

Прикладная биохимия и микробиология, 2019, T. 55, № 1, стр. 17-24
Роль аларминов в синтезе тканевых цитокинов
Д. Б. Чудаков 1, М. А. Шевченко 1, Г. В. Фаттахова 1, Е. В. Свирщевская 1, *
1 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
117997 Москва, Россия
* E-mail: esvir@.ibch.ru
Поступила в редакцию 30.03.2018
После доработки 14.07.2018
Принята к публикации 25.07.2018
Аннотация
Первичной реакцией врожденной иммунной системы на инвазию патогенов или травму тканей является синтез тканевых цитокинов интерлейкинов (ИЛ) 25, 33 и тимического стромального лимфопоэтина (ТСЛП) эпителиальными или мышечными клетками в ответ на фрагменты патогенов, называемые PAMP (pathogen associated molecular patterns), или эндогенные факторы клеточного стресса, называемые DAMP (danger associated molecular patterns). В работе проведено сравнение способности бактериального пептида N-формил-метионил-лейцил-фенилаланина (fMLP) и эндогенного медиатора зуда аминокислоты β-аланина стимулировать экспрессию генов и синтез тканевых цитокинов. Методом количественной ПЦР показали, что ex vivo в культурах эксплантов трахеи мышей fMLP стимулировал экспрессию генов ТСЛП и ИЛ-25, а β-аланин также экспрессию ИЛ-33. Уровень цитокинов ТСЛП и ИЛ-33 in vivo оценивали методом ИФА в гомогенатах легких мышей после интратрахеального введения факторов. β-аланин и fMLP вызывали выброс из внутриклеточного депо ИЛ-33 и ТСЛП. Впервые показано, что β-аланин и fMLP являются активаторами синтеза тканевых цитокинов.
Переключение В-клеток на синтез иммуноглобулинов Е-класса (IgE) к белкам, попадающим из пищи и окружающей среды, происходит под действием интерлейкинов (ИЛ) 4 и 13, активирующих транскрипционный фактор STAT6 [1]. Источником ИЛ-4 и 13 могут быть как Т-хелперы 2 типа (Тх2), принадлежащие адаптивному иммунному ответу [2], так и лимфоидные тканевые клетки 2 типа (ЛТК2), относящиеся к врожденному более древнему иммунитету [3, 4]. Показано, что ЛТК2 активируются факторами, продуцируемыми клетками барьерного эпителия, к которым относят ИЛ-25, 33 и тимический стромальный лимфопоэтин (ТСЛП) [3, 4]. ИЛ-25, 33 и ТСЛП продуцируются многими типами иммунных клеток. Эти цитокины получили название тканевых из-за того, что клетки различных тканей, не относящиеся к иммунной системе (эпителиальные, эндотелиальные, мышечные, жировые клетки), также способны их синтезировать. Значительное количество исследований посвящено анализу роли ИЛ-25, 33 и ТСЛП в аллергическом иммунном ответе [5], а также возможности блокирования этих факторов с терапевтической целью [6]. Так, в модели аллергического ответа на мышах введение блокирующего антитела к ИЛ-25 уменьшало число Тх2 в легких, продукцию ИЛ-4, 5 и 13, подавляло воспаление и фиброз ткани бронхов, снижало продукцию тотального и специфического IgE [7–9]. Похожий эффект оказывало введение блокирующего антитела к ИЛ-33 в модели респираторной аллергии на овальбумин у мышей, что приводило к снижению гиперреактивности бронхов, уменьшению числа эозинофилов в лаважах и легочной ткани, подавлению продукции специфического IgE [8–10]. Введение блокатора ТСЛП за 30 мин до инъекции овальбумина мышам подавляло синтез ИЛ-4, 5 и 13, снижало число макрофагов и эозинофилов в лаважах, уменьшало продукцию специфического IgE [9–11]. Аналогичные данные были получены и в клинике [10–12].
Известно, что тканевые цитокины высвобождаются эпителиальными клетками под действием повреждающих факторов как экзогенного (PAMP, pathogen associated molecular patterns), так и эндогенного (DAMP, danger associated molecular patterns) происхождения [5, 11]. За исключением спор грибов, остальные аллергены не содержат PAMP. Соответственно, источником первичного ответа на аллергены могут служить эндогенные DAMP. К эндогенным DAMP, высвобождаемым обычно при некротическом повреждении клеток, относят АТФ, мочевую кислоту, белки теплового шока HSP70 и HSP90 [6, 7]. Выброс указанных DAMP может осуществляться под действием протеаз [7], например гриба Alternaria [7] или клещей домашней пыли Dermatophagoides [13]. Протеазы стимулируют выброс DAMP, активируя G-белковые активируемые протеазой рецепторы (PAR, protease-activated receptor), в основном PAR-2 [12]. Активация PAR даже в отсутствие некроза клеток приводит к выбросу ими АТФ, которая, в свою очередь, стимулирует по аутокринному механизму через активацию ионотропного P2X-рецептора выброс теми же клетками преформированного ИЛ-33 [7], и, возможно, его синтез de novo, а также к выбросу еще одного DAMP – мочевой кислоты, активирующей de novo синтез ТСЛП [12]. Одновременно сигнальные пути, запускаемые при активации PAR-2 протеазами, напрямую стимулируют продукцию ИЛ-25 и ТСЛП de novo [12].
Феномен активации продукции тканевых цитокинов за счет высвобождения DAMP, в частности АТФ, может быть использован для разработки новых методов терапии и профилактики с использованием низкомолекулярных фармакологических ингибиторов пуринергических рецепторов. В одной из последних работ показана способность блокатора пуринергических рецепторов сурамина, вводимого за неделю до аллергена (овальбумина) мышам C57BL/6 интраназально, частично уменьшать интенсивность воспаления ткани легких [14]. Действительно, в запуске продукции тканевых цитокинов могут участвовать и другие алармины, отличные от АТФ по строению и не воспринимаемые пуринергическими рецепторами. В связи с этим актуальным становится поиск новых DAMP.
Спектр DAMP может быть значительно шире – например N-формилпептиды митохондиального происхождения [15], сходные по строению с пептидом бактериального происхождения N-формил-метионил-лейцил-фенилаланином (fMLP) [16], известным как хемоаттрактант нейтрофилов [15], или эндогенная аминокислота β-аланин, являющаяся медиатором зуда [17] и содержащаяся в виде дипептидов карнозина и ансерина в мышечной ткани, часто повреждаемой при инвазии макропаразитами [5], и другие. Поскольку зуд является одним из основных симптомов аллергии, было выдвинуто предположение, что β-аланин может быть одним из основных DAMP, вызывающих запуск аллергического ответа I типа.
Выявление новых DAMP – стимуляторов синтеза тканевых цитокинов – поможет дальнейшей разработке низкомолекулярных ингибиторов их рецепторов для противоаллергической терапии.
Цель работы − анализ влияния потенциального DAMP β-аланина, а также бактериального пептида fMLP на продукцию тканевых цитокинов в условиях ex vivo и in vivo.
МЕТОДИКА
Стимуляция барьерного эпителия ex vivo. В работе использовали коллагеназу I, трипсин, β-аланин, fMLP (все реактивы “Sigma”, США). Все эксперименты на животных проведены в соответствии с утвержденным протоколом Комиссии по работе с животными ИБХ РАН.
У интактных мышей линии CD1 (питомник “Андреевка” Научного центра биомедицинских технологий Федерального медико-биологического агентства, Россия) выделяли трахею (пул от 4 мышей), фрагменты уха (кожа и хрящ), а также легкие. Ткань разрезали на фрагменты (примерно 10 мг/пробу) и помещали в лунки 48-луночного планшета (“Costar”, США) в 400 мкл физраствора (ФР) без или с различными стимуляторами в концентрации по 1 мМ. Экспланты инкубировали при 37°С 4 ч на шейкере (50 об./мин). После инкубации экспланты гомогенизировали в реагенте TRizol (“Invitrogen”, Россия) для дальнейшего анализа экспрессии гена ИЛ-33 и контрольного гена глицеральдегид 3-фосфат дегидрогеназы (GAPDH).
Выделение мРНК и определение экспрессии генов ИЛ-25, ИЛ-33 и ТСЛП в эксплантах тканей методом количественной ПЦР. Тотальную матричную РНК выделяли из гомогенатов тканей с использованием набора RNeasy Mini Kit (“Qiagen”, США) и очищали от примеси ДНК обработкой ДНКазой I (“Fermentas”, США). Концентрацию мРНК определяли наприборе NanoDrop 2000 (“Thermo Scientific”, США), качество чистоту мРНК оценивали электрофоретическим методом в 1%-ном агарозном геле. Синтез кДНК проводили с использованием набора First Strand cDNA Synthesis (“Thermo Scientific”, США) и oligo(dT) 18 праймеров. Полученную кДНК использовали в качестве матрицы для стандартного ПЦР и ПЦР в реальном времени (кПЦР) со специфическими праймерами (табл. 1). Для кПЦР использовали смесь qPCRmix-HS SYBR (“Евроген”, Россия). Анализ проводили на приборе DTprime real-time PCR machine (“DNA-Technology”, Россия). Реакционная смесь включала по 1 мкл кДНК, праймеры в концентрации 0.1 мкМ и смесь qPCRmix-HS SYBR (“Евроген”, Россия). Праймеры с температурой отжига 60°С подбирали в программе Nucleotide Blast. Праймеры синтезированы компанией “Евроген” (Россия). Реакцию проводили в режиме HotStart (95°С, 300 c), затем циклами по 30 с – 95°С; 45 с – 60°С; 30 с – 72°С. Обработку результатов кПЦР осуществляли в программе RealTime_PCR (“DNA-Technology”, Россия). Анализ экспрессии каждого гена проводили в трех повторах, контрольные образцы – в 6 повторах.
Таблица 1.
Последовательности праймеров
| Ген | Последовательности праймеров | Длина продукта | t, °С отжига |
|---|---|---|---|
| GAPDH прямой | GGTGCTGAGTATGTCGTGGA | 610 | 59.5 |
| GAPDH обратный | TGGAAGAGTGGGAGTTGCTG | 59.6 | |
| TSLP прямой | CTGCCTGAATCAAACCTCACAA | 498 | 59.1 |
| TSLP обратный | TGACTGCCCGAACTGTCATT | 59.6 | |
| ИЛ-25 прямой | CCCAGCAAAGAGCAAGAACC | 632 | 59.4 |
| ИЛ-25 обратный | ATCCTCTAGCAGCACAAGCG | 60.2 | |
| ИЛ-33 прямой | GTCTCCTGCCTCCCTGAGTA | 737 | 60.0 |
| ИЛ-33 обратный | GTGGTGCCTGCTCTTCTGAA | 60.3 |
Данные кПЦР были нормированы по экспрессии мРНК гена GAPDH и приведены как относительная экспрессия генов, рассчитанная в формате 2–ddCT [18]. Использование данного метода обсчета данных кПЦР позволяет оценить, во сколько раз изменилось количество копий гена при активации аларминами по сравнению с контролем, нормированное относительно экспрессии гена GAPDH.
Стимуляция барьерного эпителия in vivo. Интратрахеальное введение стимуляторов мышам линии CD1 (n = 3–4) осуществлялось как описано ранее [19] в объеме 50 мкл ФР. Использовали 1, 50 и 250 нмоль коллагеназы I, fMLP и β-аланина на мышь [20]. В качестве контроля использовали ФР. У мышей под ингаляционным наркозом отбирали образцы крови из орбитального синуса глаза. Мышей убивали методом цервикальной дислокации через 1 и 6 ч. Поскольку ИЛ-33 содержится в эпителии в преформированном виде, анализ высвобождения цитокинов определяли в динамике через 1 и 6 ч. Легкие мышей гомогенизировали в буфере, содержащем 150 мМ NaCl, 1 мМ CaCl2, 1 мМ MgCl2, 0.5% тритон Х-100, что приводило к высвобождению внутриклеточных цитокинов [21]. Гомогенаты центрифугировали 10 мин при 13 500 g, супернатанты до анализа хранили при –20°С.
Иммуноферментный анализ. Для определения содержания тканевых цитокинов использовали иммуноферментный сэндвич-анализ (ИФА) и антитела компании BioLegend (Англия). В 96-луночные планшеты (MaxiSorb, “Costar”, США) на ночь наносили антитела к ИЛ-33 (клон Poly5165, “BioLegend”, Англия) или ТСЛП (клон 28F12) в концентрации 1 мкг/мл в 50 мкл ФБ. Между стадиями планшет промывали 5 раз 0.05%-ным твин-20 в ФБ. Неспецифическое связывание блокировали с помощью 1%-ного раствора бычьего альбумина (БСА) в ФБ, в него же добавляли образцы в разведении ½, а также различные разведения стандартов рекомбинантных мышиных ИЛ-33 и ТСЛП. После промывки добавляли меченые биотином антитела к ИЛ-33 (клон Poly5165) и ТСЛП (клон 65B12) в разведении 1/1000 и инкубировали 3 ч с последующим внесением конъюгата стрептавидин-пероксидаза хрена в разведении 1/1000 и инкубацией в течение 1 ч. После отмывки вносили субстрат 3,3',5,5'-тетраметилбензидин. Реакцию останавливали 10%-ной H2SO4 и анализировали на ридере (“ThermoScientific”, США) при длине волны 450 нм. Концентрацию рассчитывали по калибровочной кривой.
Статистический анализ. Среднее и стандартное отклонения рассчитывались с помощью программного обеспечения Excel. Достоверность различий рассчитывали по непараметрическому критерию Манна−Уитни. Результат считался статистически значимым, при p < 0.05.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Стимуляция экспрессии генов тканевых цитокинов в эксплантах барьерных тканей. Поскольку классическими DAMP являются протеазы, то в работе использовали коллагеназу и трипсин в качестве положительных контролей, наряду с потенциальными аларминами fMLP и β-аланином. Методом кПЦР оценивали экспрессию генов ИЛ-25, ИЛ-33, ТСЛП под действием аларминов в клетках трахеи, кожи и легких. Ранее было установлено методом кПЦР, что ИЛ-33 конститутивно экспрессируется во многих эпителиальных тканях, в том числе и в клетках трахеи, кожи и легких [22]. В настоящей работе показано, что во всех анализируемых тканях экспрессируются гены тканевых цитокинов ТСЛП, ИЛ-25 и ИЛ-33 (рис. 1а). Уровень экспрессии генов падал в ряду ТСЛП > ИЛ-25 > ИЛ-33.
Рис. 1.
Экспрессия генов тканевых цитокинов: а – конститутивная экспрессия генов GAPDH и цитокинов в трахее (1), коже (2) и легких (3); б–г –ОЭ генов ТСЛП (б), ИЛ-25 (в) и ИЛ-33 (г) в трахее (I), коже (II) и легких (III) через 4 ч после добавления коллагеназы (1), β-аланина (2) или fMLP (3). Звездочками отмечены достоверные отличия по критерию Манна−Уитни.
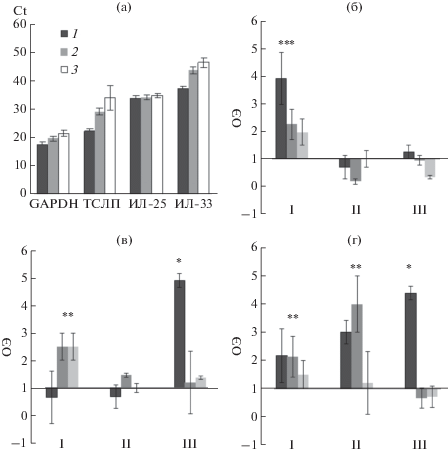
Добавление протеаз и коллагеназы I в культуры эксплантов трахеи и легких приводило к усилению экспрессии всех трех генов цитокинов (рис. 1б–1г). Эффект коллагеназы на экспрессию ИЛ-25 в трахее не был достоверным. В то же время коллагеназа стимулировала экспрессию ИЛ-33 и ТСЛП в эксплантах трахеи. Действие fMLP и β-аланина было выражено, в основном, на клетках трахеи, в которых β-аланин стимулировал экспрессию всех трех изученных генов, а fM-LP – ТСЛП и ИЛ-25 (рис. 1б–1г). В эксплантах кожи β-аланин стимулировал экспрессию гена ИЛ-33. Эффект fMLP и β-аланина в эксплантах легких отсутствовал.
Стимуляция продукции тканевых цитокинов в условиях in vivo. Для тканевых цитокинов важно было подтвердить синтез белков, поскольку индукция экспрессии гена и накопление продукта внутри клетки не обязательно означает активацию его секреции. Так, экспрессия гена ИЛ-33 на определенном уровне идет в барьерном эпителии конститутивно, однако высвобождение его возможно только после стимуляции PAMP, DAMP или в результате некротической гибели клеток. Кроме того, определенный уровень синтеза тканевых цитокинов может также осуществляться конститутивно как базовый уровень активации эпителия.
С целью подтвердить наблюдаемую in vitro стимуляцию экспрессии генов цитокинов мышам CD1 интратрахеально вводили коллагеназу I, fMLP или β-аланин и анализировали синтез тканевых цитокинов in vivo. Оценивали продукцию ИЛ-33 и ТСЛП в крови, бронхо-легочном лаваже и легких мышей. В качестве контроля использовали интактных мышей и мышей, которым вводили только ФБ. Достоверных различий между интактными мышами и группой, которой вводили ФБ, не было. На рис. 1 приведены результаты только для последней группы. Поскольку известно, что ИЛ-33 содержался в эпителии в преформированном виде, определяли его высвобождение под действием стимуляторов через 1 и 6 ч. Содержание тканевых цитокинов ИЛ-33 и ТСЛП определяли методом ИФА.
Было показано, что под действием коллагеназы I ТСЛП появлялся в легких и лаваже уже через 1 ч, а к 6 ч его уровень увеличивался в 2 раза и одновременно в крови появлялся ТСЛП (рис. 2а, 2б).
Рис. 2.
Уровень тканевых цитокинов в легких и крови мышей CD1 после интратрахеального введения DAMP. Уровень ИЛ-33 (а, б) и ТСЛП (в, г) в бронхолегочном лаваже (I), гомогенате легких (II) и крови (III) на 1 (а, в) и 6 (б, г) ч после введения фосфатного буфера (ФБ, 1), коллагеназы 1 нМ (2), β-аланина 250 нМ (3) или fMLP 50 нМ (4). Приведены средние значения и стандартная ошибка и в пересчете на мг ткани или мл крови. Звездочками отмечены достоверные отличия по критерию Манна−Уитни от группы, которой вводили только ФБ.
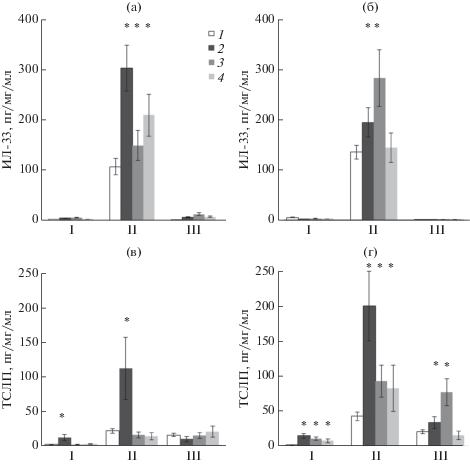
После стимуляции fMLP и β-аланином ТСЛП появлялся только к 6 ч (достоверно по сравнению с 1 ч, р < 0.05), что показывает более медленное действие DAMP в сравнении с протеазой (рис. 2б). Под действием β-аланина ТСЛП появлялся также и в крови через 6 ч.
Уровень ИЛ-33 под действием коллагеназы I был максимальным через 1 ч и на 30% уменьшался к 6 ч (рис. 2в). Введение fMLP и β-аланина стимулировало выброс ИЛ-33 на 1 ч на уровне 50 и 70% по сравнению с коллагеназой I. β-аланин, но не fMLP, усиливал в 2 раза выброс ИЛ-33 к 6 ч (рис. 2г, достоверно при р < 0.05, по сравнению с 1 ч). Достоверных различий по уровню ИЛ-33 в образцах с коллагеназой I и fMLP не было. В лаваже и крови ИЛ-33 практически отсутствовал.
Согласно литературным данным протеазы действуют через активацию G-белкового рецептора PAR-2 при частичном протеолизе его внеклеточного домена [13]. Активируемые сигнальные пути, в том числе MAP-киназные, стимулируют выброс клетками барьерного эпителия АТФ нецитолитическим путем [23]. АТФ действует на ионотропные P2X-рецепторы, открывая ионные каналы, через которые кальций поступает в клетки. Именно поступающий в клетки кальций стимулирует выброс тканевых цитокинов, в частности ИЛ-33, а также вызывает экспрессию их генов de novo [12, 19, 23]. Основные работы по влиянию PAMP/DAMP на синтез тканевых цитокинов выполнены для протеаз аллергенов [12, 13]. Эффект коллагеназы I и трипсина на экспрессию гена ИЛ-33 был показан, вероятно впервые, но является вполне ожидаемым. Впервые показан аналогичный эффект пептида fMLP и не участвующей в синтезе белка аминокислоты β-аланина на экспрессию генов ИЛ-25, ИЛ-33 и ТСЛП. Интерес к DAMP вызван необходимостью анализа роли тканевых цитокинов в индукции аллергического ответа [24–26]. Введение мышам протеаз, имеющих относительно большую молекулярную массу, будет вызывать иммунный ответ на сами протеазы, что не позволит проанализировать ответ на аллергены без протеазной активности.
Анализ влияния коллагеназы I, fMLP и β-аланина на экспрессию генов тканевых цитокинов показал, что наибольший эффект проявляла коллагеназа в трахее и легких, что можно объяснить взаимодействием одной молекулы фермента со многими рецепторами PAR-2 в отличие от классических DAMP, одна молекула которых способна взаимодействовать только с одним рецептором на клетке. Некоторые различия эффектов коллагеназы и трипсина могут объясняться вовлечением в активацию иных PAR, кроме PAR-2, поскольку коллагеназа I отличается по субстратной специфичности от трипсина. Действие fMLP и β‑аланина было выражено в клетках трахеи и отсутствовало в клетках легких.
Достаточно неожиданным было отсутствие влияния протеаз на кератиноциты, в то время как β-аланин вызывал значительную экспрессию гена ИЛ-33. Известно, что кератиноциты синтезируют все изучаемые тканевые цитокины [27, 28]. Литературные данные по эффекту протеаз, fMLP и β-аланина на экспрессию генов или синтез тканевых цитокинов кератиноцитами отсутствуют.
Анализ данных по продукции тканевых цитокинов ТСЛП и ИЛ-33 под действием коллагеназы I показал, что выброс ИЛ-33 и ТСЛП наблюдался уже через 1 ч после введения и сохранялся до 6 ч, что согласуется с литературными данными, согласно которым при активации клетки барьерного эпителия высвобождают преимущественно преформированный ИЛ-33 [23]. Пик высвобождения ТСЛП наблюдали позднее [23], что соответствовало полученным данными. Наличие ТСЛП в лизате легких через 1 ч показало, что и ТСЛП находился в преформированном виде, поскольку для синтеза de novo 1 ч недостаточно. Наличие преформированного ТСЛП в клетках легких было показано впервые.
Действие низкомолекулярных стимуляторов fMLP и β-аланина было более медленным по сравнению с коллагеназой I и проявлялось только к 6 ч, что может быть объяснено их диффузией через эпителий, с дальнейшим действием на другие клетки, находящиеся под ним (гладкомышечные клетки, фибробласты, макрофаги) [29–31].
Было показано, что ТСЛП, но не ИЛ-33 диффундирует из тканей в кровь и межтканевую жидкость (появляется в лаваже). Отсутствие ИЛ-33 в жидкостях организма через 6 ч может быть связано с недостаточным временем для созревания ИЛ-33, который синтезируется в виде предшественника и превращается в зрелый белок под действием каспазы 1. Белок-предшественник ИЛ-33 имеет двойную активность и действует как фактор транскрипции и супрессор воспаления благодаря наличию сигнала ядерной локализации в пропептиде [32].
Можно предположить, что коллагеназа I, fMLP и β-аланин взаимодействуют с различными клетками. Так коллагеназа I, имеющая молекулярную массу ~115 кДа, контактирует, в первую очередь, с эпителием бронхов, полярного по строению, высвобождающего продукты секреции через апикальную мембрану во внутрибронхиальное и внутриальвеолярное пространство. Низкомолекулярные β-аланин (89 Да) и fMLP (450 Да) способны проникать через слой эпителия, достигая клеток гладкой мускулатуры бронхов, фибробластов [29] и макрофагов [30, 31].
Первично β-аланин был охарактеризован как метаболит карнозина и ансерина – мышечных дипептидов, а уже затем как медиатор зуда, действующий через рецептор MrgprD (Mas-related G-protein coupled receptor type D ) [17]. Семейство Mrgpr-рецепторов, названное так вследствие гомологии их генов с протоонкогенным рецептором Mas1, вначале идентифицировали на сенсорных нейронах, ответственных за восприятие боли, и на тучных клетках. В дальнейшем было показано, что они экспрессируются значительно шире [33]. Так, MrgprD экспрессируют клетки дорсальных корешковых ганглий, мозжечка, а также структуры вне нервной системы – клетки мочевого пузыря, семенников, стенки артерий [33]. Показана на низком уровне экспрессия MrgprD в эпителии кожи, легких, пищеварительного тракта. Роль MrgprD во врожденном иммунном ответе пока не показана. Таким образом, мишенью β-аланина, скорее всего, являются мышечные, эндотелиальные или тучные клетки, с чем связана его более поздняя по сравнению с коллагеназой I активность.
Пептид fMLP является продуктом бактерий и служит хемоаттрактантом для клеток врожденного иммунитета [16]. Формильная группа метионина используется бактериями для синтеза белка и отсутствует в организме млекопитающих (за исключением некоторого количества в митохондриях эукариотических клеток). Миелоидные клетки распознают формильную группу с помощью рецепторов FPR (formyl-peptide receptor) и FPLR (formyl peptide-like receptor). Связывание fMLP [34] или подобных ему N-формильных пептидов, происходящих из митохондрий эукариотических клеток [15], с FPR вызывает активацию клетки, сопровождающуюся перестройкой цитоскелета, что приводит к ее миграции по градиенту пептида. Соответственно, мишенью fMLP могут быть макрофаги и дендритные клетки [34].
Участие fMLP и β-аланина в индукции тканевых цитокинов показано впервые. Приведенные данные вносят существенный вклад в понимание механизмов развития локального иммунитета.
Впервые показано, что не только протеазы, но и низкомолекулярные стимуляторы β-аланин и fMLP усиливают экспрессию генов тканевых цитокинов. Действие β-аланина и fMLP на экспрессию генов выражено слабее, чем коллагеназы I, что может быть связано как с разной природой стимулов, так и с действием на разные типы клеток. Как коллагеназа I, так и низкомолекулярные алармины β-аланин и fMLP вызывают высвобождение тканевых цитокинов ИЛ-33 и ТСЛП из внутриклеточных депо эпителиальных клеток, при этом действие β-аланина и fMLP также менее выражено и развивается медленнее, чем эффект коллагеназы I. Таким образом, не только протеазы, но низкомолекулярные алармины β-аланин и fMLP являются стимуляторами продукции тканевых цитокинов.
Работа выполнена при финансовой поддержке Программы фундаментальных исследований Президиума РАН “Наноструктуры: физика, химия, биология, основы технологий”.
Список литературы
Poulsen L.K., Hummelshoj L. // Ann. Med. 2007. V. 36. № 6. P. 440–456.
Licona-Limón P., Kim L.K., Palm N.W., Flavell R.A. // Nat. Immunol. 2013. V. 14. № 6. P. 536–542.
Moro K., Yamada T., Tanabe M., Takeuchi T., Ikawa T., Kawamoto H., Ohtani M., Fujii H., Koyasu S. // Nature. 2010. V. 463. № 7280. P. 540–544.
Neill D.R., Wong S.H., Bellosi A., Flynn R.J., Daly M., Langford T.K., Bucks C., Kane C.M., Fallon P.G., Pannell R., Jolin H.E., McKenzie A.N. Nature. 2010. V. 464. № 7293. P. 1367–1370.
Lambrecht B.N., Hammad H. // J. Allergy Clin. Immunol. 2014. V. 134. № 3. P. 499–507.
Al-Sajee D., Oliveria J.P., Sehmi R., Gauvreau G.M. // Curr Opin Pulm Med. 2018. V. 24. № 1. P. 32–41.
Gregory L.G., Jones C.P., Walker S.A., Sawant D., Gowers K.H., Campbell G.A., McKenzie A.N., Lloyd C.M. // Thorax. 2013. V. 68. № 1. P. 82–90.
Kim Y.H., Park C.-S., Lim D.H., Ahn S.-H., Son B.K., Kim J.H., Jang T.Y. // J. of Asthma. 2012. V. 49. № 7. P. 738–743.
Zhang F., Huang G., Hu B., Song Y., Shi Y. // Clin. Exp. Immunol. 2011. V. 164. № 2. P. 256–264.
Gauvreau G.M., O’Byrne P.M., Boulet L.P., Wang Y., Cockcroft D., Bigler J., FitzGelard J.M., Boedighelmer M., Davis B.E., Dias C., Gorski K.S., Smith L., Bautista E., Corneau M.R., Leigh R., Parmes J.R. // N. Engl. J. Med. 2014. № 370. P. 2102–2110.
Chan J.K., Roth J., Oppenheim J.J., Tracey K.J., Feldmann M., Horwood N., Nanchahai J. // J. Clin. Invest. 2012. V. 122. № 8. P. 2711–2719.
Hara K., Iijima K., Elias M.K., Seno S., Tojima I., K-obayashi T., Kephart G.M., Kurabayashi M., Kita H. // J. Immunol. 2014. V. 192. № 9. P. 4032–4042.
Cunningham P.T., Elliot C.E., Lenzo J.C., Jarnicki A.G., Zosky G.R., Holt P.G., Thomas W.R. // Int. Arch. Allergy Immunol. 2012. V. 158. № 4. P. 347–358.
Zhang F., Su X., Huang G., Xin X.-F., Cao E.-H., Shi Y., Song Y. // J. Immunol Res. 2017. 2017. P. 5358647. doi 10.1155/2017/5358647
Wenceslau C.F., McCarthy C.G., Scasz T., Spitler K., Goulopoulou S., Webb R.C. // Eur. Heart J. 2014. V. 35. № 18. P. 1172–1177.
Palm N.W., Rosenstein R.K., Medzhitov R. // Nature. 2012. V. 484. № 7395. P. 465–472.
Han L., Dong X. // Annu. Rev. Biophys. 2014. № 43. P. 331–355.
Pfaffl M.W. // Nucleic Acids Res. 2001. V. 29. № 9. P. e45.
Rao G.V., Tinkle S., Weissman D.N., Antonini J.M., Kashon M.L., Salmen R., Battelli R.A., Willard P.A., Hoover M.D., Hubbs A.F. // J. Toxicol. Env. Heal. A. 2003. V. 66. № 15. P. 1441–1452.
Kamijo S., Takeda H., Tokura T., Suzuki M., Inui K., Hara M., Matsuda H., Matsuda A., Oboki K., Ohno T., Saito H., Nakae S., Sudo K., Suto H., Ichikawa S., Ogawa H., Okumura K., Takai T. // J. Immunol. 2013. V. 190. № 9. P. 4489–4499.
Whitehead G.S., Grasman K.A., E.C. Kimmel // Toxicology 2003. V. 183. № 1–3. P. 175–197.
Schmitz J., Owyang A., Oldham E., Song Y., Murphy E., McClanahan T.K., Zurawski G., Moshrefi M., Qin J., Li X., Gorman D.M., Bazan J.F., Kastelein R.A. // Immunity. 2005. V. 23. № 5. P. 479–490.
Kouzaki H., Iijima K., Kobayashi T., O’Grady S.M., Kita H. // J. Immunol. 2011. V. 186. № 7. P. 4375–4387.
Chauhan A., Singh M., Agarwal A., Paul N. // J Asthma. 2015. V. 52. № 9. P. 868–872.
Mahmutovic Persson I., Menzel M., Ramu S., Cerps S., Akbarshahi H., Uller L. // Respir Res. 2018. V. 19. № 1. P. 16–25.
Al-Sajee D., Oliveria J.P., Sehmi R., Gauvreau G.M. // Curr Opin Pulm Med. 2018. V. 24. № 1. P. 32–41.
Asahina R., Nishida H., Kamishina H., Maeda S. // Vet Dermatol. 2018. doi 10.1111/vde.12531
Suto H., Nambu A., Morita H., Yamaguchi S., Numata T., Yoshizaki T., Shimura E., Arae K., Asada Y., Motomura K., Kaneko M., Abe T., Matsuda A., Iwakura Y., Okumura K., Saito H., Matsumoto K., Sudo K., Nakae S. // J. Allergy Clin Immunol. 2018. doi 10.1016/j.jaci.2017.12.1007
Kakkar R., Hei H., Dobner S., Lee R.T. // J. Biol. Chem. 2012. V. 287. № 9. P. 6941–6948.
Ohno T., Oboki K., Kajiwara N., Morii E., Flavell R.A., Okumura K., Saito H., Nakae S. // J. Immunol. 2009. V. 183. № 12. P. 7890–7897.
Qi F., Bai S., Wang D., Xu L., Hu H., Zeng S., Chai R., Liu B. // Mol. Immunol. 2017. № 87. P. 284–292.
Lee E.J., So M.W., Hong S., Kim Y.G., Yoo B., Lee C.K. // Cytokine. 2016. № 77. P. 35–43.
Solinski H., Gudermann T., Breit A. // Pharmacol. Rev. 2014. V. 66. № 3. P. 570–597.
Murphy P., Wang J. // Trends Immunol. 2002. V. 23. № 11. P. 241–248.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология