

Поверхность. Рентгеновские, синхротронные и нейтронные исследования, 2021, № 5, стр. 95-101
Количественный анализ рентгеноспектральных данных для смеси соединений методами машинного обучения
А. С. Алгасов b, *, С. А. Гуда a, b, А. А. Гуда a, **, Ю. В. Русалёв a, А. В. Солдатов a
a Международный исследовательский институт интеллектуальных материалов,
Южный федеральный университет
344090 Ростов-на-Дону, Россия
b Институт математики, механики и компьютерных наук, Южный федеральный университет
344090 Ростов-на-Дону, Россия
* E-mail: alexander.algasov@gmail.com
** E-mail: guda@sfedu.ru
Поступила в редакцию 27.03.2020
После доработки 20.05.2020
Принята к публикации 25.05.2020
Аннотация
На основе алгоритмов машинного обучения разработан метод определения структурных параметров компонентов смеси по данным рентгеновских спектров поглощения. Строится база данных спектров для всевозможных предполагаемых деформаций структуры каждого компонента. Метод машинного обучения, реализованный в программном комплексе PyFitIt, позволяет быстро рассчитать спектр для деформаций структур из рассматриваемого семейства и оптимизировать структурные параметры смеси путем подгонки теоретического спектра под экспериментальный. Возможности метода рассмотрены на примере анализа изменений структурных характеристик и концентраций компонентов смеси для бис-диоксоленового комплекса кобальта с функционализированными иминопиридиновыми лигандами при его валентно-таутомерной интерконверсии в зависимости от температуры.
ВВЕДЕНИЕ
Для определения структурных параметров компонентов смеси и их концентраций на основе анализа околопороговой тонкой структуры рентгеновских спектров поглощения (XANES – X-ray Absorption Near Edge Structure) существует несколько подходов. Исторически первым был подход моделирования смеси линейной комбинацией заданных спектров и поиск соответствующих коэффициентов [1–3]. Когда неизвестно количество компонентов и их спектры, применяют анализ главных компонентов (PCA – Principal Component Analysis), фактор-анализ [4] и в последнее время метод анализа многомерных кривых путем среднеквадратичной вариации (MCR-ALS – Multivariate Curve Resolution using Alternating Least Squares) [5, 6].
Рассматриваемая в настоящей работе задача имеет свою специфику, которую нельзя учесть перечисленными методами анализа смесей. В случае, когда с большой вероятностью известно, к какому семейству принадлежат атомные структуры компонентов смеси, для определения каждого компонента можно воспользоваться классическим, зарекомендовавшим себя подходом – подбором геометрических параметров атомной структуры так, чтобы ее расчетный (теоретический) спектр был как можно ближе к экспериментальному. Первая программа MXAN, автоматизирующая данный процесс, появилась в 2001 г. [7–10]. Впоследствии многие другие программы, рассчитывающие спектр, включили в себя функции автоматического подбора параметров. Чтобы один раз посчитать значение оптимизируемой функции, необходимо рассчитать теоретический спектр, что требует значительного времени. Автоматическая оптимизация часто работает дольше недели и иногда приводит к физически маловероятным или даже некорректным структурам. Требуется ручное вмешательство человека в процесс оптимизации. Чтобы сделать удобной ручную оптимизацию, нужно обеспечить высокую скорость расчета спектра. В программе FitIt [11] применяют предварительный расчет множества спектров для набора геометрических параметров и их последующую интерполяцию. В настоящее время процедура аппроксимации спектров, реализованная в программе FitIt, была значительно улучшена методами машинного обучения в приложении PyFitIt [12]. Функции PyFitIt были также расширены для решения задач определения структурных параметров на основе анализа рентгеновских спектров поглощения в случае смеси веществ. Наряду с интерфейсом пользователя, в котором можно менять атомные структуры компонентов смеси, добиваясь наилучшего совпадения с экспериментальными спектрами, полученными для разных температур, в PyFitIt также была встроена процедура автоматического подбора геометрических параметров компонентов смеси и их концентраций равномерно по всему интервалу температур.
Предлагаемый подход определения параметров компонентов смеси является альтернативой существующим алгоритмам – PCA, фактор-анализу, MCR-ALS – и может быть использован совместно с ними для подтверждения полученных результатов. Этот подход имеет ряд преимуществ. В частности, для анализа смеси достаточно всего одного экспериментального спектра, также алгоритм хорошо работает в случае спектров с высоким шумом.
В настоящей работе преимущества метода машинного обучения продемонстрированы на примере исследования валентно-таутомерной интерконверсии в комплексе кобальта (diox)2Co(imPy-TEMPO). Ранее [13] было показано, что этот бис-диоксоленовый кобальтовый комплекс, включающий функционализированый иминопиридиновый лиганд, претерпевает спиновый переход в интервале температур 200–300 К в твердом состоянии. Согласно данным магнитной восприимчивости, такая интерконверсия, скорее всего, вызвана валентной таутомерией. Однако все попытки изучить структурные изменения, связанные с наблюдаемым превращением, методом рентгеновской дифракции монокристаллов не увенчались успехом, поскольку кристалл разрушается при его охлаждении ниже 220 К, и остаются неизвестными особенности молекулярной структуры низкотемпературного изомера. Метод анализа спектров XANES за K-краем поглощения Co для (diox)2Co(imPy-TEMPO) в широком температурном интервале от 30 до 300 К с использованием алгоритмов машинного обучения позволил определить параметры локальной атомной структуры различных изомеров и их концентрации в зависимости от температуры.
МЕТОДЫ
Следуя подходу, реализованному в приложении PyFitIt [12], для каждого компонента смеси строят модели машинного обучения, позволяющие быстро рассчитывать спектр для заданной геометрической структуры. В интерактивном приложении пользователь с помощью слайдеров меняет параметры геометрии структур компонентов смеси и тут же видит результирующий спектр (рис. 1), что позволяет удобно проводить подгонку теоретического спектра под экспериментальный в ручном режиме или запустить автоматический оптимизатор.
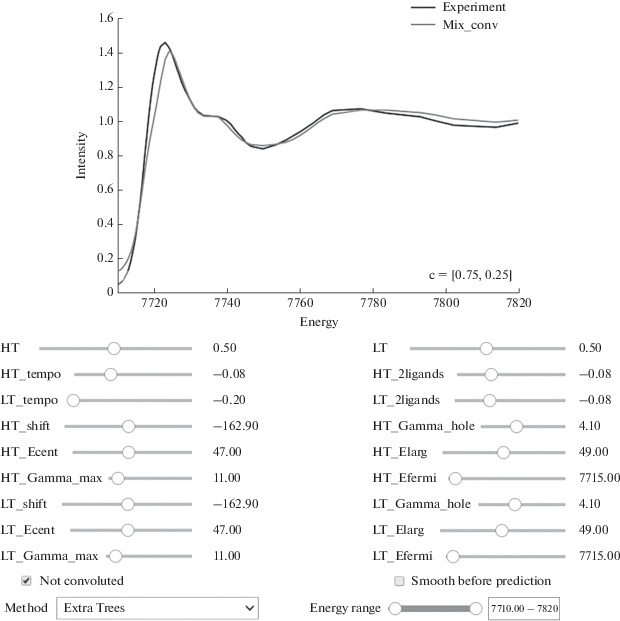
Для работы данного подхода пользователю программы необходимо для каждого предположительного i-го компонента смеси определить вектор геометрических параметров gi и составить функцию Mi, которая создает атомную структуру по заданному вектору gi, i = 1, …, n, n – число компонентов. В качестве координат вектора gi могут выступать смещения групп атомов в некоторых направлениях, повороты частей структуры, смещения отдельных атомов или изменение координат всех атомов структуры. Необходимо учитывать, что большое количество геометрических параметров потребует долгого времени при построении обучающей выборки.
Полученные параметры атомной структуры модельных соединений затем вводят в программу расчета спектров рентгеновского поглощения, например, FDMNES [14, 15]. Обозначим процедуру расчета спектра по заданной атомной структуре через S. Точное вычисление спектра требует много времени, что не позволяет использовать его в интерактивном режиме. Поэтому используем аппроксимацию методами машинного обучения. Построим обучающую выборку, рассчитав спектры XANES i-го компонента для набора значений вектора gij, j = 1, …, mi:
(1)
$\begin{gathered} {\text{XANE}}{{{\text{S}}}_{{ij}}} = S({{M}_{i}}({{g}_{{ij}}})), \\ j = 1,{\text{\;}} \ldots ,{\text{\;}}{{m}_{i}},\,\,\,\,i = 1,{\text{\;}} \ldots ,{\text{\;}}n. \\ \end{gathered} $Рассчитанная база данных спектров позволяет построить аппроксимацию Ai суперпозиции S∘Mi, для быстрого получения приближения спектра по заданному произвольному вектору геометрических параметров gi:
(2)
${\text{ApproxXANE}}{{{\text{S}}}_{i}} = {{A}_{i}}({{g}_{i}}),\,\,\,\,i = 1,{\text{\;}} \ldots ,{\text{\;}}n.$Итоговый спектр XANES получаем суммированием приближенных спектров, умноженных на некоторые веса Ci(T), зависящие в случае рассматриваемого комплекса кобальта от температуры T:
(3)
${\text{XANES(}}T{\text{)}} = {\text{\;}}\mathop \sum \limits_{i = 1}^n {{C}_{i}}(T){\text{Approx\;XANE}}{{{\text{S}}}_{i}}.$Выбор точек gij оказывает существенное влияние на точность построенной аппроксимации. Практика показала, что хорошее качество дает улучшенный метод выбора точек на основе латинского гиперкуба [16] (IHS – Improved Hypercube Sampling). В отличие от метода выбора точек в узлах координатной сетки, при IHS-подходе к генерации векторов g получают точки с неповторяющимися координатами и равномерно распределенными в пространстве. Это позволяет лучше аппроксимировать функции нескольких переменных при условии, что от одной или нескольких переменных функция зависит слабо.
В качестве модели аппроксимации допустимо использовать любые методы восстановления регрессии. Представленное на рис. 1 приложение PyFitIt позволяет осуществлять выбор между следующими методами: лес деревьев с повышенной случайностью [17], гребневая регрессия (линейная/квадратичная) [18], радиальные базисные функции [19]. Последний метод является интерполяцией и часто дает самые хорошие результаты, хотя и не всегда.
Итоговая схема аппроксимации спектров и подбора геометрических параметров приведена на рис. 2. Помимо графического интерфейса пользователя PyFitIt предоставляет возможность полностью автоматического подбора геометрических параметров атомных структур компонентов и зависимости их концентраций от температуры. Этот подход оптимизирует функцию F(g1, …, gn) структурных параметров:
(4)
$F({{g}_{1}}, \ldots ,{\text{\;}}{{g}_{n}}) = \mathop \sum \limits_T \begin{array}{*{20}{c}} {\mathop {{\text{min}}}\limits_{\{ {{C}_{1}} \ldots {{C}_{n}}\} } \left| {{\text{TheorXANES}} - \mathop \sum \limits_{i = 1}^n {{C}_{i}}{\text{ApproxXANE}}{{{\text{S}}}_{i}}} \right|} \end{array}.$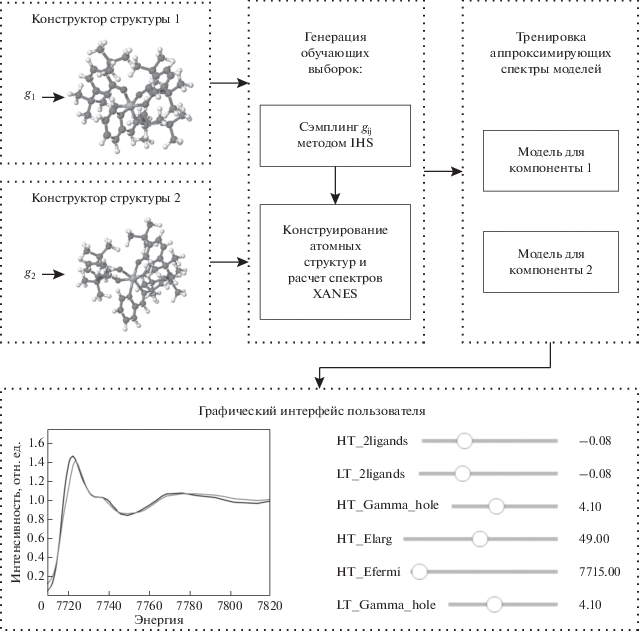
Так как оптимизация может сойтись к локальному минимуму, то PyFitIt делает несколько попыток запуска минимизации F(g1, …, gn) для различных начальных конфигураций атомной структуры. Получаемые зависимости концентраций от температуры Ci(T) в некоторых случаях могут оказаться негладкими. Для устранения этого недостатка в PyFitIt имеется возможность задать узловые точки по температуре, для которых происходит поиск концентраций Ci(T) и суммирование по T в (4). Концентрации в промежуточных точках вычисляются путем интерполяции сплайном.
Спектры XANES K-краев поглощения Co для предполагаемых структур (diox)2Co(imPy-TEMPO) при его валентно-таутомерной интерконверсии были рассчитаны с помощью метода конечных разностей полного потенциала, реализованного в коде FDMNES [14, 15]. Была использована конечно-разностная сетка с расстоянием 0.2 Å между соседними точками внутри сферы с радиусом 6 Å вокруг поглощающего атома кобальта. Теоретические спектры были дополнительно сглажены для учета уширения пиков из-за эффекта туннелирования и инструментальных погрешностей (для моделирования энергетической зависимости ширины лоренциана использован арктангенс). Затем был проведена подгонка спектров XANES с помощью программного обеспечения PyFitIt [12]. Начиная с теории функционала плотности оптимизированной структуры была применена вариация двух структурных параметров в комплексе: расстояния между атомами кобальта и азота лиганда imPy-TEMPO и расстояния между атомом кобальта и четырьмя атомами кислорода двух диокс-лигандов. Для каждой точки в двумерном пространстве структурных параметров, сгенерированной алгоритмом IHS, был рассчитан спектр XANES K-края поглощения Co. На основе полученной обучающей выборки спектры затем аппроксимировали в каждой точке двумерного пространства структурных параметров методом радиальных базисных функций.
Экспериментальные спектры поглощения кобальта за K-краем были измерены на станции структурного материаловедения Курчатовского источника синхротронного излучения при использовании монохроматора Si(111) и геометрии на прохождение.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
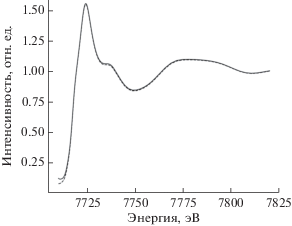
Для интерполяции спектров рентгеновского поглощения за K-краем поглощения кобальта комплекса (diox)2Co(imPy-TEMPO) по обучающей выборке наилучшими оказались радиальные базисные функции. Для проверки качества аппроксимации была применена десятиблочная кросс-валидация (перекрестная проверка): обучающая выборка была разбита на десять блоков, каждый из которых по очереди использовали в качестве проверочной выборки, тогда как все остальные – в качестве тренировочной выборки. Средняя относительная погрешность интерполяции спектра в результате оказалась равной 2.3% по отношению к ошибке аппроксимации средним спектром, что является достаточно хорошим показателем. Для наглядности на рис. 3 представлен наихудший случай аппроксимации – теоретический спектр, для которого получена самая большая ошибка аппроксимации при обучении модели по остальным спектрам.
Рис. 3.
Качество аппроксимации спектров. Представлен наихудший случай аппроксимации (штриховая линия) теоретического спектра (сплошная линия), получена самая большая ошибка аппроксимации при обучении модели по остальным спектрам.
Подгонка двумя компонентами
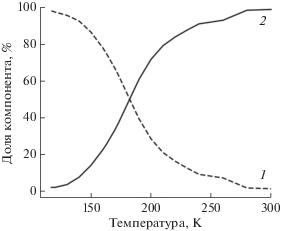
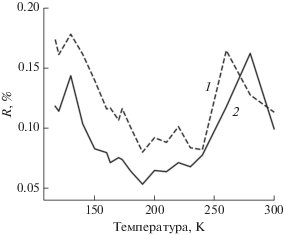
Первой попыткой моделирования эксперимента была подгонка двумя компонентами, полученными из одного семейства структур с различными геометрическими параметрами. В результате процесса подгонки под экспериментальные спектры для каждой отдельной температуры был получен график концентраций компонентов (рис. 4). Даже не накладывая искусственных ограничений на концентрации компонентов для крайних температур, получили результат с чистыми веществами для T = 117 и 300 К. Структурные параметры компонента при 117 К: среднее расстояние между атомом кобальтом и двумя атомами азота лиганда imPy-TEMPO равно 2.08 Å, (Co–N = = 2.1080, 2.1443 Å при 240 К [13]); среднее расстояние между атомом кобальта и четырьмя атомами кислорода двух диокси-лигандов 2.03 Å (Co–O = = 2.0062–2.0659 Å при 240 К [13]). Для компонента при T = 300 К среднее расстояние между атомом кобальта и двумя атомами азота лиганда imPy-TEMPO равно 2.18 Å (Co–N = 2.1226, 2.1586 Å [13]), среднее расстояние между кобальтом и четырьмя атомами кислорода двух диокс-лигандов 2.03 Å (Co–O = 2.0150–2.0747 Å [13]). Спектры двух компонентов, которые использовали при подгонке серии спектров, и соответствующие экспериментальные спектры приведены на рис. 5. Зависимость качества подгонки от температуры представлена на рис. 6 (сплошная линия). Для крайних температур R-фактор чуть больше, чем для температур, при которых концентрации обеих компонентов ненулевые. Причиной этого, видимо, является уширение спектра смеси в случае нескольких компонентов.
Рис. 4.
Зависимость концентраций компонентов от температуры. Результат получен с чистыми веществами для T = 117 (1) и 300 К (2).
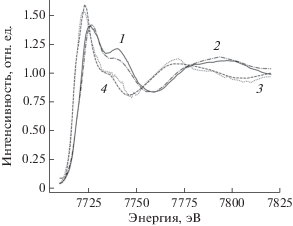
Подгонка одним компонентом
Описываемые в работе методы позволяют реализовать другой тип моделирования происходящих в эксперименте изменений. Будем проводить подгонку экспериментальных спектров одним компонентом, непрерывно изменяющим свою структуру в зависимости от температуры. Результаты такой подгонки представлены на рис. 7. Полученные для крайних температур 117 и 300 К параметры согласуются с параметрами соответствующего компонента в многокомпонентной подгонке. Для промежуточных значений, чтобы воспроизвести наблюдаемые в эксперименте уширения спектральных полос, метод выбирает несимметричные смещения лигандов, что косвенно моделирует сосуществование двух фаз. График зависимости R-фактора от температуры (рис. 6, штриховая линия) похож на график в случае многокомпонентной подгонки, только располагается выше. Это говорит о том, что результаты двухкомпонентной подгонки лучше согласуются с экспериментом.
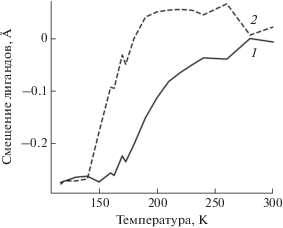
ЗАКЛЮЧЕНИЕ
В работе описан метод определения структурных параметров компонентов смеси из анализа рентгеновских спектров поглощения методами машинного обучения. На основе программного комплекса PyFitIt было создано приложение, позволяющее вычислять структурные параметры компонентов смеси по заданному набору экспериментальных спектров. Данный подход является альтернативой уже известным методам – анализу основных компонентов, фактор-анализу, MCR-ALS, так как имеет возможность подбора параметров смеси по единственному экспериментальному спектру. Разработанный метод использован для определения структурных параметров компонентов смеси и вариации их концентраций при температурной валентно-таутомерной интерконверсии в комплексе кобальта (diox)2Co(imPy-TEMPO).
Список литературы
Soldatov M.A., Martini A., Bugaev A.L. et al. // Polyhedron. 2018. V. 155. P. 232.
Frenkel A.I., Kleifeld O., Wasserman S.R., Sagi I. // J. Chem. Phys. 2002. V. 116. P. 9449.
Piovano A., Agostini G., Frenkel A.I. et al. // J. Phys. Chem. C. 2011. V. 115. P. 1311.
Fernandezgarcia M., Alvarez C.M., Haller G.L. // J. Phys. Chem. 1995. V. 99. P. 12565.
Jaumot J., de Juan A., Tauler R. // Chemom. Intell. Lab. Syst. 2015. V. 140. P. 1.
Jaumot J., Gargallo R., de Juan A., Tauler R. // Chemom. Intell. Lab. Syst. 2005. V. 76. P. 101.
Della Longa S., Arcovito A., Girasole M. et al. // Phys. Rev. Lett. 2001. V. 87. P. 155501.
Benfatto M., Congiu-Castellano A., Daniele A., Longa S.D. // J. Synchr. Rad. 2001. V. 8. P. 267.
Benfatto M., Della Longa S., Natoli C.R. // J. Synchr. Rad. 2003. V. 10. P. 51.
Hayakawa K., Hatada K., D’Angelo P. et al. // J. Am. Chem. Soc. 2004. V. 126. P. 15618.
Smolentsev G., Soldatov A.V. // Comp. Matt. Sci. 2007. V. 39. № 3. P. 569.
Martini A., Guda S.A., Guda A.A. et al. // Comp. Phys. Commun. 2020. V. 250. P. 107064. https://doi.org/10.1016/j.cpc.2019.107064
Zolotukhin A.A., Bubnov M.P., Arapova A.V. et al. // Inorg. Chem. 2017. V. 56. P. 14751. https://doi.org/10.1021/acs.inorgchem.7b02597
Bunau O., Joly Y. // J. Phys.: Condens. Matt. 2009. V. 21. P. 345 501.
Guda S.A., Guda A.A., Soldatov M.A. et al. // J. Chem. Theory Comp. 2015. V. 11. P. 4512.
Beachkofski B.K., Grandhi R.V. Improved Distributed Hypercube Sampling // 43rd AIAA/ASME/ASCE/ AHS/ASC Structures, Structural Dynamics, and Materials Conf. Denver, Colorado, 2002. https://doi.org/10.2514/6.2002-1274
Geurts P., Ernst D., Wehenkel L. // Machine Learning. 2006. V. 63. P. 3.
Тихонов А.В. // ДАН СССР. 1963. Т. 151. № 3. С. 501.
Fasshauer G.E. Meshfree Approximation Methods with Matlab. World Scientific, 2007. 520 p. https://doi.org/10.1142/6437
Дополнительные материалы отсутствуют.
Инструменты
Поверхность. Рентгеновские, синхротронные и нейтронные исследования