макроскопических свойств катализатора, может оказывать содействие различным механизмам дезактивации. Поэтому очень важно, чтобы катализаторы изучали в диапазоне промышленно значимых технологических параметров, на реальном зерне (2–3 мм) катализатора, с одновременным контролем всех взаимодействующих факторов. Долгосрочные прогоны катализатора и определение условий, при которых скорость дезактивации наименьшая, будут полезны для взаимодействия промышленности и науки.
ВЫВОДЫ
1. Эксплуатация Co–Al2O3/SiO2-катализатора в течение 1000 ч при 6.0 МПа в условиях циркуляции реакционного газа в процессе получения длинноцепочечных углеводородов в СФТ сопровождается потерей активности (дезактивацией), что выражается в снижении степени конверсии СО. Установлено, что в интенсивном синтезе длинноцепочечных углеводородов С35+ при 6.0 МПа, ОСГ 1000 ч–1 и циркуляции газа ожидаемое время работы Co−Al2O3/SiO2-катализатора до его регенерации составляет около 2341 ч.
2. Механизм дезактивации Co–Al2O3/SiO2-катализатора обусловлен такими факторами как: зауглероживание, снижение удельной поверхности, вследствие закупоривания пор тяжелыми углеводородами, агломерацией кристаллитов кобальта, а также влиянием диффузионных ограничений.
3. Скорость дезактивации, а, следовательно, и продолжительность работы катализатора СДУ можно регулировать, воздействуя на тот или иной механизм дезактивации. Повышение температуры синтеза сдвигает селективность процесса в сторону УВ с короткой цепью, которые понижают вязкость продуктов СФТ, конденсирующихся в порах, и улучшают доступность реагентов к активным центрам. Увеличение нагрузки и кратности циркуляции, особенно при высоких значениях ХСО, способствует росту интенсивности перемешивания реагентов, тем самым вероятность внешнедиффузионного торможения процесса СФТ уменьшается.
4. Скорость дезактивации следует учитывать при оптимизации технико-экономических показателей промышленного производства церезина и определения жизненного цикла кобальтсиликагелевого катализатора.
Список литературы
Rauch R., Hofbauer H., Neuling U., Kaltschmitt M. Biokerosene Status and Prospects. Springer-Verlag GmbH, Germany, 2018. P. 497.
Dry M.E. // Applied Catalysis A: General. 2004. V. 276. P. 1.
Khodakov A.Y., Chu W., Fongarland P. // Chemical Reviews. 2007. V. 107. P. 1692.
Zhang Q., Kang J., Wang Y. // ChemCatChem. 2010. V. 2. P. 1030.
Palma V., Ruocco C., Martino M., Meloni E., Ricca A. Bioenergy Systems for the Future: Prospects for Biofuels and Biohydrogen, 2017. P. 217.
Jacobs G., Davis B.H. // J. of Natural Gas Science and Engineering. 2018. V. 52. P. 549.
https://www.grandresearchstore.com/materials-and-chemicals/north-america-fischer-tropschft-wax-industry-2016-market-research-report (дата обращения 01.04.2019)
Kungurova O.A., Shtertser N.V., Chermashentseva G.K., Simentsova I.I., Khassin A.A. // Catalysis in Industry. 2017. V. 9. P. 23.
Iglesia E., Soled S., Fiato R., Via G. // J. of Catalysis. 1993. V. 143. P. 345.
Tsakoumis N.E., Ronning M., Borg O., Rytter E., Holmen A. // Catalysis Today. 2010. V. 154. P. 162.
Carvalho A., Ordomsky V.V., Luo Y., Marinova M., Muniz A.R, Marcilio N.R., KhodakovA.Y. // J. of Catalysis. 2016. V. 344. P. 669.
Narochnyi G.B., Yakovenko R.E., Savost’yanov A.P., Bakun V.G. // Catalysis in Industry. 2016. V. 8. P. 139.
Savost’yanov A.P., Yakovenko R.E., Sulima S.I., Bakun V.G., Narochnyi G.B., Chernyshev V.M., Mitchenko S.A. // Catalysis Today. 2017. V. 279. P. 107.
Savost’yanov A.P., Narochnyi G.B., Yakovenko R.E., Mitchenko S.A., Zubkov I.N. // Petrol. Chemistry. 2018. V. 58. No 1. P. 76.
Savost’yanov A.P., Narochnyi G.B., Yakovenko R.E., Bakun V.G. // Catalysis in Industry. 2014. V. 6. P. 292.
Савостьянов А.П., Нарочный Г.Б., Яковенко Р.Е., Лапидус А.Л. // Химия твердого топлива. 2015. V. 6. P. 19.
Savost’yanov A.P., Yakovenko R.E., Narochnyi G.B., Sulima S.I., Bakun V.G., Soromotin V.N., Mitchenko S.A. // Catalysis Communications. 2017. V. 99. P. 25.
Яковенко Р.Е. // Дис. ... канд. техн. наук. Новочеркасск: ЮРГПУ(НПИ) имени М.И. Платова, 2017.
PDF-2.The powder diffraction file TM. International Center for Diffraction Data (ICDD), PDF-2 Release 2012, web site: www.icdd.com (2014).
Kashin A.S., Ananikov V.P. // Russian Chemical Bulletin. 2011. V. 60. P. 2602.
Kachala V.V., Khemchyan L.L., Kashin A.S., Orlov N.V., Grachev A.A., Zalesskiy S.S., Ananikov V.P. // Russian Chemical Reviews. 2013. V. 82. P. 648.
Anderson V.R.B., Friedel R.A., Storch H.H. // J. of Chemical Physics. 1951. V. 19. P. 313.
Biesinger M.C., Payne B.P., Grosvenor A.P., Lau L.W.M., Gerson A.R., Smart R.St.C. // Applied Surface Science. 2011. V. 257. P. 2717.
Peña D., Griboval-Constant A., Diehl F., Lecocq V., Khodakov A.Y. // ChemCatChem. 2013. V. 5. P. 728.
Keyvanloo K., Fisher M.J., Hecker W.C., Lancee R.J., Jacobs G., Bartholomew C.H. // J. of Catalysis. 2015. V. 327. P. 33.
Saib A.M., Moodley D.J., Ciobîca I.M., Hauman M.M., Sigwebela B.H., Weststrate C.J., Niemantsverdriet J.W., de Loosdrecht J. // Catalysis Today. 2010. V. 154. P. 271.
Moodley D., Claeys M., van Steen E., van Helden P., Kistamurthy D., Weststrate K-Jan, Niemantsverdriet H., Saib A., Erasmus W., van de Loosdrecht J. // Catalysis Today. 2019. In Press doi:10.1016/j.cattod.2019.03.059
Savost’yanov A.P., Narochnyi G.B., Yakovenko R.E., Soromotin V.N., Zubkov I.N. // Catalysis in Industry. 2018. V. 10. P. 181.
Дополнительные материалы отсутствуют.