

Журнал неорганической химии, 2020, T. 65, № 3, стр. 309-319
Синтез кальцийфосфатных порошков в неводных средах для стереолитографической 3D-печати
Д. С. Ларионов a, М. А. Кузина a, П. В. Евдокимов a, b, А. В. Гаршев a, b, Н. К. Орлов a, В. И. Путляев a, b, *
a Московский государственный университет им. М.В. Ломоносова, Факультет наук о материалах
119991 Москва, Ленинские горы, 1, с. 73, Россия
b Московский государственный университет им. М.В. Ломоносова, Химический факультет
119991 Москва, Ленинские горы, 1, с. 3, Россия
* E-mail: valery.putlayev@gmail.com
Поступила в редакцию 26.09.2019
После доработки 28.10.2019
Принята к публикации 01.11.2019
Аннотация
Приведены методики синтеза трикальцийфосфата Са3(РО4)2 (ТКФ), пирофосфата кальция Са2Р2О7 (ПФК) и композитов ТКФ/ПФК в неводных средах в диапазоне температур 25–300°С. Показано, что сольво- и ионотермальные методы синтеза позволяют получать фосфаты сложного состава. Описан фазовый состав, микроморфология и гранулометрия полученных порошков, а также их эволюция в процессе синтеза. Синтез при 110°С и медленном добавлении гидрофосфата аммония к этиленгликоляту кальция в этиленгликоле дает нанокристаллический порошок β-ТКФ со средним размером агрегатов пластинчатых кристаллов 150 нм и шириной распределения 50 нм. Морфологические характеристики и активность к спеканию порошков, синтезированных из растворов в этиленгликоле, позволяют использовать их для изготовления макропористой биокерамики методами стереолитографической 3D-печати.
ВВЕДЕНИЕ
Создание макропористой биокерамики на основе различных фосфатов кальция для замены и регенерации костной ткани с регулируемыми характеристиками биодеградации в организме (резорбции) – одно из важных направлений медицинского материаловедения [1, 2]. При имплантации в костную ткань такой материал первоначально функционирует как опорная и направляющая конструкция, а в дальнейшем контролируемо деградирует в растворной среде организма с одновременной заменой нативной костью. Другими непременными атрибутами биокерамики нового поколения являются остеокондуктивность – способность материала обеспечивать проходимость биологических потоков, прорастание в имплантат кровеносных сосудов (васкуляризация), адгезию и связывание остеогенных клеток, а также остеоиндуктивность – активное воздействие на процессы образования и роста костной ткани [3].
Традиционно применяемая керамика из гидроксиапатита – Са10(РО4)6(ОН)2 (ГА) практически не подвергается деградации в организме и не проявляет столь важной для имплантата способности индуцировать образование новой костной ткани [1, 2, 4]. Улучшение резорбционных свойств наблюдают у керамики, сформированной из трикальцийфосфата β-Са3(РО4)2 (β-ТКФ) [1, 5–7]. Повышение предела и скорости резорбции связано с уменьшением отношения Са/Р в материале имплантата. Это означает, в частности, переход к фосфатам с конденсированным анионом, в том числе к пирофосфату Са2Р2О7 (ПФК) и полифосфатам (полиФК), например к метафосфату Са(РО3)2 (метаФК) [8]. Контроль предела и скорости резорбции за счет изменения отношения Са/Р может быть обеспечен соотношением орто- и полифосфатов кальция в двухфазном керамическом композите.
Остеокондуктивные свойства определяет связная система макропор (диаметром не менее 100 мкм) [1, 9]. Расположение пор в пространстве (архитектура материала) должно обеспечивать максимальные проницаемость и прочность. Создание макропористой керамики с заданной архитектурой немыслимо без использования технологии 3D-печати, в частности стереолитографии [1, 10]. Данная технология выдвигает определенные требования к фазовому и гранулометрическому составу исходных фосфатных порошков: оптимальный средний размер частиц в диапазоне 100–300 нм, минимальная ширина распределения частиц по размерам, регулярная микроморфология – в идеале правильная огранка частиц [11]. Высокая основность ортофосфатного аниона ${\text{PO}}_{4}^{{3 - }}$ приводит к его гидролизу в водных растворах. Вследствие этого в водном растворе могут быть синтезированы лишь кислые ортофосфаты кальция и ГА [1, 4]. Синтез порошка трикальциевого фосфата Са3(РО4)2 из солевых прекурсоров в любом случае требует высоких температур. Рекристаллизация ТКФ при высоких температурах закономерно приводит как к увеличению среднего размера частиц, так и к значительному уширению распределения частиц по размерам. Синтез микрокристаллических фосфатных порошков с узким распределением кристаллов по размерам и определенной микроморфологией путем осаждения из растворов возможен, таким образом, лишь в неводных средах.
В литературе синтез фосфатов кальция в неводных растворах освещен достаточно плохо и в основном в отношении осаждения ТКФ [12–14]. В считанных работах рассматривается синтез ТКФ в водно-метанольных [12] и водно-этиленгликольных растворах [13, 14]. Из результатов подобных работ следует, что синтез микрокристаллических (0.2–1 мкм) порошков ТКФ из растворов в принципе возможен, однако присутствие воды не позволяет сформировать порошки с регулярной морфологией. Эти синтезы нельзя назвать в полной мере неводными, поскольку на первом этапе авторы получают плохо закристаллизованную фазу, которую при дальнейшем нагревании, оформленном как отдельный этап синтеза, кристаллизуют в безводном этиленгликоле. Отсутствует мотивация выбора растворителя и попытки выйти за рамки протонных растворителей.
Цель настоящей работы – разработка способов синтеза порошков фосфатов кальция с определенной гранулометрией в сольво- и ионотермальных условиях для нужд стереолитографического получения макропористой биокерамики. Поставлены задачи выбора неводных растворителей и ионных жидкостей для проведения сольво- и ионотермальных синтезов орто- и полифосфатов кальция; выбора исходных кальций- и фосфорсодержащих реагентов для получения искомых фосфатов кальция и/или их смесей; выявление условий проведения сольво- и ионотермальных синтезов для получения порошков орто- и полифосфатов кальция с определенным фазовым составом и гранулометрией, а также оценка активности полученных порошков в процессе спекания.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе был использован достаточно широкий круг растворителей, критерии выбора которых традиционно включали сольватационные, диэлектрические и диффузионные характеристики [15]. Ниже указаны методики практически значимых синтезов, проведенных в среде этиленгликоля (ЭГ), наиболее удачной, на наш взгляд, для разработки практически приемлемой методики осаждения фосфатов кальция.
Синтез этиленгликолята кальция
Навеску металлического кальция (>99.1%, CAS 7440-70-2, Carl Roth GmbH + Co KG) помещали в безводный этиленгликоль (“ч. д. а.”, >99.5%, ГОСТ 10164-75, 200–1000 ppm воды по Фишеру). Синтез проводили по реакции:
(1)
${\text{Ca}} + 2{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}{{\left( {{\text{OH}}} \right)}_{2}} \to {{\left( {{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}}\left( {{\text{ОН}}} \right){\text{О}}} \right)}_{2}}{\text{Ca}} + {{{\text{H}}}_{2}}{\kern 1pt} \uparrow $Синтез ортофосфата натрия, безводной ортофосфорной кислоты (исходя из [17–19]), ортофосфата кальция в этиленгликоле из CaCl2 и Na3PO4 · 12H2O (способ 1), а также из этиленгликолята кальция и безводной Н3РО4 (способ 2) описан в Дополнительных материалах.
Синтез ортофосфата кальция в этиленгликоле (способ 3)
Для проведения синтеза в качестве источника фосфат-ионов использовали (NH4)2HPO4 (≥99.99%, Sigma-Aldrich). Источником ионов кальция служил полученный ранее 0.15 М раствор этиленгликолята кальция. Гидрофосфат аммония растворяли в этиленгликоле при температуре 60–80°С (0.1 М). В реакцию вступал, скорее всего, не гидрофосфат, а дигидрофосфат аммония. При нагревании гидрофосфата аммония выше 80°С в водном растворе или более 110°С в виде твердого реактива на воздухе происходит потеря аммиака с превращением:
(2)
${{\left( {{\text{N}}{{{\text{H}}}_{{\text{4}}}}} \right)}_{{\text{2}}}}{\text{HP}}{{{\text{O}}}_{4}} \to {\text{N}}{{{\text{H}}}_{{\text{4}}}}{{{\text{H}}}_{{\text{2}}}}{\text{P}}{{{\text{O}}}_{4}} + {\text{N}}{{{\text{H}}}_{3}}{\kern 1pt} \uparrow {\kern 1pt} .$С этим связано резкое увеличение растворимости фосфата (0.6 г при 25°C и 1.8 г в 100 г ЭГ при 80°С). Однако при температурах >180°С на воздухе наряду с удалением аммиака происходит и удаление воды [12]. Последнее означает поликонденсацию ортофосфатного аниона с образованием сначала пиро-, а затем и полифосфатов. По этой причине синтез ортофосфата кальция ограничен температурой 150–180°С. Таким образом, независимо от выбора фосфата аммония в горячем растворе идет реакция с дигидрофосфатом. При температурах 25–150°С растворы сливали или медленно прикапывали (∼0.5 мл/мин) друг к другу:
(3)
$\begin{gathered} {\text{3}}{{\left( {{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}}\left( {{\text{ОН}}} \right){\text{О}}} \right)}_{{\text{2}}}}{\text{Ca}} + 2{\text{N}}{{{\text{H}}}_{{\text{4}}}}{{{\text{H}}}_{{\text{2}}}}{\text{P}}{{{\text{O}}}_{4}} \to \\ \to {\text{C}}{{{\text{a}}}_{{\text{3}}}}{{\left( {{\text{P}}{{{\text{O}}}_{{\text{4}}}}} \right)}_{{\text{2}}}}{\kern 1pt} \downarrow + \,6{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}{{\left( {{\text{OН}}} \right)}_{2}} + 2{\text{N}}{{{\text{H}}}_{3}}{\kern 1pt} \uparrow {\kern 1pt} . \\ \end{gathered} $Осадок перемешивали при температуре сливания на протяжении 4–5 ч, осаждали центрифугированием, а затем высушивали на нагретой металлической пластине в течение 1 ч при температуре 90°С.
В ряде случаев в растворы солей в этиленгликоле добавляли воду, чтобы оценить пороговые значения количества воды, влияющие на образование промежуточной фазы гидроксиапатита. При pH 5.5–7 в присутствии воды происходит реакция:
(4)
$\begin{gathered} \left( {10 - x} \right){{\left( {{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{4}}\left( {{\text{ОН}}} \right){\text{О}}} \right)}_{2}}{\text{Ca}} + \\ + \,\,6{\text{N}}{{{\text{H}}}_{{\text{4}}}}{{{\text{H}}}_{{\text{2}}}}{\text{P}}{{{\text{O}}}_{4}} + \left( {2\, - \,x} \right){{{\text{H}}}_{{\text{2}}}}{\text{O}} \to \\ \to {\text{C}}{{{\text{a}}}_{{10--x}}}{\kern 1pt} {{\left( {{\text{HP}}{{{\text{O}}}_{4}}} \right)}_{x}}{\kern 1pt} {{\left( {{\text{P}}{{{\text{O}}}_{4}}} \right)}_{{6 - x}}}{\kern 1pt} {{\left( {{\text{OH}}} \right)}_{{2 - x}}}{\kern 1pt} \downarrow + \\ + \,\,6{\text{N}}{{{\text{H}}}_{3}}{\kern 1pt} \uparrow + {\text{ }}\left( {20 - 2x} \right){{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{4}}{{\left( {{\text{OН}}} \right)}_{2}}. \\ \end{gathered} $Установка для синтеза ТКФ в этиленгликоле способом 3 состоит из трехгорлой колбы с обратным холодильником, капельной воронки для добавления фосфатного реагента и термометра.
Рентгенофазовый анализ (РФА) проводили на рентгеновском дифрактометре Rigaku D/Max-2500 с вращающимся анодом (Япония). Съемку осуществляли в режиме на отражение (геометрия Брегга–Брентано) в кварцевых кюветах без усредняющего вращения с использованием CuKα-излучения. Качественный анализ полученных рентгенограмм выполняли с использованием базы данных ICDD PDF-2.
Распределение частиц по размерам оценивали с помощью принципа динамического рассеяния света на приборе Zetasizer Nano ZS в диапазоне 0.3 нм–10 мкм, а также на приборе лазерной дифракции Fritsch Analysette-22 в диапазоне 0.2–100 мкм.
Растровую электронную микроскопию (РЭМ) проводили на растровом электронном микроскопе с автоэмиссионным источником LEO SUPRA 50VP (Carl Zeiss, Германия). Ускоряющее напряжение электронной пушки составляло 5–20 кВ. Изображения получали во вторичных электронах (SE2) при увеличениях до 100 000×.
Просвечивающую электронную микроскопию (ПЭМ) осуществляли на микроскопе Libra 200 (Zeiss, Германия) с катодом с термополевой эмиссией при ускоряющем напряжении 200 кВ. Изображения детектировали с помощью CCD-камеры Ultra Scan 4000 (Gatan, США). Обработку полученных изображений производили в свободном программном пакете Gwyddion.
Содержание воды в растворителях определяли на кулонометрическом титраторе 756 KF Coulometer (metrohm ion analysis) по методу Фишера.
Термогравиметрический и дифференциальный термический анализ (ТГ и ДТА) проводили на термоанализаторе Perkin Elmer Pyris (Perkin Elmer, США) и STA 409 PC (Netzsch, Германия). Скорость нагрева составляла 5 и 10 град/мин, интервал температур 20–1500°С. Для исследования отходящих газов использовали квадрупольный масс-спектрометр Aerolos QMS 403 C (Netzsch, Германия) с нагревом капиллярной системы входного отверстия.
Дифференциальную сканирующую калориметрию (ДСК) проводили с помощью дифференциального сканирующего калориметра Netzsch DSC 200 F3 Maia в интервале температур 28–175°С с шагом нагревания 2 град/мин.
ИК-спектры поглощения образцов регистрировали на ИК-спектрофотометре Spectrum One (Perkin–Elmer, США) с использованием приставки диффузного отражения.
Дилатометрию прессованных образцов проводили в политермическом режиме до t = 1100 и 1200°С со скоростью нагрева 5 град/мин в горизонтальном дилатометре DIL 402 C (Netzsch, Германия), нагрузка составляла 30 сН.
Для расчета геометрической плотности высушенных на воздухе столбиков (после проведения шлифовки для механических испытаний) измеряли массу образцов (с точностью до 10–4 г) и линейные размеры (с точностью до 10 мкм, микрометр). Относительную ошибку измерения оценивали сверху как сумму относительных ошибок измеряемых величин.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В качестве неводных растворителей в сольвотермальных синтезах использовали этанол,
этиленгликоль, диэтиленгликоль, бутандиол-1,4, гександиол-1,6, глицерин, а в случае
ионотермального синтеза – два типа ионных жидкостей: а) на основе имидазольного катиона
– [emim]Br (tпл = 77°С, tразл ∼ 270°С, гидрофильный анион) и [emim][NTf2] (tпл = –3°С, tразл ∼ 400°С, слабогидрофильный анион), где  ≡ [emim] – катион 1-этил-3-метилимидазола; [(CF3SO2)2N]– ≡ ≡ [NTf2] – анион бис(трифторметил)сульфонилимида; б) расплав нитратов натрия и калия. Фактически растворители
принадлежали двум группам: низкокипящие с температурой кипения <100°С (этанол) и высококипящие
с температурой кипения >100°С (остальные). В конечном итоге эта характеристика, коррелирующая
с вязкостью и скоростью диффузии ионов, а не только полярность и диэлектрические свойства,
влияющие на сольватацию ионов, определяет практические результаты синтезов. Результаты
синтезов приведены в табл. S1
. Плохая растворимость реагентов (особенно фосфатного) в большинстве растворителей
и быстрое добавление реагентов друг к другу приводят к очень большим пересыщениям
растворов и, как следствие, к выпадению аморфных осадков даже при повышенных температурах.
При отсутствии оснований, способных связывать протоны гидро- и дигидрофосфатных реагентов,
образуется конкурирующая фаза монетита СаНРО4. Только в случае синтезов в этиленгликоле удается реализовать приемлемую с практической
точки зрения методику синтеза достаточных количеств (до 20 г в единичном синтезе с
выходом, близким к 100%) кристаллических порошков заданного состава и морфологии.
≡ [emim] – катион 1-этил-3-метилимидазола; [(CF3SO2)2N]– ≡ ≡ [NTf2] – анион бис(трифторметил)сульфонилимида; б) расплав нитратов натрия и калия. Фактически растворители
принадлежали двум группам: низкокипящие с температурой кипения <100°С (этанол) и высококипящие
с температурой кипения >100°С (остальные). В конечном итоге эта характеристика, коррелирующая
с вязкостью и скоростью диффузии ионов, а не только полярность и диэлектрические свойства,
влияющие на сольватацию ионов, определяет практические результаты синтезов. Результаты
синтезов приведены в табл. S1
. Плохая растворимость реагентов (особенно фосфатного) в большинстве растворителей
и быстрое добавление реагентов друг к другу приводят к очень большим пересыщениям
растворов и, как следствие, к выпадению аморфных осадков даже при повышенных температурах.
При отсутствии оснований, способных связывать протоны гидро- и дигидрофосфатных реагентов,
образуется конкурирующая фаза монетита СаНРО4. Только в случае синтезов в этиленгликоле удается реализовать приемлемую с практической
точки зрения методику синтеза достаточных количеств (до 20 г в единичном синтезе с
выходом, близким к 100%) кристаллических порошков заданного состава и морфологии.
В итоге предложена схема кислотно-основного взаимодействия в среде этиленгликоля (оптимального, на наш взгляд, протонного растворителя):
(5)
$3{{\left( {{{{\text{С}}}_{{\text{2}}}}{{{\text{Н}}}_{{\text{4}}}}{\kern 1pt} \left( {{\text{ОН}}} \right){\text{О}}} \right)}_{{\text{2}}}}{\kern 1pt} {\text{Ca}} + 2{\text{A}}{{{\text{H}}}_{{\text{2}}}}{\text{P}}{{{\text{O}}}_{4}} \to {\text{C}}{{{\text{a}}}_{{\text{3}}}}{{\left( {{\text{P}}{{{\text{O}}}_{{\text{4}}}}} \right)}_{{\text{2}}}}{\kern 1pt} \downarrow ,$Сразу после быстрого осаждения образуются в основном аморфные порошки вне зависимости от температуры сливания и времени дальнейшего перемешивания, даже при 150°С (рис. 1а, 1б, 1в), однако обжиг при 900°С (1 ч) дает кристаллический β-ТКФ без примесей (рис. 1г). Помимо мелких рентгеноаморфных частиц в осадке есть отдельные крупные (1–2 мкм) аморфные пластинчатые частицы, возникающие в результате расслаивания раствора. Анализ рентгенограммы (рис. 1в) указывает на отличия в ближней координационной сфере по сравнению с гидратированным аморфным ТКФ (называемым также аморфным фосфатом кальция – АФК), осаждаемым из водных растворов. Для гидратированного ТКФ максимум гало соответствует межатомному расстоянию 0.29 нм (Са–Са), структурно его описывают как совокупность кластеров Познера Са9(РО4)6 [20]. Структура осажденного из ЭГ аморфного ТКФ иная – он более “рыхлый” (максимум гало соответствует 0.39 нм). Возможное объяснение состоит в сильной сольватации ионов Са ЭГ за счет образования комплексов, в которых Са координирует молекулы растворителя [21, 22]. Термический анализ регистрирует заметное количество растворителя в осадке, который не удаляется при температуре кипения ЭГ (197°С), а пиролизуется и остается вплоть до 800°С (рис. S1); при этом порошок приобретает светло-коричневый цвет. Что касается водного ТКФ, то вода содержится как в пустотах между кластерами Познера (сильно связанная, кристаллогидратная вода), так и в системе мезопор, пронизывающих частицы фосфата [20, 23, 24]. Следы воды оказывают очень сильное влияние на морфологию продукта синтеза в ЭГ: размер частиц вырастает до 1–1.5 мкм с исходных 80–90 нм; это, по-видимому, вызвано гидратацией аморфного ТКФ в нанокристаллический апатит. По данным ПЭМ (рис. 2), быстро осажденные образцы представлены пластинчатыми мезопористыми частицами, как и в случае аморфного ТКФ водного синтеза, что говорит об общем механизме образования таких частиц – расслаивании исходного высококонцентрированного раствора [25] (рис. 2в, 2г). Указанные частицы в дальнейшем формируются в более компактные аморфные равноосные частицы (агломераты), частично наследующие мезопоры (в случае осаждения из водного раствора – это кристаллы наногидроксиапатита) (рис. 2б). Эти компактные частицы, являющиеся, по-видимому, агломератами первичных частиц с размерами не более 10–20 нм (рис. 2а), при дальнейшем росте становятся менее сферичными с тенденцией к уплощению и огранке (рис. 2а, врезка). Следует также отметить, что, как и в случае аморфного ТКФ водного синтеза, в мезопорах, вероятно, содержится раствор, в данном случае на основе ЭГ.
Рис. 1.
РФА осадка ТКФ, осажденного в растворе ЭГ при 110°С (а). Микроморфология осадка ТКФ после выдерживания в растворе ЭГ в течение 5 ч при 110°С представлена крупными (1–2 мкм) аморфными пластинчатыми частицами, возникшими в результате расслаивания раствора (б). РФА осадка ТКФ после обжига при 900°С в течение 1 ч (в). Микроморфология осадка ТКФ после обжига при 900°С в течение 1 ч (г).
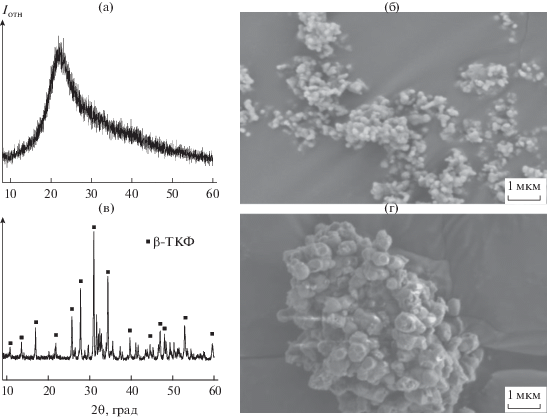
Рис. 2.
ПЭМ-микрофотографии ТКФ, осажденного из ЭГ при 110°С в течение 4 ч. Удаление ЭГ из пасты после центрифугирования путем высушивания при 120°С (пластинчатые и изометричные частицы). На врезке видно, что изометричные частицы имеют тенденцию к огранке (а). Мезопористость пластинчатых и изометричных частиц, на врезке – электронная дифракция (б). Система мезопор в пластинчатых частицах: мелкие (10 нм) поры – результат синтеза, крупные (50–200 нм) – результат нагревания, в том числе под электронным пучком (в). Слоистая агрегация пластинчатых частиц (г).

В целом, эволюция полидисперсной системы в сторону огрубления в ЭГ гораздо более медленная, чем в воде. Это, несомненно, связано: а) с худшей растворимостью ТКФ в ЭГ за счет очень плохой сольватации ортофосфатного аниона и невозможности его протонирования, б) в ситуации, когда процессы роста частиц перестают определяться растворением–осаждением, одним из вариантов роста становится ориентированное присоединение первичных частиц к агломерату, однако большая вязкость растворителя затрудняет этот процесс.
Влияние температуры и времени осаждения
Для уменьшения значительного пересыщения относительно кристаллизации фосфата кальция, которое возникает при быстром сливании растворов, раствор фосфата аммония медленно прикапывали к раствору этиленгликолята кальция в ЭГ при 25–150°С, а затем перемешивали осадок для лучшего формирования в течение 1–3 ч. Повышение температуры, как и быстрое сливание растворов, вносило свой вклад в уменьшение пересыщения. Однако ключевым фактором оказалась скорость добавления раствора фосфата аммония. При медленном прикапывании (∼0.5 мл/мин) порошки ТКФ успевают закристаллизоваться в растворе. Таким образом, нам впервые удалось в растворной среде низкотемпературным неводным методом получить β-ТКФ с подходящей морфологией и гранулометрией для стереолитографической печати (рис. 3).
Рис. 3.
Данные РФА (а), динамического светорассеяния осадков, полученных медленным прикапыванием растворов солей в этиленгликоле при 110, 130 и 150°С с последующим перемешиванием в течение 1 ч (б). Микроморфологии осадков ТКФ, полученных при 110 (в), 130 (г) и 150°С (д) с последующим перемешиванием в течение 1 ч; масштабный отрезок на снимках соответствует 1 мкм.
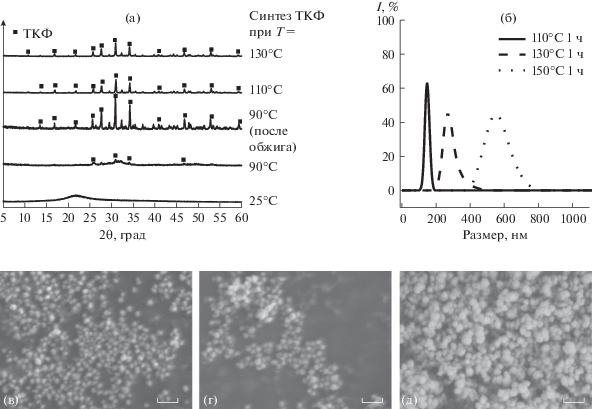
Стоит отметить, что, как и в синтезах с быстрым сливанием растворов солей, температурная граница 100°С важна для формирования итоговой фазы. Как видно на рис. 3а, при 90°С осадок не успевает полностью закристаллизоваться за 1 ч (возможно, для этого требуется большее время), а при 25°С осадок полностью рентгеноаморфен. Для определения температуры начала кристаллизации образца ТКФ, полученного прикапыванием растворов при 25°С (рентгеноаморфный осадок), проводили сравнительную дифференциальную сканирующую калориметрию образца рентгеноаморфного ТКФ в ЭГ и чистого ЭГ и вычитали тепловой вклад испарения ЭГ из тепловых эффектов, происходящих в системе ТКФ–ЭГ с учетом потери массы. На разностной кривой ДСК хорошо заметен тепловой эффект выше 100°С (рис. S3). При этой же температуре в ходе синтеза образуется кристаллический β-ТКФ в растворе, следовательно, данную температуру можно условно считать минимальной для кристаллизации ТКФ в этиленгликоле. Помимо температуры осаждения на размеры частиц ТКФ влияет время перемешивания после осаждения ТКФ. С увеличением времени перемешивания в горячем растворе распределение частиц по размерам становится шире, но их средний размер меняется незначительно.
Влияние следов воды на морфологию и состав осадка
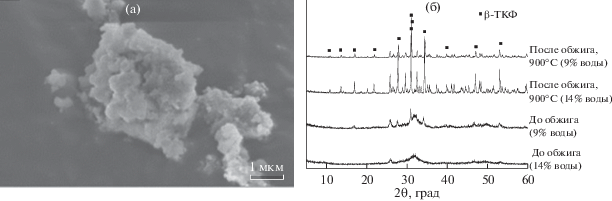
С целью изучения влияния порционных количеств воды на состав полученного ортофосфата кальция синтезы проводили в системе этиленгликоль–вода. На рис. 4а представлены данные РФА осадков, полученных в растворе ЭГ с добавлением воды в соотношении 10 : 1 и 6 : 1 соответственно. При наличии воды в растворе кристаллизация ТКФ замедляется, в то же время с увеличением содержания воды увеличивается примесь нестехиометрического ГА. После обжига осадок представляет собой кристаллический ТКФ. На рис. 4б представлены данные РЭМ осадков, полученных в растворе ЭГ : вода = 10 : 1. После осаждения порошки представляют собой пластинчатые частицы нестехиометрического гидроксиапатита, таким образом, даже маленькие порции воды в растворе приводят к нарушению морфологии осадка. После обжига при 900°С порошок представляет собой кристаллический ТКФ. Это соответствует результату реакции разложения нестехиометрического ГА с Са/Р = 1.5. Таким образом, даже небольшое количество воды в растворе ведет к искажению состава и морфологии порошка, и дальнейшее получение ТКФ возможно только обжигом. Смещение стехиометрии состава осадка в сторону стехиометрического ГА (Сa/P = 1.67) не позволяет получить чистый кристаллический β-ТКФ даже после термической обработки. По данным ИК-спектроскопии, в осадках, полученных при 110 и 130°С, меньше OH-групп, которые могут относиться к воде или этиленгликолю, чем в осадках из раствора ЭГ–вода и осадка, полученного при 90°С (рис. S4). Однозначный вывод о влиянии воды на состав осадка на основании спектров сделать нельзя, так как наблюдается сильное перекрывание полос этиленгликоля, ${\text{HPO}}_{{\text{4}}}^{{{\text{2}}--}}$ (в работе [26] сообщается о частичном протонировании аниона в неводном ТКФ) и воды, а также полос этиленгликоля и ${\text{PO}}_{{\text{4}}}^{{{\text{3}}--}}.$ Заметных пиков валентных колебаний ОН-групп и воды в области 3000 см–1 не обнаружено. Спектры образцов, синтезированных при 110 и 130°С, отвечают β-ТКФ, а спектры образцов, полученных при 90°С, с добавками воды в ЭГ – аморфному ТКФ или нестехиометрическому ГА.
Реальная структура кристаллического осадка ТКФ
Исследование осадка методом ПЭМ (рис. 5) указывает на то, что равноосные частицы, наблюдаемые на РЭМ-изображениях, представляют собой агрегаты тонких кристаллических пластин с неровными краями. В некоторых случаях заметен характерный пятнистый контраст, который можно отнести к мезопорам или остатку аморфной фазы (рис. 5а). Такую морфологию следует соотнести со структурой аморфного осадка (рис. 2). При кристаллизации наследуется его пластинчатое строение, размеры пластин в плане становятся меньше. Можно предположить, что слоистость осадков связана с особенностями координации кальция молекулами ЭГ: относительно прочными координационными связями в слое по типу мостиковая молекула ЭГ между двумя атомами Са и более слабыми, по-видимому, водородными связями между слоями (строение, присущее координационному полимеру [21]). Эта ситуация резко отличается от изотропного строения осадка аморфного фосфата кальция в водном растворе, состоящего из изометричных ионных пар Са9(РО4)6 – кластеров Познера. Следует отметить и разницу с описанной в литературе морфологией ТКФ, осажденного из ЭГ со значительным содержанием воды, в виде агрегатов в форме пористых гексагональных пластин размерами >500 нм [13, 14]. Это напоминает морфологию агрегатов, осажденных нами в присутствии воды (рис. 4б), однако важно отметить, что в этом случае осадок всегда загрязнен примесями ГА или монетита СаНРО4.
Рис. 5.
ПЭМ-изображения кристаллических частиц ТКФ, осажденных из ЭГ при 130°С. Контраст вследствие неоднородности (мезопоры или аморфная фаза), на краях образца разрешена кристаллическая решетка; на врезке два агрегата частиц, один – в плане, второй (слева) виден в профиль (а). Агрегаты частиц, на врезке электронная дифракция с области, свидетельствующая о развороте слоев друг относительно друга в слоистых агрегатах (б).

Сравнение свойств порошков ТКФ твердофазного и неводного синтеза и керамик на их основе
Для сравнения использовали β-ТКФ, полученный в ЭГ медленным прикапыванием растворов при 110°С с последующим перемешиванием в течение 2 ч и сушкой при 90°С в течение 2 ч (средний размер чаcтиц 150 нм), и β-ТКФ, полученный обжигом стехиометрической смеси пирофосфата и карбоната кальция при 900°С (размер частиц после помола бисером 0.5–1 мкм). На рис. 6а представлена микроструктура керамики из порошка с ЭГ-предысторией с хорошо сформированными зернами со средним размером 1 мкм. Мелкие поры внутри зерен свидетельствуют о том, что в процессе спекания наноразмерных частиц происходит быстрая рекристаллизация и отделение движущихся высокоугловых границ от пор. Это означает, что режимы спекания для такого мелкодисперсного порошка должны быть иными. Возможно, необходим так называемый режим двухстадийного спекания, при котором основную (вторую, длительную) стадию спекания нужно проводить при более низкой температуре (вероятно, при 1050°С) по сравнению с контрольным образцом, полученным твердофазным синтезом. Зерна образца на рис. 6б имеют более широкое распределение по размерам, как и порошок до обжига. Обжиг при 1200°С приводит к дальнейшей рекристаллизации обоих типов образцов. Становятся заметными трещины, вызванные полиморфным переходом β → α в ТКФ, сопровождающимся увеличением объема. Это означает, что параметры данного превращения сохраняются для более мелкодисперсного порошка с ЭГ-предысторией. Таким образом, температура обжига керамики β-ТКФ не должна превышать 1100°С. На рис. 6в представлен характер уплотнения образцов по данным дилатометрии со смешанным полиизотермическим режимом (5 град/мин до 1100 или 1200°С, затем изотермическая выдержка).
Рис. 6.
Микрофотографии таблеток из ТКФ после обжига при 1100 и термотравления при 1000°С. Синтез в ЭГ и обжиг при 1100°С в течение 12 ч (а), твердофазный синтез и обжиг при 1100°С в течение 12 ч (б). Данные дилатометрии для образцов ТКФ, температурный интервал 40–1100 или 40–1200°С (в). Линии – график температурных режимов, линия + + квадрат – образец “твердофазного” ТКФ, нагретый до 1100 и 1200°С (серая и черная линии соответственно). Линия + + треугольник – образец “неводного” ТКФ, нагретый до 1100 и 1200°С (серая и черная линии соответственно). Изменение плотности при спекании керамики из ТКФ (г). 1, 3 – порошок, полученный синтезом в ЭГ при 110°С (перемешивание 2 ч, сушка при 90°С 2 ч), до и после обжига при 1100 и 1200°С соответственно (6 ч); 2, 4 – порошок, полученный твердофазным спеканием с последующей дезагрегацией и добавлением 5%-ного полиэтиленгликоля, до и после обжига при 1100 и 1200°С соответственно (12 ч).
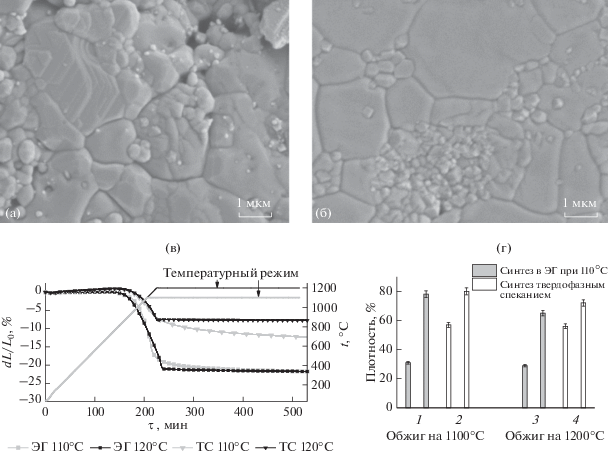
Дилатометрический анализ образцов показывает, что морфология частиц ТКФ, полученного в неводной среде, позитивно влияет на процесс спекания, в результате чего образец “неводного” ТКФ претерпевает существенно бóльшую усадку (причем подавляющая ее часть происходит на политермическом участке), чем образец “твердофазного” ТКФ. Замедление усадки при выходе на изотерму 1200°С несомненно связано с превращением β → α ТКФ, которое сопровождается расширением образца. По данным дилатометрии можно утверждать, что “неводный” образец ТКФ демонстрирует большую скорость уплотнения.
На рис. 6г представлено изменение плотности образцов из “неводного” и “твердофазного” ТКФ после обжига при 1100 и 1200°С. Первоначальная плотность таблетки из “неводного” ТКФ гораздо ниже, чем из “твердофазного”, так как в первом случае порошок монодисперсный с узким распределением по размерам, а во втором – распределение по размерам достаточно широкое. Однако в процентном соотношении изменение плотности первого образца гораздо больше. Это связано не только с наличием в осадке остаточного этиленгликоля даже после сушки (что подтверждают данные ТГ), но и с большей активностью нанокристаллических частиц, сформировавшихся в растворе.
Это коррелирует с картиной рекристаллизации порошков, представленной выше. В порошках “неводного” ТКФ первоначально происходит быстрая рекристаллизация, которая сопровождается относительно быстрым удалением значительной доли пор. Однако в дальнейшем поры оказываются “запертыми” внутри зерен, и их удаление требует очень длительных обжигов. Таким образом, при заметно лучшей способности к рекристаллизации порошки с этиленгликольной предысторией демонстрируют сопоставимую с твердофазными порошками способность к уплотнению.
Нами также апробирован синтез порошков пирофосфата кальция Са2Р2О7 (ПФК), композитов ПФК/ТКФ и ПФК/полиФК, а также замещенных ортофосфатов с общей формулой Ca(3– х)Na2х(PO4)2. Отмечено, что синтез кристаллических порошков ПФК и ПФК/ТКФ подобными методами сильно затруднен. В случае полифосфатов кальция (полиФК) и композитов ПФК/полиФК не удается добиться даже заметного количественного осаждения, что в определенной мере воспроизводит ситуацию синтеза из водных растворов. В случае замещенных ортофосфатов не удается воспроизвести стехиометрию раствора в соответствующем осадке. Более детальное описание представлено в Дополнительных материалах.
В целом, разработанные методики синтеза позволили получить достаточное количество порошков ТКФ, ПФК и ТКФ/ПФК (до 20 г), что дало возможность апробировать стереолитографическую печать макропористой биокерамики с архитектурой Кельвина на их основе. Эти результаты будут сообщены в последующих публикациях.
ЗАКЛЮЧЕНИЕ
Разработана методика синтеза фосфатов кальция в среде этиленгликоля, которая заключается в медленном добавлении этиленгликольного раствора кислых фосфатов аммония к раствору этиленгликолята Са в диапазоне температур 110–150°С и использовании: а) кислых фосфатов аммония для обеспечения эффективной сольватации фосфата за счет образования водородных связей с протонным растворителем и, в конечном итоге, высокой растворимости фосфатного реагента, б) сильного сопряженного основания С2Н4(ОН)О– для связывания протонов кислых фосфатов аммония. Показано, что этот способ синтеза обладает достаточно большой универсальностью, что позволяет разрабатывать синтез фосфатов сложного состава.
При осаждении ТКФ из раствора в ЭГ первоначально выпадает слоистый аморфный продукт, который кристаллизуется выше 100°С в фазу β-Са3(РО4)2. Медленное добавление фосфатного реагента к этиленгликоляту кальция позволяет осадить из раствора кристаллический ТКФ. Синтез при 110°С и медленном прибавлении гидрофосфата аммония к этиленгликоляту кальция дает микрокристаллический порошок β-Са3(РО4)2 со средним размером агрегатов пластинчатых кристаллов 150 нм и шириной распределения 50 нм. Средний размер частиц и ширина распределения по размерам быстро возрастают с ростом температуры и в меньшей степени с увеличением времени синтеза.
Синтезированные из растворов в ЭГ микрокристаллические порошки обладают высокой активностью в процессах спекания по сравнению с порошками, синтезированными твердофазным методом. Это выражается в их более быстрой рекристаллизации при сопоставимом характере уплотнения, что требует разработки для них специальных режимов спекания, направленных на подавление рекристаллизационных явлений. Морфологические характеристики и активность к спеканию порошков β-Са3(РО4)2, синтезированных из растворов в этиленгликоле, делают их приемлемыми для изготовления макропористой биокерамики методами стереолитографической 3D-печати.
Список литературы
Habraken W., Habibovic P., Epple M. et al. // Mater. Today. 2016. V. 19. № 2. P. 69. https://doi.org/10.1016/j.mattod.2015.10.008
Suchanek W., Yashimura M. // J. Mater. Res. 1998. V. 13. № 1. P. 94. https://doi.org/10.1557/JMR.1998.0015
Baron R. // Anat. Rec. 1989. V. 224. № 2. P. 317. https://doi.org/10.1002/ar.1092240220
Сафронова Т.В., Путляев В.И. // Наносистемы: физика, химия, математика. 2013. Т. 4. № 1. С. 24.
Joeris A., Ondrus S., Planka L. et al. // Eur. J. Pediat. Surg. 2010. V. 20. № 1. P. 24. https://doi.org/10.1055/s-0029-1241866
LeGeros R.Z., Lin S., Rohanizadeh R. et al. // J. Mater. Sci.-Mater. Med. 2003. V. 14. № 3. P. 201. https://doi.org/10.1023/A:1022872421333
Seidenstuecker M., Orcid L.K., Bernstein A. et al. // Materials. 2017. V. 11. № 1. P. 1. https://doi.org/10.3390/ma11010013
Dorozhkin S.V., Epple M. // Angew. Chem. Int. Ed. 2002. V. 41. № 17. P. 3130. https://doi.org/10.1002/1521-3773(20020902)41:17<3130:: AID-ANIE3130>3.0.CO;2-1
Eggli P.S., Mueller W., Schenk R.K. // Clin. Orthop. Relat. Res. 1988. V. 232. P. 127.
Ievlev V.M., Putlyaev V.I., Safronova T.V. et al. // Inorg. Mater. 2015. V. 51. № 13. P. 1297. https://doi.org/10.1134/S0020168515130038
Sun C., Zhang X. // Sens. Actuators, A. 2002. V. 101. № 3. P. 364. https://doi.org/10.1016/S0924-4247(02)00264-9
Jong-Shing Bow, Sz-ChianLiou, San-YuanChen // Biomaterials. 2004. V. 25. № 16. P. 3155. https://doi.org/10.1016/j.biomaterials.2003.10.046
Jinhui Tao, Haihua Pan, Halei Zhai et al. // Cryst. Growth Des. 2009. V. 9. № 7. P. 3144. https://doi.org/10.1021/cg801130w
Galea L., Bohner M., Thuering J. et al. // Acta Biomater. 2014. V. 10. № 9. P. 3922. https://doi.org/10.1016/j.actbio.2014.02.044
Marcus Y. // Chem. Soc. Rev. 1993. V. 22. № 6. P. 409. https://doi.org/10.1039/CS9932200409
Turova N.Y., Turevskaya E.P., Kessler V.G. et al. The Chemistry of Metal Alkoxides. Springer US, 2002.
Брауэр Г. Руководство по неорганическому синтезу. M.: Мир, 1985. Т. 2.
Ключников Н.Г. Руководство по неорганическому синтезу. M.: Химия, 1965.
Kirk-Othmer Encyclopedia of Chemical Technology. John Wiley & Sons, 1982. V. 17. P. 433.
Posner A.S., Betts F. // Accounts Chem. Res. 1975. V. 8. P. 273.
Fromm K.M. // Coord. Chem. Rev. 2008. V. 252. № 8–9. P. 856. https://doi.org/10.1016/j.ccr.2007.10.032
Ruttink P.J.A., Dekker L.J.M., Luider T.M. et al. // J. Mass. Spectrom. 2012. V. 47. № 7. P. 869. https://doi.org/10.1002/jms.3039
Calderin L., Dunfield D., Stott M.J. // Phys. Rev. B. 2005. V. 72. P. 224 304. https://doi.org/10.1103/PhysRevB.72.224304
Tadic D., Veresov A., Putlayev V. et al. // Materialwiss. Werkstofftech. 2003. V. 34. № 12. P. 1048. https://doi.org/10.1002/mawe.200300702
Вересов А.Г. Направленный синтез высокодисперсных материалов на основе гидроксилапатита. Дис. … канд. хим. наук. М., 2003.
Stähli C., Thüring J., Galea L. et al. // Acta Crystallogr. 2016. V. B72. P. 875. https://doi.org/10.1107/S2052520616015675
Дополнительные материалы
- скачать LarionovRJIC.docx
- Дополнительные материалы
Инструменты
Журнал неорганической химии