

Петрология, 2021, T. 29, № 4, стр. 429-448
Методика определения содержания воды в природных расплавах риолитового состава методами спектроскопии комбинационного рассеяния и электронно-зондового микроанализа
А. А. Котов a, *, **, С. З. Смирнов b, П. Ю. Плечов c, e, Э. С. Персиков f, Н. В. Черткова f, И. А. Максимович b, d, Н. С. Карманов b, П. Г. Бухтияров f
a Graduate School of Environmental Studies, Tohoku University
Sendai, Japan
b Институт геологии и минералогии им. В.С. Соболева СО РАН
Новосибирск, Россия
c Минералогический музей им. А.Е. Ферсмана РАН
Москва, Россия
d Новосибирский национальный исследовательский государственный университет
Новосибирск, Россия
e Институт геохимии и аналитической химии им. В.И. Вернадского РАН
Москва, Россия
f Институт экспериментальной минералогии им. Д.С. Коржинского РАН
Черноголовка, Московская обл., Россия
* E-mail: kotov@igm.nsc.ru
** E-mail: kotov.alexey.s1@dc.tohoku.ac.jp
Поступила в редакцию 21.12.2020
После доработки 18.01.2021
Принята к публикации 29.03.2021
Аннотация
Представлены результаты разработки методики определения содержания воды в сильно полимеризованных риолитовых и риодацитовых стеклах расплавных включений. Она предполагает одновременное определение содержания воды двумя методами: рентгеноспектрального микроанализа и спектроскопии комбинационного рассеяния (КР-спектроскопия, рамановская спектроскопия) для взаимной верификации одного метода другим. Калибровка метода КР-спектроскопии была проведена с использованием набора стандартов синтетических стекол гаплогранитного состава (SGS) с содержанием воды от 1.8 до 5.9 мас. %. Полученная калибровка была верифицирована путем анализа эталонных стекол на основе природного риолитового обсидиана (NRS) с содержанием воды от 4.7 до 9.9 мас. %. В качестве калибруемых параметров были выбраны два соотношения: 1) площадей линии колебаний в диапазоне 2900–3800 см–1 к площади линий решеточных колебаний (Si–O и Al–O связей) в диапазоне 850–1200 см–1 (Aw/As), 2) соотношение интенсивностей линии колебаний О–Н связей в области 3550–3560 см–1 и линии 480 см–1 в области решеточных колебаний (I3550/I480). В результате получены калибровочные уравнения ${{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.8458$\frac{{{{A}_{w}}}}{{{{A}_{s}}}}$(σ) ± 0.17 мас. % и ${{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 11.494$\frac{{{{I}_{{3550}}}}}{{{{I}_{{480}}}}}$(σ) ± ± 0.21 мас. %. Сопоставление концентрации воды в эталонных стеклах, определенной методом КР-спектроскопии с применением соотношений Aw/As и I3550/I480 и измеренной методом рентгеноспектрального микроанализа, показало хорошую сходимость. Метод рентгеноспектрального микроанализа использовался для определения содержания воды в стеклах путем пересчета избыточного содержания кислорода. Методика применялась для определения содержания воды в стеклах непрогретых стекловатых расплавных включений в минералах из пемз дацитового и риолитового составов кальдерных извержений оз. Курильского (Камчатка), перешейка Ветрового и залива Львиная Пасть (о. Итуруп, Курильские острова) и вулканического центра Окатаина (Новая Зеландия). Использование различных отношений Aw/As и I3550/I480 позволяет минимизировать влияние наложений линий минерала-хозяина на низкочастотную часть КР-спектров стекол расплавных включений. Установлено, что использование отношения I3550/I480 для расплавных включений в пироксенах дает более надежные результаты, чем отношение Aw/As, в то время как для включений в кварце и плагиоклазе предпочтительнее использование Aw/As. Содержание воды во включениях риолитового расплава крупных кальдерных извержений варьировало от 0.5 до 7.9 мас. %. Описанная в работе методика может использоваться в рутинном режиме для надежного измерения содержания воды в стеклах расплавных включений, размером не менее 10 мкм, содержащих как минимум 0.5 мас. % H2O.
ВВЕДЕНИЕ
Магматические расплавы являются одной из важнейших сред, обеспечивающих эндогенную циркуляцию воды и других летучих компонентов в различных тектонических обстановках. Если в глубинных зонах Земли эту роль играют расплавы базитовых и андезитовых магм, то в верхних горизонтах земной коры особую важность приобретают магмы кислого состава. Они способны растворять большие количества воды, фтора и хлора. По этой причине именно с эволюцией кислых магм в большинстве случаев связано развитие очагов катастрофических извержений вулканов и образование месторождений полезных ископаемых. Летучие компоненты, среди которых главную роль играют вода, углекислота и соединения серы, транспортируемые кислыми магмами к поверхности Земли при извержениях вулканов, попадают в атмосферу и таким образом влияют на ее экологию и климат. Из этого следует, что прямое определение содержания воды в кислых расплавах имеет большое значение для правильного понимания процессов транспорта летучих компонентов в недрах планеты и моделирования влияния эндогенных флюидов на климат, состав атмосферы и условия обитания на ее поверхности.
Разработано множество методов определения содержания воды в природных и искусственных силикатных расплавах. Для исследования природных объектов, таких как вулканы или рудоносные интрузивы, обычно используют стекла основной массы породы или микровключений в минералах. Как правило, они обладают микроскопическими размерами и методы валового анализа для них не применимы. Чаще всего концентрации воды в микровключениях расплавов в минералах в настоящее время измеряются с помощью вторичной ионной масс-спектрометрии (ВИМС), инфракрасной спектроскопии с преобразованием Фурье (Фурье-ИКС) и рентгеноспектрального микроанализа (РСМА) (Devine et al., 1995; Zajacz et al., 2005). Наиболее чувствительными методами являются титрование по методу Карла Фишера (КФТ) и инфракрасная спектроскопия с преобразованием Фурье (Behrens et al., 1996). При использовании метода КФТ исследуемое вещество полностью расходуется во время анализа. Высокоточные измерения с помощью Фурье-ИКС требуют очень тщательной и трудоемкой подготовки образцов.
Для непосредственного определения воды в природных стеклах и расплавных включениях все большую популярность приобретает метод спектроскопии комбинационного рассеяния (КР-спектроскопия), впервые примененный для этого независимо друг от друга Р. Томасом (Thomas et al., 1999) и А. Шабиро (Chabiron, 1999) и впоследствии подробно описанный в (Thomas, 2000). За последующие годы он стал применяться и усовершенствоваться многими другими исследователями, например (Chabiron et al., 2004; Holtz et al., 2005; Zajacz et al., 2005; Behrens et al., 2006; Le Losq et al., 2012; Plechov et al., 2015). Этот метод активно используется для изучения геологических материалов и позволяет анализировать небольшие (до 2–3 мкм) расплавные включения (РВ) и фрагменты стекол основной массы.
В настоящей работе предлагается новая методика определения содержания воды в сильно полимеризованных силикатных стеклах с высоким содержанием SiO2, предполагающая комбинирование метода КР-спектроскопии с рентгеноспектральным микроанализом.
Особенности спектра комбинационного рассеяния стекол с высоким содержанием SiO2
Спектр водосодержащего силикатного стекла условно может быть представлен в виде двух независимых частей: низкочастотной (0–1300 см–1) и высокочастотной (2900–4000 см–1). Низкочастотная часть спектра, которую также называют решеточной, отвечает колебаниям Si–O–Si, Al–O–Si и Si–O в структуре алюмосиликатных стекол. Она отражает состав силикатного стекла и степень его полимеризации (Mysen et al., 1982; Neuville, Mysen, 1996; Matson et al., 1983; McMillan, 1984). Известно, что по мере увеличения содержания SiO2 и других катионов, входящих в тетраэдры каркаса (Al, Ti, Fe3+), увеличивается степень полимеризации, и, с точки зрения структуры, силикатный расплав становится похож на сильно полимеризованный расплав чистого кремнезема. В его КР-спектре наиболее интенсивной является широкая линия с максимумом между 400 и 500 см–1, которая приписывается симметричным колебаниям Si–O–Si связей в кремнекислородном каркасе (Matson et al., 1983). В современной литературе эта линия интерпретируется по-разному (см. Schiavi et al., 2018 и ссылки в ней). Однако все авторы сходятся на том, что ее интенсивность и частота зависят от степени полимеризации и связаны с колебаниями структурных единиц Q4 (цифра обозначает число мостиковых атомов кислорода, приходящихся на один кремнекислородный тетраэдр). Она доминирует в сильно полимеризованных стеклах и имеет более низкую интенсивность в стеклах с меньшей степенью полимеризации (Zajacz et al., 2005; Le Losq et al., 2012; Schiavi et al., 2018). Некоторые авторы отмечают, что увеличение степени полимеризации приводит к увеличению ее волнового числа (Matson et al., 1983; Анфилогов, Быков, 1998; Rossano, Mysen, 2012). Геометрия пика 400–500 см–1 может быть осложнена плечом за счет наложения линии в районе 570–580 см–1. Ее связывают с деформационным или смешанным валентно-деформационным колебанием в структурных единицах Q2 (Furukawa et al., 1981). Она, по-видимому, должна лучше проявляться по мере снижения степени полимеризации.
В спектре кварцевого стекла в области около 600–800 см–1 выделяются малоинтенсивные линии, которые также фиксируются в спектрах высококремнистых стекол сложного состава. Линию в районе 800 см–1 связывают с колебаниями сильно полимеризованного кремнекислородного каркаса. В работах (Mysen et al., 1980; Furukawa et al., 1981; McMillan, 1984; Rossano, Mysen, 2012; Matson et al., 1983; Le Losq et al., 2012) отмечается, что она характерна для стекол с наиболее высоким содержанием SiO2 и ее интенсивность сильно увеличивается с увеличением степени полимеризации. В других исследованиях, однако, эту линию связывают с валентными колебаниями концевых атомов кислорода (McMillan et al., 1994; Neuville et al., 2006).
Линии в диапазоне 800–1200 см–1 связывают с Si–O и Si–O–Si колебаниями в структурных единицах Q1, Q2, Q3, Q4 (Mysen et al., 1980; Matson et al., 1983; McMillan, 1984; Wang et al., 1993; Zotov, Keppler, 1998; Mysen, 1997, 1999; Rossano, Mysen, 2012). В этом диапазоне в спектре кварцевого стекла выделяется две достаточно сильных линии 1060 и 1190 см–1. Их относят к ассиметричным валентным колебаниям связей O–Si–O в полимеризованном каркасе (Matson et al., 1983; McMillan, 1984). По мнению (Анфилогов, Быков, 1998), наиболее интенсивная полоса с центром 1050–1100 отвечает валентным колебаниям концевых атомов кислорода в структурных единицах Q3. Полосы с максимумом в областях 950–980 и 900 см–1 связаны с колебаниями атомов кислорода в структурных единицах Q2 и Q1 соответственно (Анфилогов, Быков, 1998; McMillan, 1984).
Присутствие железа в стеклах способствует появлению еще одного пика с центром около 940–970 см–1, который отвечает Fe–O–Si колебаниям в структурной единице Q3 (Wang et al., 1993, 1995; Sharma et al., 1997). В работе (Behrens et al., 2006) отмечается также наличие линии 913 см–1, которую они относят к колебаниям Si–ОН.
Соотношение интенсивностей и волновых чисел разных линий в диапазоне решеточных колебаний напрямую зависит от безводного состава стекла. В частности, от соотношения Al/(Si + Al) (Neuville, Mysen, 1996), содержания щелочей (Matson et al., 1983) и железа (Wang et al., 1993, 1995). Хотя присутствие воды напрямую не сказывается на характере спектра стекол в диапазоне 200–1500 см–1, содержание воды влияет на степень полимеризации расплава и, соответственно, на вариации соотношения интенсивностей линий, отвечающих колебаниям Q1–Q4.
В высокочастотной части спектра проявлены колебания связей O–H (3200–3700 см–1) в молекулах воды и гидроксильных группах, входящих в состав силикатных стекол (Анфилогов, Быков, 1998). Также наличие молекулярной воды в составе силикатных стекол может проявляться в виде линии 1630 см–1 (Thomas, 2000). Однако слабая интенсивность этой линии не позволяет ее использовать для получения надежных количественных измерений (Behrens et al., 2006).
МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Калибровочные стандарты
Исходя из анализа спектральных характеристик силикатных стекол, мы применили для калибровки два набора стандартов: синтетические стекла гаплогранитного состава (SGS) с содержанием воды от 1.8 до 5.9 мас. % H2O и стекла на основе природного риолитового обсидиана (NRS) с содержанием воды от 4.7 до 9.9 мас. %. Составы эталонных стекол приведены в табл. 1. Набор SGS использовался для построения калибровочных кривых, а набор NRS – для их верификации.
Таблица 1.
Составы эталонных стекол наборов SGS и NRS и результаты измерения калибруемых отношений методом КР-спектроскопии
| Компоненты | GL-1 | GL-2 | GL-3 | GL-4 | GL-5 | Che-n2 | Che-n3 | Che-n4 | Che-n9 |
|---|---|---|---|---|---|---|---|---|---|
| Cинтетические стекла гаплогранитного состава (SGS) | Природный обсидиан риолитового состава (NRS) | ||||||||
| n | 22 | 14 | 15 | 15 | 14 | 6 | 4 | 3 | 4 |
| SiO2 | 75.08 | 72.86 | 74.94 | 71.49 | 71.17 | 72.92 | 73.88 | 74.57 | 72.31 |
| TiO2 | НПО | НПО | НПО | НПО | НПО | 0.08 | 0.08 | 0.09 | 0.14 |
| Al2O3 | 13.19 | 13.17 | 13.29 | 12.76 | 12.57 | 12.47 | 12.55 | 12.66 | 12.30 |
| FeO | НПО | НПО | НПО | НПО | НПО | 0.10 | 0.17 | 0.48 | 0.05 |
| MnO | НПО | НПО | НПО | НПО | НПО | 0.06 | 0.06 | 0.05 | 0.05 |
| MgO | НПО | НПО | НПО | НПО | НПО | 0.06 | 0.06 | 0.07 | 0.06 |
| CaO | НПО | НПО | НПО | НПО | НПО | 0.49 | 0.52 | 0.50 | 0.47 |
| Na2O | 4.75 | 4.63 | 4.79 | 4.43 | 4.36 | 2.63 | 3.13 | 3.17 | 2.54 |
| K2O | 5.23 | 5.24 | 5.46 | 5.12 | 4.88 | 2.28 | 3.37 | 3.95 | 2.27 |
| P2O5 | НПО | НПО | НПО | НПО | НПО | НПО | 0.01 | 0.02 | 0.01 |
| SO3 | НПО | НПО | НПО | НПО | НПО | 0.01 | НПО | НПО | НПО |
| Cl– | НПО | НПО | НПО | НПО | НПО | 0.04 | 0.05 | 0.04 | 0.05 |
| Сумма | 98.25 | 95.90 | 98.49 | 93.80 | 92.97 | 91.14 | 93.87 | 95.60 | 90.23 |
| H2O КФТ | 2.12 | 3.84 | 1.84 | 5.83 | 5.92 | 8.88 | 6.06 | 4.71 | 9.93 |
| H2O РСМА | 2.65 | 4.28 | 2.07 | 5.93 | 6.70 | НО | НО | НО | НО |
| 100NBO/T1 | 16 | 29 | 14 | 45 | 46 | 62 | 42 | 33 | 70 |
| Aw/As (1σ) | 2.85 ± 0.16 | 4.70 ± 0.18 | 2.24 ± 0.19 | 6.68 ± 0.40 | 6.91 ± 0.32 | 9.35 ± 0.08 | 7.58 ± 0.31 | 4.54 ± 0.19 | 9.93 ± 0.19 |
| I3550/I480 (1σ) | 0.183 ± 0.005 | 0.331 ± 0.020 | 0.151 ± 0.008 | 0.458 ± 0.030 | 0.558 ± 0.012 | 0.679 ± 0.015 | 0.53 ± 0.032 | 0.416 ± 0.033 | 0.707 ± 0.022 |
Примечание. Здесь и в табл. 2: n – количество измерений методом рамановской спектроскопии, НПО – ниже предела обнаружения, НО – не определялось. 1100NBO/T по (Персиков, 1984).
Стекла SGS были синтезированы Б. Грохау в Германии (Институт минералогии, Университет Ганновера, Германия) в 1993 г. и предоставлены в распоряжение В.П. Чупина (Институт геологии и минералогии СО РАН, г. Новосибирск). Метод получения стекол не раскрывался. Содержание воды в нем было измерено методом КФТ и заверено методом рентгеноспектрального микроанализа, который будет описан ниже.
Для синтеза водосодержащего риолитового стекла набора NRS использовался однородный образец обсидиана (Артени, Армения), который изначально содержал около 0.47 мас. % H2O (Степанов и др., 2016). Цилиндрические фрагменты обсидиана выдерживали в присутствии чистой воды в течение 48 ч при температуре выше температуры ликвидуса водосодержащего риолита (1000–1100°С) и различном давлении (от 4.2 до 10 кбар) в аппаратах высокого давления типа “газовая бомба” (IHPV) и “цилиндр–поршень” в Институте экспериментальной минералогии РАН, г. Черноголовка. В ходе синтеза происходило полное плавление риолита и его насыщение водой. Содержание воды и однородность синтезированных стекол оценивались несколькими независимыми методами – КФТ, ИК‑спектроскопия, КР-спектроскопия (Степанов и др., 2016).
Расплавные включения
Для проверки применимости разработанного комплексного метода определения содержания воды были исследованы расплавные включения (РВ) во вкрапленниках пемз из пемзово-пирокластических толщ крупных кальдерообразующих извержений Курило-Камчатской островной дуги (оз. Курильское (7600 14С) (Южная Камчатка) (Ponomareva et al., 2004), перешейка Ветрового (ПВ) (20 000 лет назад (метод термолюминесценции)) (Smirnov et al., 2019), залива Львиная Пасть (ЛП) (13000 14С (Дегтерев и др., 2015) (о. Итуруп, Курильские острова)) и Новозеландской островной дуги (вулканический центр Окатаина в зоне Таупо (ТП) (пемзы моложе 65 тыс. лет (Davidson, Kamenetsky, 2007))). Все вкрапленники в пемзах содержат большое количество природно-закаленных расплавных включений. В большинстве случаев они содержат прозрачное бесцветное стекло. Реже помимо стекла во включениях присутствует небольшой усадочный пузырек (рис. 1а–1г).
Рис. 1.
Расплавные включения (РВ) в минералах пемз кальдерных извержений оз. Курильского, п-ов Камчатка (a), перешейка Ветровой, о. Итуруп (б), залива Львиная Пасть, о. Итуруп (в), вулканического центра Окатаина, Новая Зеландия (г). Qz – кварц, Pl – плагиоклаз.

Зерна кварца, плагиоклаза и пироксенов из пемз перечисленных выше извержений монтировались в шашки из эпоксидной смолы. Перед измерением включения выводились на поверхность стачиванием на алмазных пастах таким образом, чтобы большая их часть оставалась в минерале-хозяине, а на поверхности обнажалась только небольшая площадка, которая необходима для последующего микрозондового анализа включения. Это позволяет уменьшить степень наложения линий минерала-хозяина при регистрации КР-спектров.
Метод съемки эталонов и расплавных включений при калибровке КР-спектрометра
При определении содержания воды в силикатных стеклах методом КР-спектроскопии существует два подхода к калибровке: калибровка с внешним и внутренним стандартом. Калибровка с внешним стандартом обычно подразумевает определение соотношений площади или интенсивности линии воды в исследуемом стекле с ее интенсивностью или площадью в спектрах стекол с известным содержанием воды (Behrens et al., 2006; Thomas et al., 2008).
Базовый принцип “внутренней” калибровки состоит в измерении отношений площадей или интенсивностей линий в высокочастотной (водной) и низкочастотной (решеточной) областях спектра. Эти отношения напрямую коррелируют с содержанием воды в изучаемом стекле. В аналитической химии под использованием “внутреннего” стандарта подразумевается нормирование измеряемых характеристик, в данном случае соотношения площадей линии воды и суммы площадей индивидуальных линий решеточных колебаний на содержание одного из компонентов стекла, которое было определено иным способом. Однако предлагаемые в работах (Chabiron et al., 2004, Zajacz et al., 2005; Le Losq et al., 2012) методики калибровки не предполагают соотнесения площадей отдельных пиков или их суммы в области решеточных колебаний с содержанием компонентов, участвующих в построении каркаса, а именно Si и Al. По сути, они предполагают использование внешнего стандарта с известным содержанием воды, но не для отдельной линии, а для соотношения площадей пиков или групп пиков.
В разное время исследователями предлагались различные отношения в качестве калибруемого параметра. Так, например, в (Behrens et al., 2006) использовано отношение площади линии воды к суммарной площади линий в области 20–1500 см–1, в (Chabiron et al., 2004) использовано отношение площади линии воды к площади линии в области 490 см–1, а (Zajacz et al., 2005; Plechov et al., 2015) предлагают использование отношение площади линии воды к суммарной площади линий в области 850–1300 см–1.
В качестве калибруемого параметра в настоящей работе были выбраны две величины: 1) соотношение площади линии, отвечающей колебаниям связей О–Н в диапазоне 2900–3800 см–1 к сумме площадей линий решеточных колебаний в области 850–1200 см–1 (Aw/As), подобно методике (Zajacz et al., 2005); 2) отношение интенсивности линии воды в диапазоне 3550–3560 см–1 к интенсивности линии вблизи 480 см–1 в низкочастотной области спектра (I3550/I480).
КР-спектр каждого стандартного стекла был снят несколько раз в различных точках. Измерения проводились с использованием спектрометра Horiba LabRam HR800 в Институте геологии и минералогии СО РАН, г. Новосибирск. Позиционирование лазерного луча производилось с помощью встроенного поляризационного микроскопа Olympus BX-41. Для возбуждения использовался твердотельный лазер с длиной волны 532 нм и выходной мощностью 75 мВт. Регистрация проводилась с помощью CCD детектора, охлаждаемого до –70°С элементом Пельтье. Анализ проводился в геометрии обратного рассеяния. Для сбора рассеянного света использовался объектив 100× с числовой апертурой 0.9. Спектры были получены в диапазонах 100–1300 и 2900–4000 см–1. Калибровка спектрометра проводилась по линии кремния 520.7 см–1. Спектральное разрешение дифракционной решетки составляло 1800 штр/мм.
Для проверки влияния величины конфокального отверстия на воспроизводимость результата были проведены измерения с различными размерами диафрагмы (50–300 мкм) при неизменности других параметров в течение одного цикла. Также для проверки воспроизводимости результата были проведены измерения одних и тех же эталонных стекол с различием во времени накопления сигнала и количестве циклов накопления. Время и количество циклов накопления в пределах спектрального окна варьировалось от 5 с × 5 циклов до 20 с × 20 циклов. Для проверки стабильности калибровки было сделано 10 серий измерений эталонных стекол с различным интервалом в течение одного года.
Особое внимание было уделено фокусировке лазера. Известно, что чем глубже расположен фокус лазера, тем сильнее рассеянное излучение поглощается стеклом. При этом степень поглощения для высоко- и низкочастотных частей спектра на разных глубинах значительно различается (Schiavi et al., 2018). При этом (Schiavi et al., 2018) показано, что наиболее подходящей глубиной фокусировки является та, на которой достигается максимальная интенсивность полосы, отвечающей колебаниям О–Н, и минимальное отношение площади этой полосы к суммарной площади низкочастотной части спектра (Aw/As). Следуя этим выводам, при изучении стекол регистрация спектра проводилась при фокусировке лазера на глубине 5–7 мкм. В Supplementary, ESM_2.pdf22 приведены результаты эксперимента для эталона GL-4, показывающие зависимость глубины фокусировки и величин Aw, As, I3550, I480, Aw/As, I3550/I480, а также Aw/As(total), где As(total) – суммарная площадь всех пиков в области 100–1200 см–1 после вычитания базовой линии.
Измерения стандартных стекол показали, что такие параметры, как величина конфокального отверстия и различия во времени накопления сигнала, не оказывают существенного влияния на полученные результаты. Разброс значений для одной и той же точки измерения, обусловленный различными размерами конфокального отверстия и изменением времени накопления, не превышал ±0.15 мас. %. Проверки стабильности измерений для одного и того же стандарта в течение нескольких месяцев показали, что различия в оценках содержания воды варьируют в пределах ±0.18 мас. %.
КР-спектры стекол РВ снимались при тех же параметрах, что и эталонные стандарты. Исключение составляли только размер конфокальной диафрагмы и время накопления спектра, которые регулировались в зависимости от размера включения. Минимальный размер конфокального отверстия составил 30 мкм для включений 5–10 мкм, максимальный – 300 мкм для включений >100 мкм. Время накопления сигнала изменялось от 25 с/спектральное окно для крупных РВ до 400 с/спектральное окно для мелких РВ.
Определение содержания воды в силикатных стеклах методом КР-спектроскопии
Общепринятая процедура обработки КР-спектров проходит в три этапа: 1) введение частотно-температурной коррекции Лонга (Long, 1977), 2) вычитание базовой линии и 3) деконволюция и интегрирование интересующих частей спектра.
Частотно-температурная коррекция Лонга заключается в коррекции интенсивности необработанного спектра с учетом длины волны возбуждающего излучения и окружающей температуры. Эта поправка необходима для сравнения спектров, полученных с разными длинами волн или при разных температурах (Long, 1977; Neuville, Mysen, 1996). В настоящем исследовании все спектры снимались с одной длиной волны возбуждающего лазера и без значительных колебаний температуры в ходе анализа. Попытки использовать коррекцию Лонга для спектров эталонных стекол набора SGS продемонстрировали незначительное влияние ее применения на итоговые значения Aw/As или I3550/I480. Аналогичный результат был получен ранее (Zajacz et al., 2005; Mercier et al., 2009). Поэтому обработка полученных данных в нашем исследовании проводилась без введения коррекции Лонга.
Вычитание базовой линии для учета влияния фона проводилось отдельно для каждого из диапазонов. В настоящей работе использовалось вычитание ломанной линии, проведенной по минимумам спектров в точках: 100, 180 ± 5, 700 ± 10, 850 ± 5, 1200 см–1 для области решеточных колебаний. В области колебаний связей О–Н в качестве базовой была выбрана прямая линия, соединяющая точки с волновыми числами 2900 см–1, 3800 см–1 (рис. 2а, 2б). Аналогичный подход к вычитанию базовой линии использовался, например, в работе (Severs et al., 2007).
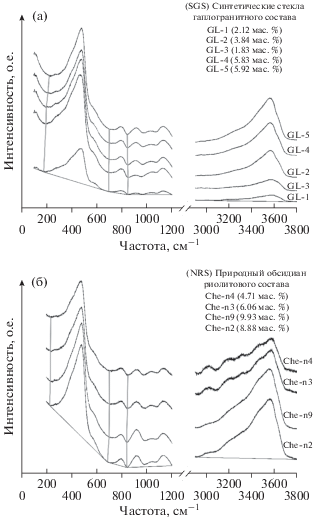
Рис. 2.
Спектры комбинационного рассеяния эталонных стекол SGS (a) и NRS (б). Серые вертикальные линии проведены через промежуточные минимумы, используемые при построении базовой линии (пояснения см. текст).
При изучении стекол РВ в различных минералах наиболее сложным остается третий этап. Он заключается в выборе того участка области спектра решеточных колебаний, который будет подвергаться деконволюции и интегрированию. Эта проблема не возникает при съемке стандартных стекол, но из-за малых размеров РВ на их КР-спектры в диапазоне 100–1200 см–1 неизбежно накладываются интенсивные линии спектра минерала-хозяина (рис. 3а–3г).
Рис. 3.
Спектры комбинационного рассеяния расплавных включений в кварце (a), плагиоклазе (б), авгите (в), гиперстене (г) из пемз перешейка Ветрового. На врезках показаны примеры гауссовской деконволюции низкочастотной части КР-спектров включений при отсутствии (а, б) и наличии (в, г) наложения линий минерала-хозяина. Черными сплошными линиями показаны гауссовские компоненты, полученные при деконволюции. Красной линией показан результат суммирования пиков стекла. Синим цветом показана аппроксимированная линия минерала-хозяина. Под примерами деконволюции приведены остатки вычитания аппроксимированных пиков стекла из экспериментальных спектров включений. Стрелками указаны линии минерала-хозяина в исходных КР-спектрах стекол включений: Qz – кварц, Aug – авгит, Hyp – гиперстен.
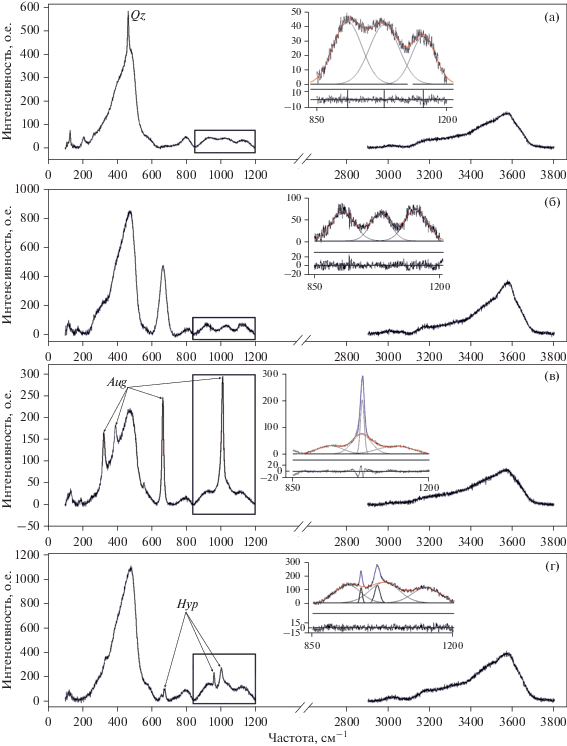
В кварце и полевых шпатах главные линии расположены в диапазоне 400–520 см–1 и близки к наиболее интенсивной линии спектра силикатного стекла с максимумом около 480 см–1. Эти линии не мешают обработке спектров РВ в области 850–1200 см–1, но могут накладываться на линию в области 480 см–1 (Chabiron et al., 2004) (рис. 3а, 3б). Зачастую в сильно полимеризованных стеклах линия кварца или полевого шпата может не проявляться из-за высокой интенсивности линии стекла в области 480 см–1. Особенно если анализируется крупное включение. Трудности могут возникать при работе с включениями небольшого размера, при анализе которых линии минерала-хозяина проявляются в низкочастотной части спектра. Высокая интенсивность линий кварца или полевого шпата зачастую делает невозможным корректную деконволюцию этой части спектра и ее дальнейшее использование.
КР-спектры темноцветных минералов (пироксены, амфиболы) содержат наиболее интенсивные линии в области 650–1030 (клино- и ортопироксены) и 670–1060 см–1 (амфиболы) (рис. 3в, 3г). Кроме этого, достаточно интенсивные линии этих минералов присутствуют в диапазоне 200–400 см–1. Таким образом, в темноцветных минералах все части спектров стекол РВ так или иначе могут быть осложнены линиями минерала-хозяина. Интенсивность линий стекла в диапазоне 850–1200 см–1 не так значительна, как интенсивность пика в области 480 см–1. По этой причине при регистрации спектра РВ в пироксенах почти неизбежно будут проявляться линии минерала-хозяина вне зависимости от размера включения.
Также частым акцессорным минералом в рассматриваемых породах является апатит. Так, например, во вкрапленниках пемз Курильского озера (п-ов Камчатка) и перешейка Ветрового (о. Итуруп, Курильские острова) зачастую встречаются комбинированные включения, содержащие стекло и апатит. В КР-спектре апатита наиболее интенсивная линия расположена около 980 см–1 и может осложнять спектр стекла в этой области. С точки зрения осложнения спектра стекла апатит подобен пироксенам и делает затруднительным работу с частью спектра в диапазоне 850–1200 см–1, однако не затрудняет работу с низкочастотной частью спектра в области основного силикатного пика 480 см–1.
Для правильного определения суммарной площади и интенсивностей линий в области решеточных колебаний и вычитания линий минерала-хозяина процедура деконволюции проводилась с помощью программы Fityk 0.9.8 (Wojdyr, 2010).
На рис. 3а–3г представлены примеры деконволюции низкочастотной части спектра для стекол РВ из кварца, плагиоклаза и пироксенов перешейка Ветрового (о. Итуруп). Как видно, для включений в кварце и плагиоклазе не возникает сложностей при деконволюции спектра в диапазоне 800–1200 см–1. Спектры РВ в пироксенах значительно осложнены линиями минерала-хозяина в этой области. В нашем случае для вычисления величины As в отношении Aw/As суммировались только компоненты, отвечающие линиям стекла, что в нашем представлении отвечает вычитанию линии минерала-хозяина из этой части спектра. Подобное вычитание приводит к увеличению общей погрешности. То же происходит, если на низкочастотную часть спектра стекла РВ в кварце накладывается интенсивный пик 465 см–1. В связи с этим для получения хороших результатов в настоящей работе использовались единичные крупные включения. РВ в кварце выбирались так, чтобы в их спектрах на силикатный пик 480 см–1 не накладывалась линия кварца.
Определение содержания воды методом рентгеноспектрального микроанализа
Для верификации данных, полученных методом КР-спектроскопии, мы воспользовались определением содержания воды в стеклах на основе прямого определения содержания кислорода методом рентгеноспектрального микроанализа. Точное определение содержания кислорода, как и других легких элементов (B, C, N, F), методом РСМА сопряжено с трудностями, обусловленными большими погрешностями поправок на матричные эффекты в длинноволновой области рентгеновского излучения, а также повышенными требованиями к качеству полировки поверхности образца и толщине токопроводящего покрытия. Важно также при анализе строго соблюдать геометрические условия: рабочее расстояние, угол отбора излучения, угол падения электронного пучка на образец. Указанные трудности в равной степени относятся как к волновым спектрометрам (ВДС), так и энерго-дисперсионным (ЭДС). Известно, что электронный пучок оказывает деструктивное воздействие на образцы с высокими содержаниями воды и/или щелочных элементов, выражающееся в изменении (дрейфе) регистрируемой интенсивности рентгеновского излучения во времени. Поэтому анализ РВ методом РСМА предпочтительно выполнять при низких токах электронного пучка с применением ЭДС, детектор которого обладает высокой чувствительностью регистрации излучения. В работе (Newbury, Ritchie, 2015) приводятся данные, в которых относительная погрешность определения кислорода методом РСМА с применением ЭДС в оксидах алюминия, кремния и железа не превышает 2%, при этом для обработки спектров используется некоммерческое программное обеспечение NIST DTSA-II EDS (Ritchie NWM, NIST DTSA-II software. www.cstl.nist.gov/div837/837.02/ epq/dtsa2/index.html) без указания используемых образцов сравнения.
В настоящей работе исследование состава РВ и сопутствующих минералов выполнялось на сканирующем электронном микроскопе MIRA-3 LMU (Tescan Orsay Holding) в Институте геологии и минералогии СО РАН (г. Новосибирск), оснащенном системой микроанализа INCA Energy 450 XMax-80 (Oxford Instruments Nanoanalysis), при ускоряющем напряжении 20 кВ и токе электронного пучка 1.5 нА с активным временем набора спектров 60 с. В качестве образцов сравнения использовались чистые металлы (Ti, V, Cr, Mn, Fe), а также простые соединения Al2O3 (Al), SiO2 (Si, O), NaCl (Cl) и минералы диопсид (Mg, Ca), альбит (Na) и ортоклаз (K). Учет матричных эффектов осуществлялся методом XPP из программного обеспечения системы микроанализа, при этом использовался режим расчета “все элементы определяемые – all elements analyzed”. При данных условиях анализа нижняя граница определяемых содержаний для кислорода составляет 0.3 мас. %, а остальных компонентов – около 0.1 мас. %. Методика анализа и ее метрологические характеристики не отличаются от таковых, приведенных в работе (Лаврентьев и др., 2015).
Препаратами для исследования служили тонкие полированные пластинки породы, помещенные в эпоксидную смолу таким образом, что их поверхности были перпендикулярны оптической оси микроскопа. Препараты покрывались слоем углерода толщиной до 15 нм. Каждое включение анализировалось в двух точках при этом для снижения дрейфа интенсивности рентгеновского излучения под воздействием электронного пучка с высокой плотностью тока (диаметр 9 нм), приводящего к недоопределению концентраций Na и K (Morgan, London, 1996, 2005), и снижения влияния микрорельефа образца в исследуемой области анализ выполнялся в режиме малого растра с размером сканируемой области до 10 × 10 мкм2 для РВ и до 20 × 20 мкм2 для стандартных стекол. Кроме этого, при анализе РВ вблизи включения анализировался минерал-хозяин. Наш опыт свидетельствует, что сумма результатов анализа стехиометричных минералов (кварц, полевые шпаты, пироксены и т.д.) при соблюдении приведенных выше условий близка к 100%, а содержание определяемого кислорода совпадает с вычисленным по стехиометрии. Значимое (более 1 мас. %) отклонение суммы результатов анализа от 100% может быть обусловлено вариациями тока зонда, негоризонтальной поверхностью образца или отклонением реальной толщины углеродного покрытия от толщины, указанной для расчета поправок на матричные эффекты. Поэтому состав минерала-хозяина можно использовать как для оценки качества анализа включений, так и их корректировки. В частности, варьированием толщины напыления при расчете матричных поправок можно подобрать такую величину, при которой расчетное по стехиометрии и определяемое содержание кислорода совпадут. Если при этом сумма результатов анализа минерала-хозяина будет значимо отличаться от 100%, то результат анализа включения необходимо нормировать на эту сумму. При вычислении матричных поправок водосодержащих стекол присутствует систематическая погрешность, обусловленная неучетом влияния содержания водорода на величину матричных поправок, но эта погрешность вряд ли превышает 1% и ее влияние можно считать несущественным.
Избыточное содержание кислорода, связанного с водородом, определяется как разность между измеренным в ходе анализа и рассчитанным по стехиометрии оксидов. Таким образом, содержание воды во включении рассчитывается в соответствии со следующим выражением:
Для проверки правильности получаемых данных и воспроизводимости результатов методом РСМА были проанализированы эталонные стекла набора SGS, содержание воды в которых определено методами КФТ и КР-спектроскопии.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Спектры комбинационного рассеяния эталонных стекол
Спектры эталонных стекол из наборов SGS и NRS, приведенные на рис. 2а и 2б, имеют много общего как в области решеточных колебаний, так и в области колебаний связей О–Н. В области решеточных колебаний доминирует линия с максимумом 470–480 см–1, демонстрирующая, что стекла характеризуются сильной степенью полимеризации. Рассматриваемая линия на всех спектрах осложнена слабым плечом в области 570–580 см–1. В области решеточных колебаний 600–1200 см–1 в спектрах обоих наборов эталонных стекол присутствуют линии около 800, 920, 1030 и 1130–1135 см–1.
Для характеристики химического состава и степени полимеризации расплава принято использовать отношение NBO/T, где NBO – количество немостиковых атомов кислорода в расплаве, а T – суммарное количество катионов, находящихся в тетраэдрической координации по кислороду. Для сильно полимеризованных кислых расплавов это отношение должно быть близко к нулю. В настоящей работе нами определялось отношение 100NBO/T, следуя работам (Персиков, 1984; Persikov et al., 1990). Предлагаемый параметр с большей чувствительностью отражает структурные особенности изучаемого расплава. 100NBO/T определяется из химического состава расплава, выраженного в мас. % оксидов, включая летучие компоненты, с использованием следующего уравнения: $\frac{{100{\text{NBO}}}}{{\text{T}}}$ = = $\frac{{200({\text{O}} - 2{\text{T}})}}{{\text{T}}},$ где O – суммарное количество грамм-ионов кислорода в расплаве, рассчитанное по формуле $\sum {\left[ {\frac{{n{\text{O}}({\text{ox}})}}{{M{\text{(ox)}}}} \times C({\text{ox}})} \right]} ,$ где nO(ox) – количество атомов кислорода в молекуле оксида, M(ox) – молекулярный вес оксида, С(ox) – концентрация оксида в мас. % при сумме анализа 100%. T = Al3+ + Si4+ + Fe3+ + Ti4+ + P5+ + B3+ – суммарное количество грамм-ионов сеткообразователей, находящихся в тетраэдрической координации по отношению к кислороду и входящих в анионную часть структуры стекла (Персиков, 1984, Persikov et al., 1990). Таким образом, нами были получены значения 100NBO/T для всех стекол, задействованных в работе, с учетом концентрации воды, установленной электронно-микрозондовым анализом по избытку кислорода для расплавных включений и методом КФТ для эталонных стекол. Для стекол набора SGS значения 100NBO/T с учетом содержания воды, определенной методом КФТ, составляют 14–46, для стекол NRS – 33–70. Стекла изученных в настоящей работе расплавных включений имеют значения 100NBO/T в диапазоне 6–67, что близко к диапазону значений для эталонных стекол и свидетельствует о схожести их структур с точки зрения степени полимеризации.
На рис. 2 видно, что соотношение интенсивностей линий в диапазоне 800–1200 см–1 приблизительно одинаково для всех эталонных стекол. Следовательно, соотношения структурных элементов Q1–Q3 в них не имеют больших различий. Наибольшей интенсивностью в этой части спектра обладает линия с волновым числом 1130–1135 см–1, близкая к диапазону 1060–1190 см–1 (Matson et al., 1983) в спектре кварцевого стекла. Их наличие можно связать с комплексным влиянием валентных колебаний в тетраэдрах Q3, формирующих бесконечные слои, и в изолированных тетраэдрах с одним концевым кислородом, возникающих при добавлении щелочных металлов (Matson et al., 1983). Волновые числа этих линий в высококремнистых щелочных стеклах несколько выше тех, которые зафиксированы в ходе нашего исследования. В работе (Neuville, Mysen, 1996) показано, что добавление Al в количестве 0.2 Al/(Al + Si) приведет к смещению волнового числа в область более низких частот около 1140 см–1, что и наблюдается в наших спектрах. Аналогично ведет себя и менее интенсивная линия, которая в спектре кварцевого стекла должна находиться в области 1060 см–1. Повышение доли алюминия от 0 до 0.2 приведет к уменьшению волнового числа до ~1030 см–1.
Линия в области 920 см–1 присутствует во всех спектрах исследованных нами стекол и ее интенсивность сопоставима или выше интенсивности линии 1030 см–1. Как уже отмечалось, на параметрах этой линии в области решеточных колебаний непосредственно может сказываться наличие растворенной в стекле воды (Behrens et al., 2006). Сопоставима с вышеупомянутыми и интенсивность линии около 800 см–1, характерная для стекол и расплавов кремнезема с высокой степенью полимеризации.
В высокочастотной части спектров эталонных стекол присутствует широкая ассиметричная полоса с максимумом около 3550–3560 см–1, приписываемая колебаниям О–Н в молекулах воды и групп ОН– в силикатных стеклах.
Уравнение калибровки
По результатам измерений суммарных площадей “водной” части спектра в диапазоне 2900–3800 см–1 и гауссовых компонент спектра стекла в диапазоне 850–1200 см–1 после вычитания базовой линии в эталонных стеклах SGS получено уравнение калибровки:
где ${{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ – содержание воды в стекле, Aw – площадь линии колебаний связей О–Н в диапазоне 2900–3800 см–1, As – площадь группы трех линий, отвечающих колебаниям Q2–Q4 в диапазоне 850–1200 см–1. Среднее стандартное отклонение калибровки (σ) ±0.21 мас. % (рис. 4а).

По результатам измерений интенсивностей линий в областях 3550 и 480 см–1 после вычитания базовой линии в эталонных стеклах SGS получено уравнение калибровки:
где ${{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ – содержание воды в стекле, I3550, I480 – интенсивности линии колебания связей О–Н в диапазоне 2900–3800 см–1 и линии около 480 см–1 соответственно. Среднее стандартное отклонение (σ) ±0.17 мас. % (рис. 4б). При построении регрессии было принято, что обе калибровочные прямые проходят через начало координат.
Спектры эталонных стекол из набора NRS были сняты как образцы с неизвестным содержанием воды для проверки правильности калибровочных уравнений, полученных по набору стекол SGS. Сопоставление результатов приведено на рис. 4в, 4г.
ПРИМЕНЕНИЕ МЕТОДИКИ К СТЕКЛАМ РАСПЛАВНЫХ ВКЛЮЧЕНИЙ ИЗ ПЕМЗ КРУПНЫХ КАЛЬДЕРНЫХ ИЗВЕРЖЕНИЙ
Спектры стекол расплавных включений
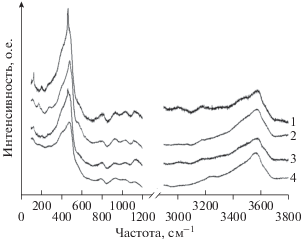
РВ из всех четырех объектов, рассматриваемых в настоящей работе, близки по составу между собой и с эталонными стеклами (табл. 2). Стекла расплавных включений перешейка Ветрового (ПВ), залива Львиная Пасть (ЛП) (о. Итуруп, Курильские о-ва) и оз. Курильского (ОК) (Камчатка) отличаются по содержанию K2O (1.6–2.1 мас. %) от эталонов (4.9–5.5 мас. % K2O) и расплавных включений в кварце пемз вулканического центра Окатаина в зоне Таупо (Новая Зеландия) (ТП) (4.0–5.2 мас. % K2O) (см. Supplementary, ESM_1.xlsx). Примеры спектров комбинационного рассеяния РВ всех четырех объектов приведены на рис. 5. В низкочастотной части спектров РВ доминирует линия 480 см–1, осложненная плечом 570–580 см–1. Спектры изученных РВ имеют характерный пик в области 795–800 см–1. В диапазоне 850–1200 см–1 интенсивности линий около 940 и 1140 см–1 могут вести себя по-разному. Как правило, после процедуры вычитания базовой линии интенсивность полосы 940 см–1 выше или равна интенсивности полосы 1140 см–1. Однако в спектрах РВ из зоны Таупо и в эталонных стеклах SGS наблюдается небольшое преобладание линии 1140 см–1 (рис. 5), что может быть следствием повышенного содержания в них K2O (Matson et al., 1983).
Таблица 2.
Представительные анализы стекол расплавных включений в минералах дацитовых и риолитовых пемз крупных кальдерных извержений Камчатки, Курильских островов и Новой Зеландии
| Компоненты | It-15-52-35 | It-15-57-9-1 3 | 54-tc-23 MI 2 | 15-60-22-1 | It-15-11-35a-1 | It-15-11-5b | It-15-16-4c | Ok-1 18-5 | Ok-1 19-1 | Ok-1 54-1 | Tau-31-1 | Tau-18 | Tau-6 1 | Tau-1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| перешеек Ветровой | залив Львиная Пасть | оз. Курильское | Окатаина, Таупо | |||||||||||
| Минерал-хозяин | Pl | Cpx | Opx | Qz | Pl | Pl | Pl | Pl | Pl | Pl | Qz | Qz | Qz | Qz |
| SiO2 | 74.78 | 70.81 | 73.78 | 75.60 | 73.73 | 73.80 | 76.82 | 73.57 | 69.75 | 70.06 | 73.61 | 75.13 | 74.54 | 73.13 |
| TiO2 | 0.32 | 0.35 | 0.40 | 0.26 | 0.29 | 0.30 | 0.23 | 0.41 | 0.35 | 0.50 | 0.12 | 0.00 | 0.13 | 0.13 |
| Al2O3 | 11.11 | 13.11 | 11.37 | 10.91 | 11.52 | 11.32 | 11.32 | 12.46 | 11.85 | 12.31 | 11.85 | 11.98 | 11.54 | 12.29 |
| FeO | 1.59 | 1.89 | 2.66 | 1.56 | 1.49 | 1.39 | 1.51 | 2.11 | 1.76 | 2.17 | 1.05 | 1.02 | 1.12 | 0.98 |
| MnO | 0.05 | НО | 0.08 | 0.05 | 0.07 | 0.04 | НПО | НПО | НПО | НПО | НПО | НПО | НПО | НПО |
| MgO | 0.27 | 0.35 | 0.28 | 0.28 | 0.32 | 0.27 | 0.27 | 0.44 | 0.35 | 0.45 | 0.02 | НПО | 0.08 | 0.00 |
| CaO | 1.55 | 2.11 | 1.49 | 1.44 | 1.74 | 1.72 | 1.57 | 1.64 | 1.55 | 1.76 | 0.58 | 0.54 | 0.61 | 0.61 |
| Na2O | 3.88 | 4.32 | 4.24 | 4.19 | 4.18 | 3.82 | 3.90 | 4.39 | 4.16 | 4.05 | 3.49 | 4.14 | 3.80 | 4.04 |
| K2O | 1.62 | 1.42 | 1.62 | 1.64 | 1.70 | 1.76 | 1.73 | 2.14 | 1.96 | 2.00 | 5.21 | 4.30 | 4.18 | 4.40 |
| Cl– | 0.26 | 0.27 | 0.25 | 0.24 | 0.25 | 0.23 | 0.20 | 0.17 | 0.17 | 0.18 | 0.23 | 0.25 | 0.24 | 0.27 |
| Сумма РСМА | 95.42 | 94.70 | 96.15 | 96.16 | 95.30 | 94.66 | 97.56 | 97.33 | 91.91 | 93.48 | 96.15 | 97.35 | 96.24 | 95.84 |
| H2O РСМА | 4.83 | 5.10 | 3.65 | 3.74 | 4.82 | 5.22 | 2.22 | 2.50 | 7.90 | 6.24 | 3.91 | 2.32 | 3.69 | 3.93 |
| Сумма РСМА + H2O | 100.25 | 99.79 | 99.80 | 99.91 | 100.11 | 99.88 | 99.78 | 99.83 | 99.81 | 99.72 | 100.06 | 99.68 | 99.93 | 99.77 |
| КР H2O (Aw/As метод) |
4.04 | 2.67 | 2.71 | 3.37 | 4.76 | 5.16 | 2.66 | 2.97 | 5.73 | 5.91 | 3.77 | 2.75 | 3.91 | 4.05 |
| КР H2O + РСМА (Aw/As метод) |
99.46 | 97.37 | 98.86 | 99.53 | 100.06 | 99.82 | 100.22 | 100.29 | 97.65 | 99.39 | 99.92 | 100.10 | 100.16 | 99.89 |
| КР H2O (I3550/I480 метод) |
4.49 | 5.36 | 3.80 | 4.61 | 5.02 | 5.24 | 2.73 | 3.13 | 7.72 | 4.51 | НО | 4.03 | 5.38 | 3.64 |
| КР H2O + РСМА (I3550/I480 метод) | 99.91 | 100.06 | 99.95 | 100.77 | 100.32 | 99.90 | 100.29 | 100.46 | 99.64 | 97.99 | – | 101.38 | 101.62 | 99.48 |
| 100NBO/T | 40 | 43 | 34 | 33 | 41 | 44 | 20 | 24 | 67 | 54 | 33 | 20 | 31 | 32 |
Рис. 5.
Спектры комбинационного рассеяния стекол расплавных включений во вкрапленниках отложений кальдерообразующих извержений: 1 – залива Львиная Пасть, о. Итуруп, 2 – оз. Курильского, п-ов Камчатка, 3 – вулканического центра Окатаина, Новая Зеландия, 4 – перешейка Ветровой, о. Итуруп. РВ (1, 3) находятся в кварце, РВ (2, 4) находятся в плагиоклазе.
Критерии отбраковки некачественных анализов стекол расплавных включений
Описанное исследование фактически представляет собой определение содержания воды методом РСМА и двумя разными способами с применением КР-спектроскопии.
Для оценки правильности данных, полученных при определении содержания воды, суммировались ее содержания, полученные методом КР-спектроскопии с содержаниями других оксидов и элементов, определенных методом рентгеноспектрального анализа. Если сумма попадала в диапазон 99–101%, содержание воды, полученное методом КР-спектроскопии, сравнивалось с концентрацией, определенной методом рентгеноспектрального анализа. Различия между этими содержаниями в указанном диапазоне сумм могли достигать 1 мас. %. Однако, если сузить интервал допустимых значений до 99.5–100.5%, различия между содержаниями воды, определенными разными методами, оказывались менее 1 мас. %, что хорошо согласуется с определенными ранее погрешностями измерения. По этой причине в настоящей работе анализами удовлетворительного качества считались те, сумма которых попадала в этот диапазон. Важно отметить, что по избранному критерию было отбраковано около 35% результатов.
Содержание воды в расплавах крупных кальдерных извержений
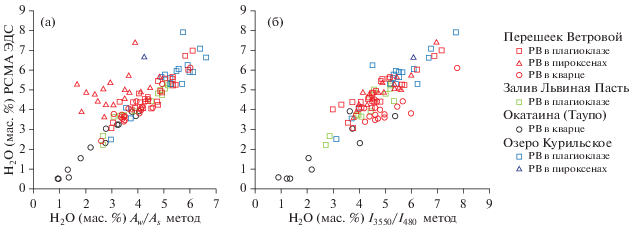
Кварц из пемз оз. Курильского содержит стекловатые природно-закаленные РВ (рис. 1а). Результаты детальных исследований отложений этого извержения и РВ приведены в (Ponomareva et al., 2004; Плечов и др., 2010). Пемзы этого извержения по составу варьируют от дацитов до риолитов, а среди игнимбритов встречаются андезитовые и андезибазальтовые составы (Ponomareva et al., 2004). Исследованные РВ в плагиоклазе имеют риолитовый состав (табл. 2). По данным (Плечов и др., 2010), содержание воды, определенное методом вторично-ионной масс-спектрометрии в этих РВ, составляет 5.96–7.44 мас. %. Согласно нашим исследованиям, в РВ из вкрапленников плагиоклаза установлено содержание воды от 2.5 до 7.9 мас. %. При этом большая часть ее установленных концентраций находится в диапазоне 5.3–6.6 мас. %.
В пемзах перешейка Ветрового на о-ве Итуруп стекловатые природно-закаленные РВ присутствуют в плагиоклазе, кварце (рис. 1б), гиперстене, клинопироксене, апатите, ильмените и магнетите. Спектры включений в плагиоклазе, кварце и пироксенах приведены на рис. 3 и 5. Детальное описание составов РВ и условий их захвата, в ходе которых отмечались значительные вариации содержания воды от 2.3 до 7.2 мас. %, приведено в (Kotov et al., 2017, Smirnov et al., 2019). Содержание воды, измеренное для РВ во вкрапленниках пемз перешейка Ветрового в ходе данного исследования, варьирует в диапазоне 2.4–7.4 мас. %. При этом большая часть полученных значений находится в диапазоне 3.5–5.5 мас. %.
Пемзы из отложений кальдерного извержения залива Львиная Пасть по химическому составу аналогичны пемзам перешейка Ветрового. Однако в отличие от них содержат в ассоциации минералов-вкрапленников амфибол, что указывает на иной флюидный режим и параметры становления магматического очага (Смирнов и др., 2017; Smirnov et al., 2019). Вкрапленники пемз залива Львиная Пасть также содержат однофазовые стекловатые включения, однако, в отличие от пемз перешейка Ветрового, они встречаются реже (рис. 1в). Зачастую РВ девитрифицированы. Среди дочерних фаз в подобных включениях встречается магнетит, а в КР-спектрах появляется линия этого минерала – 670 см–1. По составу стекла таких включений практически не отличаются от стекол РВ пемз перешейка Ветрового (табл. 2). Ранее по недостатку суммы рентгеноспектрального анализа было сделано предположение, что содержание воды в стеклах РВ составляет 3.8–6.3 мас. % (Смирнов и др., 2017). Определенные в настоящей работе концентрации воды в стекловатых РВ в плагиоклазе пемз залива Львиная Пасть находятся в диапазоне 2.2–5.3 мас. %.
Вулканический центр Окатаина в зоне Таупо представляет собой крупный кальдерный комплекс диаметром 20 × 13 км. Образование кальдер связывают с 2–4 эпизодами, произошедшими между 557 и 33 тыс. лет назад (Cole et al., 2014). Пемзы кальдерных извержений имеют риолитовый состав. Кварц содержит стекловатые природно-закаленные РВ (рис. 1г). В большинстве из них присутствуют небольшие флюидные обособления, содержащие водный раствор, дочерние солевые фазы и газовый пузырек. Подобные РВ описаны в работах (Davidson, Kamenetsky, 2007; Johnson et al., 2013; Смирнов и др., 2011). Примеры составов включений, изученных в настоящей работе, приведены в табл. 2. Все стекла имеют риолитовый состав с высоким содержанием K2O. Наши измерения показали, что они обладают самой низкой концентрацией воды, полученной в ходе данного исследования (от 0.5 до 3.9 мас. %). Измерения методом КР-спектроскопии хорошо согласуются с измерениями методом рентгеноспектрального микроанализа. Однако полученный нами диапазон концентрации оказался ниже приведенного в работах (Johnson et al., 2013; Смирнов и др., 2011) 4–6 мас. %, измеренного методами ВИМС и Фурье-ИКС. Возможно, это связано с постзахватным выделением в изученных включениях высокоплотной водно-солевой флюидной фазы, о которой говорилось выше.
Результаты сравнения концентрации воды, измеренной методами рентгеноспектрального анализа и КР-спектроскопии с использованием разных уравнений калибровки, представлены на рис. 6а, 6б.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Определение воды в стеклах с помощью КР-спектроскопии имеет явное преимущество перед другими методами, благодаря своей недеструктивности, простоте подготовки препаратов и хорошей локальности. При изучении РВ в минералах и стекол в пепловых частицах эти преимущества делают его наиболее экспрессным из существующих методов, позволяющих в течение одного дня получить десятки определений. Вероятно, при современных темпах развития аналитического оборудования КР-спектрометры скоро станут распространенным рутинным методом в минералогических и петрологических исследованиях. Возможность количественного определения летучих компонентов в невскрытых включениях с локальностью около 1 мкм однозначно позволит перейти на новую ступень в изучении флюидного режима магматических систем.
Недостатком метода является высокий предел обнаружения, который связан со сложностью выделения колебаний O–H при низких концентрациях воды. Этот предел оценивается в 0.3 мас. % Н2О (Thomas et al., 2008; Le Losq et al., 2012; Plechov et al., 2015), но может быть понижен до 0.1 мас. % за счет увеличения времени накопления сигнала (Schiavi et al., 2018). В настоящей работе изучались синтетические стекла и включения кислых расплавов, содержание воды в которых существенно выше предела обнаружения. Часть стекол РВ могут существенно или полностью обезвоживаться в результате постзахватных процессов во включении (Плечов, 2014 и ссылки в ней), однако в таких случаях обычно не стоит задача точного определения остаточного содержания воды. Вероятно, в нашей работе к таким объектам могут относиться некоторые включения в кварце из вулканической зоны Таупо (Новая Зеландия), содержащие минимальное количество воды (около 0.5 мас. %) (см. Supplementary, ESM_1.xlsx). Выбранный нами критерий отбраковки результатов РСМА и КР-спектроскопии по попаданию в диапазон суммы 99.5–100.5 мас. % допускает разницу в содержании воды не более ±0.5 мас. %, что и определяет высокий уровень пределов обнаружения.
Определение низкого содержания воды (<0.5 мас. %) в высококремнистых РВ возможно, но требует тщательного индивидуального подхода, что увеличивает временные затраты предлагаемой методики и уменьшает ее преимущества по сравнению с другими методами анализа. КР-спектроскопия позволяет оперативно выделить включения с предельно низким содержанием воды, а их анализ в случае необходимости может быть проведен другими, более прецизионными методами. Однако вышесказанное не отрицает необходимости дальнейших исследований с целью усовершенствования метода для определения предельно низкой концентрации воды и повышения точности. На данный момент, предлагаемая методика может использоваться в рутинном режиме с допустимыми погрешностями для измерения содержания воды более 0.5 мас. % в кислых стеклах РВ.
Размер РВ, используемых для получения достоверных результатов, ограничен следующими факторами: 1) соотношение количества вещества включения и минерала-хозяина, попадающих в область возбуждения; 2) возможность фокусировки лазера на оптимальной глубине 5–7 мкм (см. раздел “Метод съемки эталонов и расплавных включений при калибровке КР-спектрометра”); 3) ограничения по размерам РВ при анализе методом РСМА, чтобы избежать захвата вещества минерала-хозяина.
При попадании вещества минерала-хозяина в область возбуждения происходит наложение линий минерала на спектр заключенного во включении вещества. В нашем случае линии минералов могут осложнять разные диапазоны низкочастотной части спектра стекла и затруднять их обработку, увеличивая погрешности. Влияние этого фактора можно значительно минимизировать подбором оптимальных параметров съемки (размера конфокальной диафрагмы, время накопления сигнала, глубины фокусировки). При этом предлагаемая возможность выбирать одно из двух калибруемых соотношений позволяет и вовсе избежать необходимость работы с осложненным спектром стекла. Необходимость фокусировки лазера на глубине 5–7 мкм остается значительным ограничивающим фактором, который приводит к дополнительным ошибкам при работе с включениями маленького размера. Третий фактор остается наиболее важным. Особенности распространения электронов в объеме возбуждаемого вещества и необходимость анализа в режиме сканирования площадки накладывает существенные ограничения на размер включения. Вероятность захвата вещества минерала-хозяина при анализе включений маленького размера лишает нас возможности верифицировать оценки H2O, полученные методом КР-спектроскопии. В ходе исследования было установлено, что для получения достоверных результатов необходимо, чтобы анализируемое включение было не менее 10×10×10 мкм. Таким образом, в настоящей работе любое РВ, которое было использовано для анализа РСМА, автоматически имеет достаточный размер, чтобы учесть второй лимитирующий фактор – необходимость фокусировки на оптимальной глубине.
Предлагаемая возможность использования одного из двух калибруемых значений (Aw/As или I3550/I480) позволяет решить проблему определения содержания воды в небольших включениях из разных магматических минералов. Как уже отмечалось выше, спектр кварца и плагиоклаза осложняется в диапазоне 100–600 см–1. Это позволяет успешно использовать калибровку по отношению Aw/As, так как на параметр As эти линии не оказывают влияния. У темноцветных минералов и апатита, напротив, наиболее сильные линии осложняют диапазон 600–1200 см–1, что делает предпочтительным выбор калибровки по отношению I3550/I480.
Для изученной выборки включений в кварце, плагиоклазе и пироксене из пемз кальдерных извержений была оценена сходимость полученных результатов с оценками воды по содержанию кислорода, определенного методом РСМА (см. Supplementary, ESM_1.xlsx). Для включений в пироксене лучшая сходимость наблюдается при использовании отношения интенсивностей I3550/I480, для включений в кварце – при использовании соотношения площадей Aw/As, для плагиоклаза удачные результаты могут быть получены с применением обоих отношений.
Результаты проверки калибровки на наборе стекол NRS показывают, что при содержании воды более 8.0 мас. % наблюдается существенное отклонение от линейной регрессии для набора NRS, что, возможно, связано с различиями в составе обсидиановых эталонных стекол (NRS) и синтетических эталонных стекол (SGS) или нелинейностью калибровочной зависимости в этом диапазоне содержаний. Это показывает, что приведенная в настоящей работе калибровка может надежно применяться для измерения содержания воды в диапазоне 0.5–8.0 мас. % в синтетических и природных стеклах риолитового состава.
Кальдерообразующие извержения являются идеальным объектом для применения описанной методики, так как в большинстве случаев предшествующие процессы и развитие этих явлений связаны с очагами кислой магмы с большим содержанием воды (Wallace, Gerlach, 1994; Chesner, Luhr, 2010; Smith et al., 2010; Miyagi et al., 2017; Forni et al., 2018). Спектры стекол РВ из продуктов всех четырех рассматриваемых извержений весьма похожи (рис. 5) и соответствуют спектрам стекол, использованных для калибровки. Это позволило нам использовать одни и те же калибровочные уравнения и процедуры обработки спектров. Полученные данные хорошо согласуются с результатами исследования других объектов и показывают, что в расплавах очагов кальдерных извержений максимальное содержание воды может достигать 5–8 мас. %, что соответствует пределам насыщения для глубин 3–9 км, которые типичны для кальдерных очагов. В то же время проведенное исследование показывает, что содержание воды в расплавах варьирует в широких пределах, опускаясь до 0.5 мас. %. Причинами таких вариаций могут быть процессы дегазации в малоглубинных очагах или изменение содержания воды в расплавах включений после их захвата минералом-хозяином. Установление точных причин в каждом конкретном случае требует дополнительных тщательных исследований.
ЗАКЛЮЧЕНИЕ
В настоящей работе предложена методика, которая подразумевает одновременное определение содержания воды в силикатных стеклах риолитового состава двумя методами: рентгеноспектрального анализа и КР-спектроскопии. По своей сути этот подход предполагает проверку достоверности результатов одного метода другим.
Калибровка была проведена с использованием двух наборов стандартов, имеющих состав близкий к гранитам и риолитам. Для соотношений Aw/As и I3550/I480 получены калибровочные уравнения (1) ${{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 0.8458$\frac{{{{A}_{w}}}}{{{{A}_{s}}}}$ (±0.21 мас. %) и (2) ${{C}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = = 11.494$\frac{{{{I}_{{3550}}}}}{{{{I}_{{480}}}}}$ (±0.17 мас. %) соответственно. Сопоставление результатов определения содержания воды в эталонных стеклах методами КР-спектроскопии, с применением соотношений Aw/As и I3550/I480, и рентгеноспектрального анализа показали хорошую сходимость. Было показано, что некачественными следует считать анализы, сумма оксидов, хлора и воды для которых выходит за пределы 99.5–100.5 мас. %.
Методика была применена для определения воды в стеклах непрогретых РВ в минералах из пемз дацитового и риолитового составов кальдерных извержений оз. Курильского (Камчатка), перешейка Ветрового и залива Львиная Пасть (о. Итуруп, Курильские острова) и вулканического центра Окатаина (Новая Зеландия). Содержание воды во включениях риолитового расплава крупных кальдерных извержений варьирует от 0.5 до 7.9 мас. %. Наши исследования показали, что калибровка по I3550/I480 дает более точные результаты для РВ в пироксенах, в то время как отношение Aw/As удобнее использовать для включений в кварце и плагиоклазе. Плагиоклаз оказался наиболее благоприятным минералом-хозяином для применения предлагаемой методики. Предлагаемый подход с комбинированным использованием РСМА и КР-спектроскопии является надежным способом изучения водосодержащих РВ кислого состава во вкрапленниках вулканитов. Введение жесткого критерия отбраковки результатов компенсируется возможностью делать большое количество анализов включений за короткий срок. Однако, как было отмечено, чем ниже диапазон концентрации воды в РВ, тем более жесткими должны быть критерии, что в свою очередь ставит под вопрос использование данного подхода для массового определения воды во включениях с низким ее (<0.5 мас. %) содержанием. Вероятно, решением данного вопроса является либо разработка более сложных подходов к измерениям таких включений предложенным методом, либо использование традиционных высокопрецизионных методов (ВИМС, ИК-спектроскопия).
Благодарности. Авторы благодарны М.В. Хлестову и В.А. Даниловской за помощь в проведении анализов методом рентгеноспектрального анализа. Авторы приносят благодарность проф. Университета Тасмании (Австралия) В.С. Каменецкому за предоставленные образцы кварца из пемз вулканического центра Окатаина (Новая Зеландия). Авторы признательны Т.А. Шишкиной (ГЕОХИ РАН, Москва) и В.О. Япаскурту (МГУ, Москва) за замечания и конструктивные предложения, позволившие доработать и улучшить текст статьи и представление результатов.
Источники финансирования. Работа выполнена в рамках государственного задания ИГМ СО РАН и работ по грантам РФФИ (№№ 18-05-00819, 19‑05-00101а).
Список литературы
Анфилогов В.Н., Быков В.Н. Силикатные расплавы, термодинамика, физические свойства. Миасс: Институт минералогии УрО РАН, 1998. 160 с.
Дегтерев А.В., Рыбин А.В., Арсланов Х.А. и др. Катастрофические эксплозивные извержения Львиной Пасти (о. Итуруп): стратиграфия и геохронология // Всероссийская научная конференция с международным участием “Геодинамические процессы и природные катастрофы. Опыт Нефтегорска”. Сб. материалов в 2-х томах под ред. Б.В. Левина, О.Н. Лихачевой. Владивосток: Дальнаука, 2015. С. 210–213.
Лаврентьев Ю.Г., Карманов Н.С., Усова Л.В. Электронно-зондовое определение состава минералов: микроанализатор или сканирующий электронный микроскоп? // Геология и геофизика. 2015. Т. 56. № 8. С. 1473–1482.
Персиков Э. С. Вязкость магматических расплавов. М.: Наука, 1984. 160 с.
Плечов П.Ю. Методы изучения флюидных и расплавных включений. М.: Изд-во КДУ, 2014. 268 с.
Плечов П.Ю., Балашова А.Л., Дирксен О.В. Дегазация магмы кальдерообразующего извержения Курильского озера 7600 лет назад и ее влияние на климат // Докл. АН СССР. 2010. Т. 433. № 3. С. 1–4.
Смирнов С.З., Томас В.Г., Соколова Е.Н., Куприянов И.Н. Экспериментальное исследование герметичности включений водосодержащих силикатных расплавов при внешнем давлении D2O при 650°С и 3 кбар // Геология и геофизика. 2011. Т. 52. № 5. С. 690–703.
Смирнов С.З., Рыбин А.В., Соколова Е.Н. и др. Кислые магмы кальдерных извержений о. Итуруп: первые результаты исследования расплавных включений во вкрапленниках пемз кальдеры Львиная Пасть и перешейка Ветровой // Тихоокеанская геология. 2017. Т. 36. № 1. С. 50–68.
Степанов О.В., Черткова Н.В., Плечов П.Ю. и др. Аналитические методы определения высоких концентраций воды в обсидиановых стеклах // Тр. Всероссийского ежегодного семинара по экспериментальной минералогии, петрологии и геохимии. М.: ГЕОХИ, 2016. С. 50–51.
Behrens H., Romano C., Nowak M. et al. Near-infrared spectroscopic determination of water species in glasses of the system MAlSi2O8 (M = Li, Na, K): In interlaboratory study // Chem. Geol. 1996. V. 128. P. 41–63.
Behrens H., Roux J., Neuville D.R., Siemann M. Quantification if dissolved H2O in silicate glasses using confocal microRaman spectroscopy // Chem. Geol. 2006. V. 229. P. 96–112.
Chabiron A., Peiffert C., Pironon J., Cuney M. Determination of water content in melt inclusions by Raman spectroscopy // ECROFI XV Abstracts. Terra Nostra. 1999. V. 99. № 6. P. 68–69.
Chabiron A., Pironon J., Massare D. Characterization of water in synthetic rhyolitic glasses and natural melt inclusions by Raman spectroscopy // Contrib. Mineral. Petrol. 2004. V. 146. P. 485–492.
Chesner C., Luhr J. A melt inclusion study of the Toba Tuffs, Sumatra, Indonesia // J. Volcanol. Geotherm. Res. 2010. V. 197. P. 259–278.
Cole J.W., Deering C.D., Burt R.M. et al. Okataina Volcanic Centre, Taupo Volcanic Zone, New Zealand: A review of volcanism and synchronous pluton development in an active, dominantly silicic caldera system // Earth-Sci. Rev. 2014. V. 128. P. 1–17.
Davidson P., Kamenetsky V.S. Primary aqueous fluids in rhyolitic magmas: Melt inclusion evidence for pre- and post-trapping exsolution // Chem. Geol. 2007. V. 237. P. 372–383.
Devine J., Gardner J., Brack H. et al. Comparison of microanalytical methods for estimating H2O contents of silicic volcanic glasses // Amer. Mineral. 1995. V. 80. P. 319–328.
Forni F., Degruyter W., Bachman O. et al. Long-term magmatic evolution reveals the beginning of a new caldera cycle at Campi Flegrei // Sci. Adv. 2018. V. 4. № 11. https://doi.org/10.1126/sciadv.aat9401
Furukawa T., Fox K.E., White W.B. Raman spectroscopic investigation of the structure of silicate glasses. III. Raman intensities and structural units in sodium silicate glasses // J. Chem. Phys. 1981. V. 75. P. 3226–3237.
Johnson E., Kamenetsky V., McPhie J. The behavior of metals (Pb, Zn, As, Mo, Cu) during crystallization and degassing of rhyolites from the Okataina Volcanic center, Taupo Volcanic Zone, New Zealand // J. Petrol. 2013. V. 54. № 8. P. 1641–1659.
Holtz F., Sato H., Lewis J. et al. Experimental petrology of the 1991–1995 Unzen dacite, Japan. Part I: Phase relations, phase compositions and pre-eruptive conditions // J. Petrol. 2005. V. 46. № 2. P. 319–337.
Kotov A.A., Smirnov S., Maksimovich I. et al. Water in melt inclusions from phenocrysts of dacite pumice of the Vetrovoy Isthmus (Iturup Island, Southern Kuriles) // IOP Conference Series: Earth and Environmental Sci. 2017. V. 110. https://scholarcommons.usf.edu/geo_facpub/2162
Le Losq C., Neuville D.R., Moretti R., Roux J. Determination of water content in silicate glasses using Raman spectrometry: Implications for the study of explosive volcanism // Amer. Mineral. 2012. V. 97. № 5–6. P. 779–790.
Long D.A. Raman Spectroscopy. N.Y.: MacGraw-Hill, 1977. 276 p.
Matson D.W., Sharma S.K., Philpotts J.A. The structure of high-silicate glasses. A Raman spectroscopic investigation // J. Non-Cryst. Solids. 1983. V. 58. P. 323–352.
Mercier M., Di Muro A., Giordano D. et al. Influence of glass polymerization and oxidation on microRaman water analysis in alumino-silicate glasses // Geochim. Cosmochim. Acta. 2009. V. 73. P. 197–217.
McMillan P. Structural studies of silicate glasses and melts – applications and limitations of Raman spectroscopy // Amer. Mineral. 1984. V. 69. P. 622–644.
McMillan P., Poe B., Gillet Ph., Reynard B. A study of SiO2 glass and supercooled liquid to 1950 K via high-temperature Raman spectroscopy // Geochim. Cosmochim. Acta. 1994. V. 58. P. 3653–3664.
Miyagi I., Kita N., Morishita Y. The geochemical and petrological characteristics of prenatal caldera volcano: A case of the newly formed small dacitic caldera, Hijiori, Northeast Japan // Contrib. Mineral. Petrol. 2017. V. 172. № 79. https://doi.org/10.1007/s00410-017-1391-8
Morgan G.B., London D. Optimizing of electron microprobe analysis of hydrous alkali glasses // Amer. Mineral. 1996. V. 81. P. 1176–1185.
Morgan G.B., London D. Effect of current density on the electron microprobe analysis of alkali aluminosilicate glasses // Amer. Mineral. 2005. V. 90. P. 1131–1138.
Mysen B.O. Aluminosilicate melts: Structure, composition and temperature // Contrib. Mineral. Petrol. 1997. V. 127. № 1–2. P. 104–118.
Mysen B.O. Structure and properties of magmatic liquids: From haplobasalt to haploandesite // Geochim. Cosmochim. Acta. 1999. V. 63. № 1. P. 95–112.
Mysen B.O., Virgo D., Harrison W.J., Scarfe C.M. Solubility mechanisms of H2O in silicate melts at high pressures and temperatures: A Raman spectroscopic study // Amer. Mi-neral. 1980. V. 65. P. 900–914.
Mysen B.O., Virgo D., Seifert F.A. The structure of silicate melts: Implications for chemical and physical properties of natural magma // Rev. Geophys. Space Phys. 1982. V. 20. P. 353–383.
Neuville D.R., Mysen B.O. Role of aluminum in the silicate network: In situ, high-temperature study of glasses and melts on the join SiO2–NaAlO2 // Geochim. Cosmochim. Acta. 1996. V. 60. P. 1727–1737.
Neuville D.R., Cormier L., Massiot D. Al coordination and speciation in calcium aluminosilicate glasses: Effects of composition determined by 27Al MQ-MAS NMR and Raman spectroscopy // Chem. Geol. 2006. V. 229. P. 173–185.
Newbury D.E., Ritchie N.W.M. Performing elemental microanalysis with high accuracy and high precision by scanning electron microscopy/silicon drift detector energy-dispersive X-ray spectrometry (SEM/SDD-EDS) // J. Mater. Sci. 2015. V. 50. P. 493–518.
Persikov E.S., Zharikov V.A., Bukhtiyarov P.G., Polskoy S.F. The effect of volatiles on the properties of magmatic melts // Eur. J. Mineral. 1990. V. 2. P. 621–642.
Plechov P.Yu., Blundy J., Nekrylov N. et al. Petrology and volatile content of magmas erupted from Tolbachik Volcano, Kamchatka, 2012–13 // J. Volcanol. Geotherm. Res. 2015. V. 307. P. 182–199.
Ponomareva V.V., Kyle P.R., Melekestsev I.V. et al. The 7600 (14C) year BP Kurile Lake caldera-forming eruption, Kamchatka, Russia: Stratigraphy and field relationships // J. Volcanol. Geotherm. Res. 2004. V. 136. P. 199–222.
Rossano S., Mysen B. Raman spectroscopy of silicate glasses and melts in geological systems // Eds. J. Dubessy, M.C. Caumon, F. Rull. Raman Spectroscopy Applied to Earth Sciences and Cultural Heritage, EMU Notes in Mine-ralogy, Europ. Mineral. Union, London. 2012. V. 12. P. 319–364.
Schiavi F., Bolfan-Casanova N., Withers A. et al. Water quantification in silicate glasses by Raman spectroscopy: Correcting for the effect of confocality, density and ferric iron // Chem. Geol. 2018. V. 483. P. 312–331.
Severs M.J., Azbej T., Thomas J.B. et al. Experimental determination of H2O loss from melt inclusions during laboratory heating: Evidence from Raman spectroscopy // Chem. Geol. 2007. V. 237. P. 358–371.
Sharma S.K., Cooney T.F., Wang Z.F., VanderLaan S. Raman band assignments of silicate and germanate glasses using high-pressure and high-temperature spectral data // J. Raman Spectrosc. 1997. V. 28. № 9. P. 697–709.
Smirnov S.Z., Rybin A.V., Kruk N.N. et al. Parental melts and magma storage of a Large-volume dacite eruption at Vetrovoy Isthmus (Iturup Island, Southern Kuril Islands); Insight into the genesis of subduction-zone dacites // J. Petrol. 2019. V. 60. № 7. P. 1349–1370.
Smith V., Shane P., Nairn I. Insights into silicic melt gene-ration using plagioclase, quartz and melt inclusions from the caldera-forming Rotoiti eruption, Taupo volcanic zone, New Zealand // Contrib. Mineral. Petrol. 2010. V. 160. P. 951–971.
Thomas R. Determination of water concentrations of gra-nite melt inclusion by confocal laser Raman microprobe spectroscopy // Amer. Mineral. 2000. V. 85. P. 868–872.
Thomas R., Webster J.D., Heinrich W. Melt inclusions in pegmatite quartz: Complete miscibility between silicate melts and hydrous fluids? // ECROFI XV Abstracts. Terra Nostra. 1999. V. 99. № 6. P. 305–307.
Thomas S.M., Thomas R., Davidson P. et al. Application of Raman spectroscopy to quantify trace water concentrations in glasses and garnets // Amer. Mineral. 2008. V. 93. P. 1550–1557.
Wallace P.J., Gerlach T.M. Magmatic vapor source for sulfur dioxide released during volcanic eruptions: Evidence from Mount Pinatubo // Science. 1994. V. 265. P. 497–499.
Wang Z.F., Cooney T.F., Sharma S.K. High-temperature structural investigation of Na2O–0.5Fe2O3–3SiO2 and Na2O–Fe2O3–SiO2 melts and glasses // Contrib. Mineral. Petrol. 1993. V. 115. № 1. P. 112–122.
Wang Z.F., Cooney T.F., Sharma S.K. In situ structural investigation of iron-containing silicate liquids and glasses // Geochim. Cosmochim. Acta. 1995. V. 59. № 8. P. 1571–1577.
Wojdyr M. Fityk: A general-purpose peak fitiing program // J. Appl. Cryst. 2010. V. 43. P. 1126–1128.
Zajacz Z., Halter W., Malfait W.J. et al. A composition-independent quantitative determination of the water content in silicate glasses and silicate melt inclusions by confocal Raman spectroscopy // Contrib. Mineral. Petrol. 2005. V. 150. P. 631–642.
Zotov N., Keppler H. The influence of water on the structure of hydrous sodium tetrasilicate glasses // Amer. Mineral. 1998. V. 83. P. 823–834.
Дополнительные материалы
- скачать ESM_1.xlsx
- Табл. 1
- скачать ESM_2.docx
- Изменение интенсивности и площади линий стекла от глубины фокусировки лазера
Инструменты
Петрология