

Нефтехимия, 2020, T. 60, № 1, стр. 101-113
Получение из угля синтетических реактивных топлив, взаимозаменяемых с нефтяными топливами типа Джет А-1 и Т-8В
Л. С. Яновский 1, Н. И. Варламова 1, И. М. Попов 1, В. О. Самойлов 2, А. Б. Куликов 2, М. И. Князева 2, Р. С. Борисов 2, Д. Н. Рамазанов 2, *, А. Л. Максимов 2
1 Центральный институт авиационного моторостроения им. П.И. Баранова
111116 Москва, Россия
2 Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Россия
* E-mail: ramazanov@ips.ac.ru
Поступила в редакцию 16.08.2019
После доработки 04.09.2019
Принята к публикации 06.09.2019
Аннотация
Предложена и экспериментально проверена схема комбинированной переработки каменного угля с получением синтетического реактивного топлива. Установлено, что при применении подхода, включающего коксование угля с последующими газификацией, синтезом Фишера–Тропша и гидроизомеризацией получаемые керосиновые фракции характеризуются низкими значениями плотности (0.741–0.751 кг/дм3 при 20°С) относительно уровня, заданного техническими требованиями к используемым сегодня маркам реактивных топлив Джет А-1 и Т-8В. Применение для улучшения низкотемпературных свойств парафинистой фракции синтеза Фишера–Тропша (СФТ) процесса гидроизомеризации (Т = 330–345°С, ${{p}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ = 72 бар) в присутствии катализатора 2%Pt/Al-HMS(10) позволило получить изопарафиновую фракцию с температурой начала кристаллизации до –56°С. Получаемая при коксовании угля каменноугольная смола была подвергнута глубокому гидрированию с получением нафтенового компонента реактивного топлива, характеризующегося при пределах кипения 150–250°С плотностью около 0.873 кг/дм3 (20°С), а также низким содержанием серы и ароматических углеводородов (3 м. д. и 23 мас. %, соответственно). Компаундированием полученных изопарафиновой и нафтеновой фракций были получены экспериментальные образцы синтетических реактивных топлив, удовлетворяющие техническим требованиям, предъявляемым к топливам марок Джет А-1 и Т-8В.
В связи с неизбежным истощением запасов нефти развитие производства альтернативных топлив, в том числе для авиационных газотурбинных двигателей, является объективной необходимостью. За рубежом производятся в промышленном масштабе по стандарту ASTM D 7566-09 и допущены к применению на авиатехнике синтетические авиакеросины, производимые из угля и природного газа. Этим стандартом предусмотрено применение в качестве реактивного топлива, отвечающего требованиям стандарта ASTM D 1655 на топливо Jet A-1, смеси синтетического керосина 50 : 50 со стандартным топливом Jet A-1. Добавление стандартного нефтяного топлива в синтетический авиакеросин объясняется, главным образом, тем, что синтетический авиакеросин имеет, как правило, пониженную, по сравнению с допустимой по ASTM D 1655 для топлива Jet A-1, плотность. Кроме того, синтетический авиакеросин без соответствующих присадок обладает повышенной окисляемостью и низкими противоизносными свойствами ввиду практически полного отсутствия гетероатомных соединений (в частности, сернистых). Добавление в него стандартного нефтяного авиакеросина позволяет устранить эти недостатки.
В поправке к стандарту ASTM 7566-09 от 1 июля 2011 г. разрешено применение смеси 50 : 50 синтетического топлива из биосырья и стандартного нефтяного топлива Jet A-1. Ожидается, что уже в ближайшее время для заправки российских самолетов в зарубежных авиапортах может быть предложено альтернативное топливо.
По зарубежным данным, в настоящее время промышленное производство реактивного топлива из угля является рентабельным. Промышленное производство синтетического реактивного топлива из природного газа станет рентабельным в ближайшие годы. Промышленное производство реактивного топлива из наземного биосырья может стать рентабельным уже через несколько лет, из водорослей – примерно через 10 лет [1].
Разработка прогрессивных технологий промышленного производства альтернативных топлив и их рационального применения на транспортных средствах (в том числе в авиации) в настоящее время являются актуальными для нашей страны задачами. Несмотря на то, что Россия занимает первое место в мире по экспорту нефти, следует принимать во внимание и наличие в нашей стране значительных ресурсов альтернативного углеродсодержащего сырья – природного газа, угля и биомассы.
Принципиально известны два основных подхода к получению синтетических жидких топлив из углей, которые опираются на технологии прямого гидроожижения угля по Бергиусу либо непрямого ожижения угля через газификацию с последующим получением синтетического жидкого топлива по СФТ [2–9]. Дополнительно следует указать на описанную в работах [10, 11] возможность гидрогенизационной переработки каменноугольной смолы, получаемой при коксовании углей, с получением высококачественных реактивных топлив. Ввиду того обстоятельства, что конкурентоспособность технологии прямого гидроожижения углей в настоящее время неочевидна, целесообразным представляется построение концепции переработки угля в топливо с опорой на комбинирование технологий коксования угля (с получением каменноугольной смолы) и технологии CTL (газификация с последующим СФТ).
В России имеется определенный научно-технический задел по лабораторным технологическим регламентам получения альтернативных авиатоплив из природного газа и биомассы (биоэтанола) [12]. Учитывая актуальность вопроса, по инициативе Центрального института авиационного моторостроения им. П.И. Баранова (ЦИАМ) были проведены совместные исследования по отечественным альтернативным авиакеросинам из угля совместно с Институтом нефтехимического синтеза им. А.В. Топчиева РАН (ИНХС РАН). Опытные образцы реактивных топлив разрабатывались по техническим требованиям ЦИАМ, которые предусматривают соответствие синтезированного топлива по физико-химическим и эксплуатационным свойствам топливу ДжетА-1 (ГОСТ 32595), допущенному к применению на авиатехнике гражданского назначения, и топливу Т-8В (ГОСТ 12308), как потенциальному унифицированному реактивному топливу для перспективной авиатехники [13]. Исследования синтезированных образцов на соответствие техническим требованиям выполнялись
ЦИАМ, определяя этим доработку прототипа технологии получения топлива в нужном направлении.
Цель настоящей работы – экспериментальное установление особенностей получения синтетических реактивных топлив на основе угля путем раздельной переработки каменноугольного кокса и каменноугольной смолы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и катализаторы. Каменноугольная смола по ТУ 2453-203-00190437-2005 для исследований была предоставлена Московским коксогазовым заводом. Основные показатели качества приведены в табл. 1.
Таблица 1.
Основные показатели качества каменноугольной смолы (данные производителя)
| Показатель | Единица измерения | Значение |
|---|---|---|
| Плотность при 20°С | кг/м3 | 1180–1220 |
| Массовая доля воды | мас. % | 4.0 |
| Массовая доля золы | мас. % | <0.1 |
| Массовая доля веществ, не растворимых в толуоле | мас. % | <11 |
| Массовая доля веществ, не растворимых в хинолине | мас. % | <4 |
В качестве модельного парафинистого сырья (воспроизводящего по составу среднедистиллятную фракцию СФТ) была использована фракция н-алканов С9–С20 (“мягкий парафин”) с установки “Парекс”, полученная с ПО “Киришинефтеоргсинтез”. Состав фракции, определенный методом ГЖХ, приведен в табл 2.
Таблица 2.
Компонентный состав модельного парафинистого сырья
| Компонент | Химическая формула | Концентрация, мас. % |
|---|---|---|
| н-Нонан | С9Н20 | 0.9 |
| н-Декан | С10Н22 | 5.4 |
| н-Ундекан | С11Н24 | 9.9 |
| н-Додекан | С12Н26 | 10.8 |
| н-Тридекан | С13Н28 | 13.1 |
| н-Тетрадекан | С14Н30 | 14.2 |
| н-Пентадекан | С15Н32 | 13.9 |
| н-Гексадекан | С16Н34 | 11.7 |
| н-Гептадекан | С17Н36 | 10.8 |
| н-Октадекан | С18Н38 | 6.3 |
| н-Нонадекан | С19Н40 | 2.5 |
| Эйкозан | С20Н42 | 0.5 |
Методика приготовления бифункционального платиносодержащего катализатора гидроизомеризации парафинистых фракций была описана ранее [14]. В качестве катализатора глубокого гидрирования дистиллята каменноугольной смолы использовали катализатор НВС-А (Ангарский завод катализаторов и органического сырья), подвергнутый предварительному осернению раствором диметилдисульфида (ДМДС) (1 мас. %) в прямогонной дизельной фракции.
Оборудование. Вакуумную дистилляцию каменноугольной смолы проводили с использованием стандартных лабораторных приборов для перегонки под вакуумом, в конструкцию которых были внесены некоторые модификации, учитывающие особенности перегоняемого сырья. Так, капилляр для подачи инертного газа вводили непосредственно в куб (двугорлую колбу); сечение капилляра было увеличено относительно обычно применяемого ввиду вязкости сырья. Ввиду опасности резкого вскипания смолы, сопровождающегося выбросом жидкости из куба в приемник, вместо насадки Вюрца была использована насадка Кляйзена, горло которой, соосное с колбой, закрывали пробкой. Аллонж без отвода соединял воздушный холодильник с круглодонной двугорлой колбой, служащей приемником дистиллята. Перегонку вели в токе азота при остаточном давлении 20–25 мм рт. ст. с отбором фракции Н.К.–220°С (индекс указывает на остаточное давление); приведенная к атмосферному давлению температура конца кипения дистиллятной фракции около 380–400°С, выход дистиллятной фракции 40–45 мас. %.
Эксперименты по гидрооблагораживанию дистиллятной фракции каменноугольной смолы и по гидроизомеризации парафинистой фракции проводили с использованием проточных лабораторных установок высокого давления, снабженных реактором с неподвижным слоем гранулированного катализатора. Ректификацию катализатов, полученных на лабораторных установках, осуществляли с использованием лабораторных ректификационных колонн с регулируемым потоком флегмы.
Измерения. Состав исходных фракций и продуктов их гидрооблагораживания определяли методами ГЖХ (Кристаллюкс-4000М, газ-носитель – гелий, пламенно-ионизационный детектор, колонка SPB-1 30 × 0.25 × 0.25 (Supelco)) и ГХ-МС. Изучение состава сырья и продуктов методом ГХ-МС проводили на хромато-масс-спектрометре ThermoFocus DSQ II (капиллярная колонка Varian VF-5ms, 30 м, внутренний диаметр 0.25 мм, толщина фазы 0.25 мкм, газ-носитель – гелий, режим работы: температура инжектора 270°С, начальная температура печи хроматографа 40°С, затем нагрев со скоростью 15°С/мин до 300°С, затем изотерма в течении 10 мин; режим работы масс-спектрометра: энергия ионизации 70 эВ, температура источника 230°С, сканирование в диапазоне 10–800 Да со скоростью 2 скан/с, разрешение единичное по всему диапазону масс.
Определение физико-химических свойств катализатов, полупродуктов и конечных образцов – прототипов топлив, проводили по стандартным методикам, регламентированным для испытаний реактивных топлив Джет А-1 и Т-8В по ГОСТ 32595-2013 и 12308-2013, соответственно. Основные методы, использованные для контроля свойств полупродуктов, приведены в табл. 3.
Таблица 3.
Основные методы испытания полупродуктов и экспериментальных образцов реактивных топлив
| Показатель | Метод измерения |
|---|---|
| Плотность | ГОСТ 3900-85 |
| Фракционный состав | ГОСТ 2177-99 |
| Температура начала кристаллизации | ГОСТ 5066-91 |
| Массовая доля ароматических углеводородов | ГОСТ EN 12916-2012 |
| Массовая доля нафталиновых углеводородов | ГОСТ EN 12916-2012 |
| Массовая доля общей серы | ГОСТ 32139-2013 |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Схема переработки. Возможная схема комплексной переработки угля с получением моторного топлива, рассмотренная нами в настоящей работе, приведена на рис. 1. Согласно предложенной схеме, первой стадией является коксование каменного угля с получением каменноугольной смолы и каменноугольного кокса. Этот процесс широко известен и реализован в промышленности в крупных масштабах. Продукты коксования, полученные на первой стадии, в дальнейшем подвергают раздельной переработке.
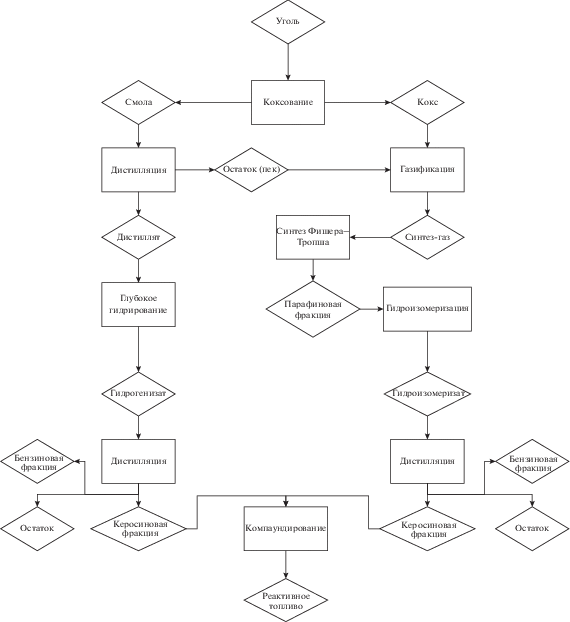
Каменноугольная смола подвергается дистилляции с отбором необходимой фракции, дистиллят отправляется на гидрооблагораживание, предполагающее глубокую гидроочистку от гетероатомных соединений и глубокую гидродеароматизацию. Следует отметить, что в отношении гидрооблагораживания каменноугольной смолы (и ее фракций) могут быть применены схемы с различным количеством технологических стадий [11]. В нашем случае было использовано одностадийное гидрооблагораживание под высоким давлением, позволяющее после ректификации продукта получать нафтеновый компонент реактивного топлива. Остаток фракционирования каменноугольной смолы (обычно называемый пеком) может быть использован для газификации наряду с коксом.
Каменноугольный кокс подвергается газификации с последующим превращением полученного синтез-газа во фракцию синтетических углеводородов по Фишеру–Тропшу. Известно, что в СФТ возможно управлять селективностью процесса (как в отношении классов получаемых соединений, так и в отношении распределения молекулярных масс продуктов) путем изменения свойств катализатора и условий процесса. СФТ при температурах 200–250°С и давлении синтез-газа 10–12 МПа позволяет получать фракции, практически нацело состоящие из парафинов нормального строения [15, 16].
Парафинистые фракции – продукты СФТ– требуют дальнейшего облагораживания с целью улучшения своих низкотемпературных свойств. В рамках рассматриваемой концепции был применен процесс гидроизомеризации с получением изопарафинового компонента реактивного топлива. Полученные компоненты могут быть компаундированы в различных соотношениях в зависимости от того, в какой марке топлива имеется необходимость.
Гидроизомеризация парафинистой фракции. Основным назначением процесса гидроизомеризации парафинистой фракции является улучшение низкотемпературных свойств продукта. В настоящем случае ввиду нацеленности на получение реактивных топлив, обеспечение низкой температуры начала кристаллизации продукта имеет критическую важность.
Гидроизомеризацию фракции нормальных парафинов С9–С20 проводили с использованием катализатора 2%Pt/Al-HMS(10), ранее эффективно примененном в процессе комбинированного гидрокрекинга–гидроизомеризации нормальных парафинов состава С19–С38 [12]. В настоящей работе указанный катализатор был использован для гидроизомеризации более легкой фракции.
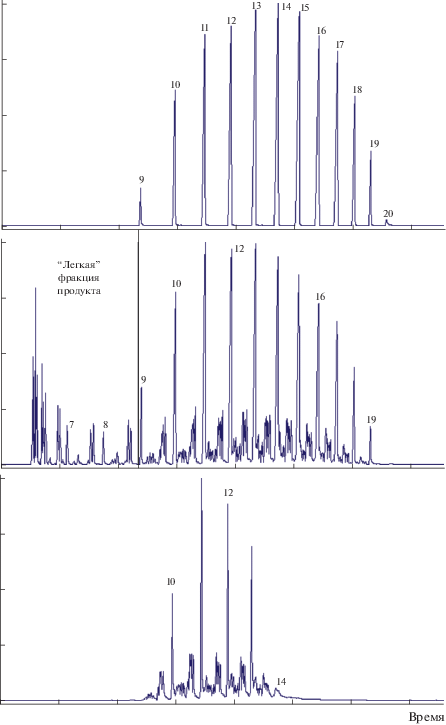
Хроматограмма сырья процесса и представительная хроматограмма стабильного гидрогенизата, позволяющие составить представления об изменениях химического состава, происходящих в результате превращения (Т = 300–350°С, ${{p}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ = 50–80 бар, v = 0.2–1.0 ч–1, водород : сырье = = 500–1000 нл/л) в присутствии катализатора 2%Pt/Al-HMS(10), приведены на рис. 2. В средней фракции гидрогенизата (С9–С20) пики, находящиеся на хроматограмме в интервалах между пиками н-алканов, отвечают алканам разветвленного строения, образуемым в результате гидроизомеризации, которая сопровождалась реакциями крекинга углеводородов (УВ), приводящих к образованию “легкой” части гидрогенизата (нормальные и разветвленные алканы С5–С8). Схожий вид хроматограммы в интервалах, отвечающих группам УВ с одинаковым числом атомов углерода (например, между пиками н-гептана и н-октана и между пиками н-октана и н-нонана), указывающий на аналогичный состав изомеров, позволяет предположить, что изоалканы легкой фракции были образованы в том числе в результате вторичной изомеризации легких н-алканов – продуктов гидрокрекинга более тяжелых УВ. Выделяемая ректификацией керосиновая фракция 150–250°С содержала алканы нормального и разветвленного строения с числом атомов углерода от С9 до С14.
Рис. 2.
Представительные хроматограммы сырья гидроизомеризации (фракции н-алканов С9–С20) (верх), стабильного гидрогенизата (середина), выделенной из гидрогенизата ректификацией фракции 150–250°С (низ). Временн${\text{ы}}'$е шкалы хроматограмм совмещены. Номерами обозначены пики н-алканов с соответствующим количеством атомов углерода в цепи. Вертикальной чертой условно отделена “легкая” фракция продукта Н.К.–150°С.
Выход газообразных продуктов крекинга при гидроизомеризации мягкого парафина в присутствии катализатора 2%Pt/Al-HMS(10) при Т = 335–345°С и ${{p}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ = 72 бар колебался в пределах 4–9 мас. % в расчете на сырье процесса (табл. 4). В целом увеличение жесткости режима (повышение температуры и снижение объемной скорости подачи сырья) приводило к росту степени изомеризации н-алканов сырья, сопровождаемому ростом выхода продуктов крекинга – как газообразных, так и легкой бензиновой фракции. Таким образом, оптимизация условий реакции в данном случае предполагает поиск режима, отвечающего максимальной степени изомеризации при минимальном выходе продуктов гидрокрекинга. С другой стороны, в составе реактивных топлив, вырабатываемых из нефти, присутствует некоторое количество алканов нормального строения – следовательно, выбор режима гидроизомеризации в данном случае должен быть основан на минимально допустимой степени изомеризации, обеспечивающей приемлемые низкотемпературные свойства целевой фракции гидрогенизата.
Таблица 4.
Влияние оперативных параметров процесса гидроизомеризации фракции н-алканов С9–С20 на выход продуктов процесса и их качество. Соотношение водород : сырье = 1000 нл/л, давление водорода 72 бар
| Показатель | Режим процесса (Т, °С/v, ч–1) | |||
|---|---|---|---|---|
| Выход в расчете на сырье процесса, мас. %: | 345/1.5 | 345/1.0 | 340/1.0 | 335/1.0 |
| Углеводородные газы С1–С4 | 6.6 | 9.1 | 7.5 | 4.8 |
| Жидкий продукт, в том числе: | 93.4 | 90.9 | 92.5 | 95.2 |
| бензиновая фракция (Н.К.–150°С) | 28.1 | 42.4 | 33.6 | 25.8 |
| средняя фракция (150+°С) | 65.3 | 48.5 | 58.9 | 69.5 |
| Соотношение изоалканы : алканы в жидком катализате | 3.02 | 3.75 | 3.21 | 2.38 |
| Плотность жидкого катализата (при 20°С), кг/дм3 | 0.721–0.725 | |||
Фракционирование катализата приводит к трем изопарафиновым фракциям: бензиновой (Н.К.–150°С), керосиновой (150–250°С) и остаточной (>250°С). Согласно приведенным в табл. 5 данным, соотношение изоалканы : н-алканы в целевой фракции гидрогенизата 150–250°С (выделяемой ректификацией) заметно выше, чем тоже соотношение, рассчитанное по результатам ГЖХ для стабильного гидрогенизата в целом: так, из катализата с упомянутым соотношением 1.9 была выделена керосиновая фракция с соотношением 2.7 и температурой начала кристаллизации –41°С. Такой уровень низкотемпературных свойств недостаточен для компонента реактивного топлива, хотя и может быть скорректирован в результате последующего компаундирования с более низкозастывающей нафтеновой фракцией. Керосиновая фракция, выделяемая из гидрогенизата с соотношением изоалканы : н-алканы = 3.0, характеризовалась температурой начала кристаллизации –51°С, что делает ее пригодной для компаундирования топлива Джет А-1 (температура начала кристаллизации согласно ГОСТ не выше –47°С).
Таблица 5.
Материальный баланс фракционирования гидроизомеризатов и свойства получаемых фракций
| Показатель | Соотношение изоалканы : н-алканы в сырье ректификации | ||
|---|---|---|---|
| 1.9 | 3.0 | 3.2 | |
| Выход (плотность) фракций при ректификации, мас. % (кг/дм3) | |||
| Н.К.–150°С | 32.9 (0.674) | 27.6 (0.674) | 30.5 (0.669) |
| 150–250°С | 53.4 (0.751) | 59.9 (0.751) | 58.5 (0.741) |
| >250°С | 13.7 (0.773) | 12.5 (0.773) | 11.1 (0.770) |
| Фракция 150–250°С | |||
| Соотношение изоалканы : н-алканы | 2.7 | 3.8 | 4.0 |
| Температура начала кристаллизации, °С | –41 | –51 | –56 |
Легкая фракция не содержит серы, олефинов и ароматических углеводородовиполностью состоит из нормальных изопарафинов. Ввиду низкого содержания гетероатомных соединений легкая часть подобных фракций может быть вовлечена в производство изомеризата; при этом оптимальным представляется их ввод через узел фракционирования катализатов изомеризации: в этом случае отделяемые изопарафины образуют изомеризат, а парафины нормального строения возвращаются на изомеризацию. “Тяжелая” часть (тяжелее н-гексана) в этом случае может быть использована для производства углеводородных растворителей.
Получаемый при ректификации катализатов остаток (>250°C) по низкотемпературным свойствам может рассматриваться как компонент летних и некоторых дизельных топлив: выделенная из гидроизомеризата с соотношением изоалканы : н-алканы = 3.2 фракция имела температуру начала кристаллизации –19°С. Также целесообразным представляется организация схемы с рециклом остатка ректификации (рис. 3), что и было реализовано нами в отдельном эксперименте: остаток ректификации >250°C был смешан с сырьем гидроизомеризации в балансовом количестве (выход остатка 12.5 мас. % на сырье гидроизомеризации при Т = 345°С, ${{p}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ = 72 бар и v = 1.5 ч–1). Полученное сырье было подвергнуто конверсии в том же режиме, что и исходная фракция н-алканов.
Рис. 3.
Схема гидроизомеризации парафиновой фракции С9–С20 с рециклом тяжелой фракции гидрогенизата.
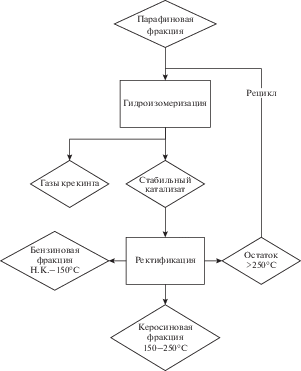
При сравнении результатов, полученных при работе на исходном сырье и сырье с добавлением остатка ректификации, нами не было обнаружено значительных различий: в обоих случаях выход углеводородных газов крекинга и жидкого катализата, фракционный состав жидкого катализата и соотношение изоалканы : н-алканы в целевой фракции 150–250°С оставалось одинаковым (в пределах колебаний значений между отдельными катализатами). Таким образом, остаток ректификации можно подавать на рецикл для получения дополнительных количеств легких фракций Н.К.–150°С и 150–250°С – или использовать как компонент летних и некоторых зимних дизельных топлив.
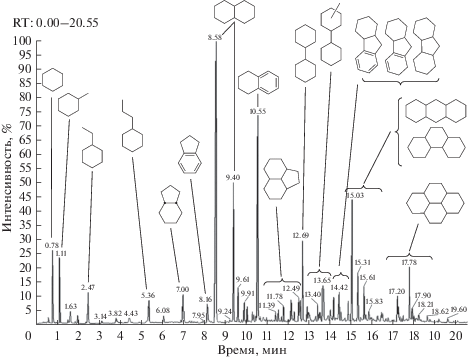
Фракционирование и гидрооблагораживание каменноугольной смолы. Выход дистиллятной фракции, отбираемой до температуры 230°С (остаточное давление 3 мм. рт. ст.), составлял 44 мас. % на исходную каменноугольную смолу. Состав полученного дистиллята приведен на рис. 4 и в табл. 6. Полученная дистиллятная фракция состоит главным образом из би- и трициклических аренов: основными компонентами являются инден, нафталин, метилнафталины, бифенил, аценафтен, флуорен, антрацен, фенантрен с некоторым содержанием тетрациклических УВ–пирена и флуорантена. Фракция богата гетероатомными соединениями серы (бензотиофен, дибензотиофен), азота (карбазол и некоторое количество хинолина) и кислорода (фенол и дибензофуран). Общее содержание серы в полученном дистилляте составило около 0.34 мас. %.
Рис. 4.
Хроматограмма дистиллята, полученного вакуумной перегонкой каменноугольной смолы (получена методом ГХ/МС).
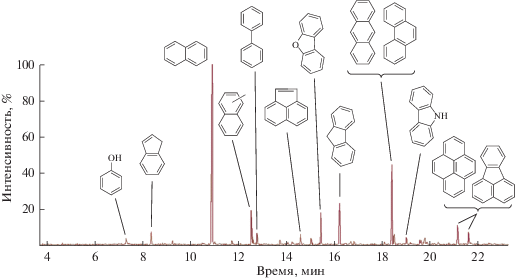
Таблица 6.
Компонентный состав дистиллята, полученного вакуумной перегонкой каменноугольной смолы (ГХ/МС, полуколичественный анализ по времени выхода, рис. 4)
| Компонент | Содержание, мас. % |
|---|---|
| Фенол | 0.6 |
| Инден | 2.0 |
| Крезол | 0.4 |
| Нафталин | 35.8 |
| Бензтиофен | 0.4 |
| Метилнафталины | 5.7 |
| Индол | 0.6 |
| Бифенил | 0.9 |
| Нафталины-С2 | 0.9 |
| Аценафтен | 1.4 |
| Винилнафталин | 1.1 |
| Дибензофуран | 3.3 |
| Флуорен | 5.5 |
| Гидроксифлуорен + фенален | 0.7 |
| Ксантен | 0.5 |
| Дибензотиофен | 0.6 |
| Фенантрен | 13.8 |
| Антрацен | 12.3 |
| Карбазол | 5.9 |
| Метилантрацен + метилфенантрен | 1.5 |
| Бензофлюорен | 1.0 |
| Фенилнафталин | 0.2 |
| Пирен + флюорантен | 4.8 |
Гидрирование дистиллятной фракции КУС было осуществлено в присутствии катализатора НВС-А, известного высокой активностью в отношении гидродеароматизациивысокоароматизированногосернистого сырья [17–18 ]. Условия процесса: Т = 390°С, ${{p}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ = 200 бар, v = 0.5 ч–1, водород : сырье = 2500 нл/л. Основные свойства стабильного катализата, выход которого составил 94 мас. %, приведены в табл. 7.
Таблица 7.
Основные свойства жидкого продукта гидрирования дистиллятной фракции КУС
| Показатель | Значение |
|---|---|
| Плотность при 20°С, кг/м3 (расчетная) | 0.901 |
| Фракционный состав по ГОСТ 2177-99: | |
| Н.К. | 112 |
| 5% | 147 |
| 10% | 168 |
| 20% | 191 |
| 30% | 202 |
| 40% | 212 |
| 50% | 222 |
| 60% | 236 |
| 70% | 256 |
| 80% | 276 |
| 90% | 296 |
| 95% | 315 |
| Групповой углеводородный состав (ВЭЖХ), мас. % | |
| насыщенные | 77.35 |
| моноароматические | 21.39 |
| диароматические | 1.19 |
| полиароматические | 0.06 |
| Содержание серы, мг/кг | 3 |
Гидрирование дистиллятной фракции каменноугольной смолы сопровождалось глубокой гидроочисткой: содержание серы в продукте составляло всего 3 м. д. (табл. 7). Полученная фракция состояла, главным образом, из насыщенных углеводородов, по фракционному составу отвечающих интервалу 112–315°С. Основные компоненты полученной фракции приведены на хроматограмме (рис. 5).
Рис. 5.
Хроматограмма жидкого продукта глубокого гидрирования дистиллятной фракции КУС (получена методом ГХ/МС).
Фракционирование катализата осуществлялось с отбором трех фракций: легкой бензиновой (Н.К.–150°С), целевой керосиновой (150–250°С) и остаточной (>250°C). Следует отметить, что в полученных дистиллятных фракциях би- и полициклические арены, являющиеся нежелательными компонентами реактивных топлив, отсутствовали, концентрируясь в остаточной фракции >250°C (табл. 8).
Таблица 8.
Материальный баланс фракционирования продукта гидрирования дистиллятной фракции КУС и свойства получаемых фракций*
| Материальный баланс | Свойства фракций | |||||
|---|---|---|---|---|---|---|
| плотность при 20°С, кг/дм3 | групповой углеводородный состав, мас. % | |||||
| взято | мас. % на сырье | парафино-нафтеновые | МАУ | ДАУ | ПАУ | |
| Гидрогенизат дистиллятной фракции КУС | 100.0 | 0.901 | 77.35 | 21.39 | 1.19 | 0.06 |
| Получено | ||||||
| Н.К.–150°С | 5.1 | 0.791 | 91.75 | 8.23 | 0.00 | 0.00 |
| 150–250°С | 62.5 | 0.873 | 77.11 | 22.89 | 0.00 | 0.00 |
| Остаток | 31.6 | 0.932 | 72.20 | 23.85 | 3.76 | 0.19 |
Гидрирование дистиллятной фракции каменноугольной смолы сопровождалось заметным облегчением фракционного состава (табл. 7): приведенная температура конца кипения дистиллятной фракции КУС составляла около 400°С, в то время как 95 об. % отгонки стабильного гидрогенизата отвечала температура 315°С. Исходя из этого, представляется целесообразным отправлять остаточную фракцию на рецикл: в этом случае достигается как получение дополнительных количеств легких фракций, так и разбавлениевысокоароматизированного сырья гидрирования фракцией, богатой насыщенными УВ.
Основными компонентами легкой фракции гидрогенизата (Н.К.–150°С) являются циклогексан и его алкилзамещенные гомологи, причем содержание серы в бензиновой фракции также не превышало 3 м. д. Такое сочетание свойств обусловливает привлекательность этой фракции как сырья для каталитического риформинга.
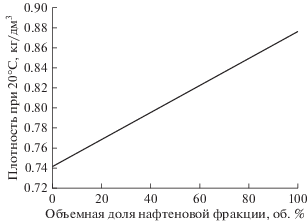
Компаундирование полученных топливных фракций. Ввиду низких значений плотностей выделенных из гидроизомеризатов керосиновых (изоалкановых) фракций, находящихся в пределах 0.741–0.751 кг/дм3 в зависимости от степени изомеризации продукта, для достижения приемлемых значений показателя необходимо компаундирование фракций-гидроизомеризатов с более плотными (нафтеновыми) керосиновыми фракциями гидрирования КУС. В зависимости от соотношения компонентов при компаундировании плотность компаунда может меняться в интервале от 0.741 до 0.880 кг/дм3 (рис. 6), что открывает возможность для выработки топлива с любой плотностью из находящихся внутри указанного интервала – как Джет А-1 (плотность при 20°С не менее 0.775 кг/дм3) так и Т-8В (плотность при 20°С не менее 0.800 кг/дм3).
Рис. 6.
Зависимость плотности компаунда, получаемого при смешении керосиновых фракций (150–250°С), выделенных из гидроизомеризата и продукта гидрирования дистиллятной фракции КУС.
Компаундированием фракций в различных соотношениях были получены два образца реактивных топлив, показатели качества которых были определены в объеме лабораторных испытаний. Результаты исследований опытных образцов на соответствие техническим требованиям приведены в табл. 9.
Таблица 9.
Показатели качества экспериментальных образцов синтетических реактивных топлив, полученных компаундированием
| № | Наименование показателя | Нормы ГОСТ 32592 для топлива ДЖЕТ A-1 | Нормы ГОСТ 12308 для топлива Т-8В | Фактические данные | |
|---|---|---|---|---|---|
| образец № 1 | образец № 2 | ||||
| 1 | Плотность при 20°С, кг/м3 | 775–840 (при 15°С) | >800 | 788 | 812 |
| 2 | Фракционный состав: | ||||
| – температура начала перегонки, °С – 10% отгоняется при температуре, °С – 50% отгоняется при температуре, °С – 90% отгоняется при температуре, °С – 98% отгоняется при температуре, °С – остаток от разгонки, % – потери от разгонки, % |
– <205 – – <300 <1.5 <1.5 |
>165 <185 – – <280 – – |
142 162 192 222 241 1.5 0.5 |
165 175 206 251 262 0.5 0.5 |
|
| 3 | Кинематическая вязкость, мм2/с, при температуре: | ||||
| 20°С –20°С |
– <8 |
>1.5 | 1.89 4.85 |
2.10 4.9 |
|
| –40°С | – | <16 | 9.94 | 10.2 | |
| 4 | Низшая теплота сгорания, кДж/кг | >42 800 | >42 900 | 43 400 | 43 190 |
| 5 | Высота некоптящего пламени, мм | >25 | >20 | 25 | 24 |
| 6 | Кислотность, мг KОН на/100 см3 топлива: | <0.1 | <0.7 | 0.27 | 0.31 |
| 7 | Иодное число, г иода на 100 г топлива | – | <0.9 | 0.13 | 0.32 |
| 8 | Температура вспышки в закрытом тигле, °С | >38 | >45 | 44 | 45 |
| 9 | Температура начала кристаллизации, °С | <–50 | <–0 | –60 | –59 |
| 10 | Термоокислительная стабильность в статических условиях: | ||||
| а) массовая концентрация осадка, мг/100 см3 топлива | – | <6 | 2.9 | 1.2 | |
| б) массовая концентрация растворимых смол, мг/100 см3 топлива | – | Не нормируется. Определение обязательно | 5.0 | 9.5 | |
| в) массовая концентрация нерастворимых смол, мг/100 см3 топлива | – | Не нормируется. Определение обязательно | 2.4 | 1.6 | |
| 11 | Массовая доля ароматических углеводородов, % | <25 | <22 | 9 | 16 |
| 12 | Содержание фактических смол, мг/100 см3 топлива | <7 | <4 | 2 | 3 |
| 13 | Массовая доля общей серы, % | <0.25 | <0.10 | Отс. | Отс. |
| 14 | Массовая доля меркаптановой серы, % | <0.003 | <0.001 | Отс. | Отс. |
| 15 | Испытание на медной пластинке | Выдерживает | Выдерживает | Выдерживает | Выдерживает |
| 16 | Содержание водорастворимых кислот и щелочей | – | Отс. | Отс. | Отс. |
| 17 | Зольность, % | – | <0.003 | 0.0018 | 0.0019 |
| 18 | Содержание механических примесей и воды | – | Отс. | Отс. | Отс. |
| 19 | Массовая доля сероводорода | – | Отс. | Отс. | Отс. |
| 20 | Массовая доля нафталиновых углеводородов, % | – | <2.0 | 0.3 | 0.9 |
| 21 | Взаимодействие с водой, баллы, не более: | ||||
| а) состояние поверхности раздела б) состояние разделенных фаз |
1b – |
1 1 |
1 1 |
1 1 |
|
| 22 | Удельная электрическая проводимость без антистатической присадки при температуре 20°С, пСм/м | <10 | <10 | 0 | 0.2 |
| 23 | Термоокислительная стабильность при контрольной температуре не ниже 275°С: | При 260°С | |||
| а) перепад давления на фильтре, кПа (мм рт. ст.) | <3.3 (25) | <3.3 (25) | 0 | 0 | |
| б) цвет отложений на трубке, баллы по цветовой шкале (при отсутствии нехарактерных отложений) | <3 | <3 | <1 | <1 | |
Опытный образец № 1 реактивного топлива, синтезированного из угля, полностью соответствует требованиям ГОСТ 32595-2013 к топливу Джет А-1. Опытный образец № 2 полностью соответствует требованиям ГОСТ 12308-2013 для топлива Т-8В.
Основываясь на полученных данных по физико-химическим и эксплуатационным показателям опытных образцов синтетических реактивных топлив, можно с большой долей вероятности предположить, что их применение на авиационной технике возможно как в смеси с товарными образцами реактивного топлива из нефтяного сырья, так и индивидуально с введенными противоизносной и антиокислительной присадками.
ВЫВОДЫ
В работе предложена схема комбинированной переработки каменного угля с получением синтетического реактивного топлива, применение которой проверено экспериментально. Показано, что при применении стандартного подхода, включающего коксование угля с последующей газификацией и синтезом Фишера−Тропша, получаемые керосиновые фракции характеризуются низкими значениями плотности (0.741–0.751 кг/дм3 при 20°С) относительно уровня, заданного техническими требованиями к используемым сегодня маркам реактивных топлив Джет А-1 и Т-8В. Для переработки парафинистой фракции синтеза Фишера–Тропша в компонент реактивного топлива был успешно применен процесс гидроизомеризации (Т = 330–345°С, ${{p}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ = 72 бар) в присутствии катализатора 2% Pt/Al-HMS (10), ранее эффективно использованного для гидрокрекинга–гидроизомеризации более тяжелых парафинистых продуктов (С19–С38) синтеза Фишера–Тропша.
Получаемая при коксовании угля каменноугольная смола была переработана с получением нафтенового компонента реактивного топлива, характеризующегося при пределах выкипания 150–250°С, плотностью около 0.873 кг/дм3 (при 20°С). Схема переработки включает фракционирование с получением дистиллятной фракции (выход 44 мас. % на исходную смолу при температуре конца отбора 2303°С), сопровождаемое глубоким гидрированием в присутствии промышленного катализатора НВС-А (Т = 390°С, ${{p}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ = 200 бар). Полученный продукт характеризуется высоким содержанием насыщенных углеводородов (77 мас. %) и низким содержанием серы (3 мг/кг).
Компаундированием полученных изопарафиновой и нафтеновой фракций были получены экспериментальные образцы синтетических реактивных топлив, исследованные в объеме лабораторных испытаний. Результаты испытаний демонстрируют принципиальную возможность получения реактивных топлив, удовлетворяющих техническим требованиям, предъявляемым к топливам марок Джет А-1 и Т-8В.Лабораторный технологический регламент получения из угля синтетических реактивных топлива типа Т-8В и Джет А-1 может служить основой для разработки промышленной технологии производства из угля реактивных топлив типа Т-8В и Джет А-1.
Список литературы
Aircraft Commerce. 2011. IV–V. Issue N75. P. 21.
Williams R.H., Larson E.D., Guangjian L., Kreutz T.G. // Energy Procedia. 2009. V. 1. № 1. P. 4379.
Hook M., Aleklett K. // Int. J. Energy Res. 2010. V. 34. P. 848.
Малолетнев А.С., Шпирт М.Я. // Рос. хим. журн. (Ж. Рос. хим. об-ва им. Д.И. Менделеева). 2008. Т. LII. № 6. С. 44.
Mochida I., Okuma O., Yoon S.-H. // Chem. Rev. 2014. V. 114. P. 1637.
Химическая технология твердых горючих ископаемых. Под ред. Макарова Г.Н. и Харламповича Г.Д. М.: Химия. 1986. 496 с.
Choudhary S., Thakur A., Gupta A., Ambekar S.B. // Int. Research J. of Engineering and Technology. 2016. V. 3. P. 2704.
Sadeqzadeh Boroujeni M. // Proceedings of the World Congress on Engineering and Computer Science 2008 (WCECS 2008), October 22–24. San Francisco, USA. 2008.
Зорина Г.И., Брун-Цеховой А.Р. Современное состояние технологии газификации угля за рубежом. М.: ЦНИИТЭнефтехим, 1986. 50 с.
Balster L.M., Corporan E., DeWitt M.J., Timothy Edwards J. et al. // Fuel Proc Technol. 2008. V. 89. № 4. P. 364.
Tao Kan, Wang H., He H., Li C., Zhang S. // Fuel. 2011. V. 90.P. 3404.
Яновский Л.С., Федоров Е.П., Варламова Н.И., Попов И.М., Бородако П.В., Пацина М.Н. // Двигатель. 2012. Т. 80. С. 6.
Федоров Е.П., Варламова Н.И., Яновский Л.С., Попов, И.М. // Двигатель. 2015. Т. 102. С. 8.
Лысенко С.В., Куликов А.Б., Онищенко М.И. и др. // Вестник Московского университета. Серия 2: Химия. 2015. Т. 56. № 6. С. 409.
Куликова М.В., Дементьева О.С., Кузьмин А.Е., Чудакова М.В. // Наногетерогенный катализ. 2016. Т. 1. № 2. С. 136.
Кузьмин А.Е., Куликова М.В., Дементьева О.С. // Наногетерогенный катализ. 2018. Т. 3. № 1. С. 40.
Мишин Н.Н., Куликов А.Б., Максимов А.Л. // Нефтехимия. 2014. Т. 54. № 5. С. 371.
Mishin N.N., Kulikov A.B., Maksimov A.L. // Petrol. Chem. 2014. V. 54. P. 366.
Дополнительные материалы отсутствуют.