

Прикладная биохимия и микробиология, 2020, T. 56, № 1, стр. 45-51
Клонирование генов нуклеозидфосфорилаз из экстремофильной бактерии Halomonas chromatireducens AGD 8-3, конструирование рекомбинантных штаммов-продуцентов этих белков и исследование их ферментативных свойств
А. Н. Антипов 1, Н. Н. Мордкович 1, Т. В. Хижняк 2, Н. А. Окорокова 1, В. П. Вейко 1, *
1 Институт биохимии им. А.Н. Баха, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” Российской академии наук
119071 Москва, Россия
2 Институт микробиологии им. С.Н. Виноградского, Федеральный исследовательский центр
“Фундаментальные основы биотехнологии” РАН
119071 Москва, Россия
* E-mail: vladveiko@yahoo.com
Поступила в редакцию 14.06.2019
После доработки 20.08.2019
Принята к публикации 30.08.2019
Аннотация
Клонированы гены тимидинфосфорилазы (deoA) и пуриннуклеозидфосфорилазы (deoD) из экстремофильной бактерии Halomonas chromatireducens AGD 8-3. Сконструированы экспрессионные плазмиды и получены высокоэффективные рекомбинантные штаммы − продуценты этих белков. Рекомбинантные нуклеозидфосфорилазы выделены методами ионообменной хроматографии в гомогенном состоянии, исследованы их физические и ферментативные свойства. Показано, что изучаемые тимидинфосфорилаза (HrТРР) и пуриннуклеозидфосфорилаза (HrPNP) формируют димерную и гексамерную формы, соответственно. Выявлена повышенная по отношению к тимидину (в сравнении c ее аналогом из Escherichia coli) удельная активность тимидинфосфорилазы из экстремофильной бактерии H. chromatireducens AGD 8-3.
Нуклеозидфосфорилазы – семейство ферментов, катализирующих обратимое расщепление N‑гликозидной связи у нуклеозидов, присутствуют в клетках практически у всех организмов и принимают участие в биосинтезе нуклеозидов. К ним относятся тимидинфосфорилаза (TPP, КФ .4.2.4), пуриннуклеозидфосфорилаза (PNP, КФ 2.4.2.1) и уридинфосфорилаза (UDP, КФ 2.4.2.3). В последние десятилетия нуклеозидфосфорилазы являются объектами пристального внимания исследователей, что определяется, по крайней мере, тремя причинами. Первая − стремление на основе исследования пространственной структуры этих белков решить фундаментальную задачу определения молекулярного механизма функционирования данного класса ферментов [1–4]. Вторая причина − возможность практического использования нуклеозидфосфорилаз в ферментативном синтезе (“зеленая химия”) аналогов нуклеозидов, находящих самое широкое применение в терапии многих заболеваний [5–10].
Не менее важным является также третий аспект, который заключается в необходимости ингибирования нуклеозидфосфорилаз при лечении различных, в том числе и онкологических, заболеваний. В настоящее время обнаружено, что эти ферменты способствуют прогрессированию злокачественных новообразований. Механизм этого процесса до конца не выяснен, но для тимидинфосфорилазы показано, что этот фермент способствует неконтролируемому ангиогенезу, вносящему существенный вклад в развитие таких заболеваний, как сердечно-сосудистые патологии, ревматоидный артрит, атеросклероз, диабетическая ретинопатия [11–13]. Указанные факты переводят, в частности, тимидинфосфорилазу, в статус несомненной мишени для воздействия на нее при терапии многих, включая и онкологические, заболеваний.
Схема противоопухолевой терапии предполагает использование антипролиферативных препаратов на основе модифицированных нуклеозидов (например, 5-фторурацил). Однако высокий уровень накопления тимидин- и уридинфосфорилаз в клетках злокачественных новообразований приводит к разрушению используемых соединений и резкому снижению их эффективности [14], что, в свою очередь, вызывает необходимость использования повышенных доз этих высокотоксичных соединений. Применение в этом случае специфических ингибиторов нуклеозидфосфорилаз позволяет проводить противораковую терапию в более щадящих для пациента условиях [15].
В ряду нуклеозидфосфорилаз особое внимание также уделяется пуриннуклеозидфосфорилазе – важнейшему ферменту метаболизма пуринов. В настоящее время ведется активный поиск высокоспецифических и эффективных ингибиторов этого фермента, что необходимо для создания селективного Т-клеточного иммунодефицитного статуса организма при проведении трансплантации тканей и отдельных органов. Кроме того, как и другие нуклеозидфосфорилазы, этот фермент нашел свое применение в стереоселективном ферментативном синтезе модифицированных нуклеозидов [16, 17].
Все указанные аспекты (изучение структурно-функциональной организации нуклеозидфосфорилаз, использование этих ферментов в биотехнологических процессах, терапия, основанная на целевом воздействии на этот класс ферментов) инициировали получение и исследование свойств нуклеозидфосфорилаз из различных источников [18–21]. На основании этих данных высказываются как предположения о строении активных центров и причинах различия в функциональной активности этих белков, так и осуществляется подбор оптимальных ферментов для получения модифицированных нуклеозидов.
В этом ряду особое внимание уделяется нуклеозидфосфорилазам из экстремофильных, в частности термофильных, бактерий [22–25]. Интерес к термостабильным нуклеозидфосфорилазам объясняется не только вопросами оптимизации биотехнологических процессов получения модифицированных нуклеозидов (например, увеличению растворимости гетероциклических оснований, используемых в этих процессах), но и фундаментальными попытками объяснить природу устойчивости самих ферментов к термальной денатурации.
Как указано выше, сравнительно низкая растворимость многих гетероциклических оснований при нейтральных значениях pH во многом затрудняет ферментативный синтез модифицированных нуклеозидов. Одновременно с этим, сами нуклеозидфосфорилазы проявляют оптимальную активность именно при нейтральных значениях этого показателя, что приводит к дисбалансу в проводимом процессе. В связи с этим, поиск ферментов, способных осуществлять синтез производных нуклеозидов в приближенных к щелочным pH, также может существенно оптимизировать способ самого ферментативного синтеза.
Цель работы − клонировать гены нуклеозидфосфорилаз из мезофильной бактерии H. chromatireducens AGD 8-3 и сконструировать рекомбинантные штаммы-продуценты этих белков, а также исследовать их ферментативные свойства.
МЕТОДИКА
В работе использовали: ДДС-Na, агарозу (Type I, Low EEO), борную кислоту, трис-основание (Трис-OH), трис-гидрохлорид (Трис-HCl), ЭДТА – (“Sigma”, США), акриламид, N,N'-метилен-бис-акрилиамид (“Serva”, Германия), бромистый этидий, персульфат аммония, N, N, N', N'-тетраметилэтилендиамин – “Fluka” (Швейцария), триптон “Bacto”, агар “Bacto”, дрожжевой экстракт “Bacto” (“Difco”, США), ампициллин “AppliChe-m” (Германия), дезоксирибонуклеозидтрифосфаты “MBI Fermentas” (Литва). Ксантиноксидаза молока коровы, тимидин, инозин и диметилсульфоксид − “Sigma” (США). Неорганические соли фирмы “Merck” (Германия), реактивы квалификации х. ч. и о. с. ч. (Россия). Белковые маркеры молекулярной массы “Unstained Protein Molecular Weight Marker” фирмы “MBI Fermentas” (Литва).
Taq-полимеразу, рестрицирующие эндонуклеазы (BamHI и HindIII) и ДНК-лигазу фага Т4 производства “MBI Fermentas” (Литва) использовали в соответствии с рекомендациями фирм-производителей.
Выделение ДНК, очистку, гидролиз эндонуклеазами рестрикции, лигирование фрагментов ДНК, а также трансформацию клеток E. coli плазмидами проводили согласно [26].
Штаммы E.coli JM110, С600ΔudpRecA- (thi thrB leuB lacY supE tonA recA Tn10) предоставлены Всероссийской коллекцией промышленных микроорганизмов (НИЦ “Курчатовский институт”-ГосНИИгенетика”, Россия). Источником рекомбинантной тимидинфосфорилазы из E. coli (референс-белок) служил полученный нами ранее штамм-продуцент этого фермента [27].
В качестве реципиентного использовали сконструированный ранее бактериальный вектор pUU18 [28, 29], содержащий в своем составе промотор-операторную область гена уридинфосфорилазы из E. coli.
Полимеразную цепную реакцию проводили в амплификаторе “Eppendorf Mastercycler gradient” (“Eppendorf”, Германия) в объеме 20–25 мкл при 2.0 мМ MgCl2; 0.2 мМ каждого из дезоксирибонуклеозидтрифосфатов (dNTP); 67 мМ Трис-HCl (pH 8.3); 0.5 ед. Taq-полимеразы; 1–10 нг ДНК; 5 пмоль каждого праймера; 5% диметилсульфоксида. Режим амплификации (°С/с): 95/60 – 1 цикл; 95/10, 58/10, 72/20 – 25 циклов; 95/10, 58/10, 72/180 – 1 цикл.
В качестве матричной использовали геномную ДНК, выделенную из биомассы клеток H. chromatireducens AGD 8-3 [30] с применением коммерческого наборa “GeneJET Genomic DNA Purification Kit” (“Thermo Fisher Scientific”, США).
Олигодезоксирибонуклеотиды, использованные в работе, приведены в табл. 1.
Таблица 1.
Структуры олигодезоксирибонуклеотидов, использованных в работе*
| Название | 5'-3' | Ген |
|---|---|---|
| C-tpp | TGCCGAAGCTTTCGAGTACCAGTACGAT | deoА |
| N-str | TGGAGGGATCCTGATGCGTTCTCAAG | deoА |
| C_str | AGTCGCCGGAAGCTTCGATGGTCCGA | deoD |
| N_str | TTTTCGGATCCATGGCGACTCCCCATAT | deoD |
Синтез олигодезоксирибонуклеотидов осуществляли с использованием автоматического синтезатора ASM‑800 (“Биоссет”, Россия) и очищали согласно [31].
Выделение плазмид проводили с использованием набора “GeneJet™ Plasmid Miniprep Kit” производства “MBI Fermentas” (Литва). Клетки E. coli, содержащие плазмиду, культивировали в течение 16–18 ч в стеклянных пробирках (или колбах) со средой LB (ампициллин – 150 мкг/мл) при 37°С и 250 об./мин в шейкере-инкубаторе “Excella E25” (“New Brunswick Scientific”, США).
Концентрацию белка определяли по методу Бредфорд [32] с окраской реагентом “Bio-Rad Protein Assay” (“Bio-Rad”, США). В качестве стандарта использовали раствор бычьего сывороточного альбумина (“Sigma”, США).
Электрофоретическое разделение белков проводили по Леммли [33].
Ферментативную активность рекомбинантных тимидинфосфорилазы (HrTPP) и пуриннуклеозидфосфорилазы (HrPNP) определяли в К+-фосфатном буфере согласно [34, 35] соответственно.
Определение четвертичной структуры рекомбинантных HrTPP и HrPNP проводили методом аналитической гель-фильтрации на колонке Tricorn 10/300 c сорбентом Superdex 200 с использованием прибора AKTA FPLC (“GE Healthcare”, Великобритания) в 10 мM Na+-фосфатном буфере, pH 7.4, содержащем 150 мM NaCl. Регистрацию осуществляли при длине волны 280 нм. Объем образца – 100 мкл, скорость элюции – 1 мл/мин. В качестве белков-маркеров использовали набор “Gel Filtration Calibration Kits” (GE Healthcare Life Sciences, Великобритания), а также рекомбинантные тимидин- и уридинфосфорилазу из E. coli.
Выделение и очистку рекомбинантных тимидин- и пуриннуклеозидфосфорилазы проводили, как описано нами ранее для уридинфосфорилазы [36].
Первичную структуру выделенных рекомбинантных белков подтверждали методом MALDI-TOF/TOF-масс-спектрометрического анализа их триптических гидролизатов.
Статистическую обработку результатов серии измерений проводили с использованием программы StatPlus2007 (http://analystsoft.com).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В представленной работе в качестве экстремофильного микроорганизма нами была выбрана гетеротрофная галоалкалофильная бактерия H. chromatireducens AGD 8-3, выделенная из солончаков Кулундинской степи (Россия) [30, 37]. Бактерии способны к росту при высоких значениях солености (2.5 М NaCl) в щелочных условиях при рН 9.5–10.1, что позволяет отнести ее к экстремофилам. Ареал обитания бактерии предполагает наличие у нее особых систем адаптации к суммарному стрессу. Способность к росту в экстремальных условиях предполагает также присутствие в клетках ферментов с измененными свойствами, в том числе и нуклеозидфосфорилаз. Поиск таких белков инициировал клонирование генов тимидин- и пуриннуклеозидфосфорилаз из данного экстремофила, конструирование соответствующих рекомбинантных штаммов-продуцентов этих белков и исследование их свойств.
В настоящее время геном H. chromatireducens AGD 8-3 раскрыт [38] и доступен в базе данных (GeneBank: NZ_CP014226.1), что позволило провести планирование структур олигодезоксирибонуклеотидных праймеров (табл. 1) для амплификации и клонирования участков ДНК, кодирующих структурную часть соответствующих генов.
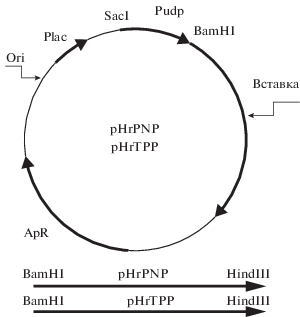
Важно отметить, что при проведении амплификации интересующих фрагментов ДНК из генома H. chromatireducens AGD 8-3 необходимо использовать диметилсульфоксид, в качестве дополнительного (на фоне температурного) фактора денатурации геномной ДНК. В ходе экспериментальной работы было обнаружено, что наиболее эффективно амплификация целевых фрагментов осуществлялась в присутствии в реакционной смеси 5% диметилсульфоксида. Этот факт нами был объяснен высоким G/C-составом ДНК из H. chromatireducens AGD 8-3 (62.8%). Полученные фрагменты были выделены, очищены и клонированы по сайтам BamHI-HindIII в составе экспрессионного вектора pUU18 под контроль промотор-операторной области гена уридинфосфорилазы E. coli. Соответствие первичной структуры клонированных фрагментов запланированной подтверждали секвенированием. Сконструированные рекомбинантные экспрессионные плазмиды были обозначены pHrTPP и pHrPNP (рис. 1).
Рис. 1.
Общая схема экспрессионных плазмид (pHrPNP и pHrTPP). Pudp – промотор-операторная область гена уридинфосфорилазы из E. coli. Вставка − структурная часть генов тимидинфосфорилазы или пуриннуклеозидфосфорилазы из H. chromatireducens AGD 8-3.
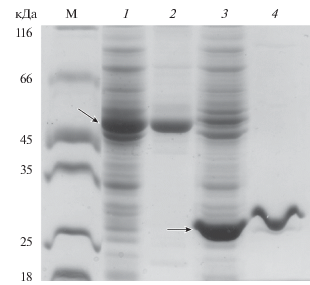
Экспрессионными векторами (pHrPNP и pHrTPP) трансформировали реципиентный штамм E. coli С600 Δudp и анализировали уровень накопления рекомбинантных белков. Полученные результаты (рис. 2) свидетельствуют о том, что рекомбинантные нуклеозидфосфорилазы накапливались в растворимой фракции клеток штаммов-продуцентов. Эти данные также указывают на то, что промотор-операторная область гена udp из E. coli способна осуществлять высокоэффективную транскрипцию гибридных генов (deoA и deoD из H. chromatireducens AGD 8-3) в условиях гетерологичной экспрессии.
Рис. 2.
Электрофоретический анализ рекомбинантных белков в клетках E. coli C600 в 12.5% ПААГ с ДСН-Na. М – маркеры молекулярной массы белков. 1, 3 – белки растворимых фракций, полученных после разрушения ультразвуком клеток-трансформантов E. coli C600. 2, 4 – очищенные рекомбинантные (тимидинфосфорилаза) (HrTPP) и (пуриннуклеозидфосфорилаза) (HrPNP) соответственно. Рекомбинантные белки обозначены стрелками.
Были также проведены отдельные эксперименты по клонированию полноразмерных генов deoA и deoD из H.chromatireducens AGD 8-3. Однако сконструированные экспрессионные векторы, в составе которых структурные части генов исследуемых нуклеозидфосфорилаз находились под контролем нативных промотор-операторных областей, практически не приводили к накоплению целевых ферментов в клетках E. coli (данные не приводятся). Этот факт следует отнести к существенному различию в функционировании промоторных областей в E. coli [39] и H. chromatireducens AGD 8-3, что может быть предметом отдельного исследования.
Рекомбинантные белки были выделены и очищены также, как в работе [36], с использованием ионообменной хроматографии (рис. 2), проведено изучение их ферментативных и физико-химических свойств. Первичную структуру ферментов подтверждали методом MALDI-TOF/TOF масс-спектрометрического анализа их триптических гидролизатов.
Нуклеозидфосфорилазы из различных организмов отличаются топологией третичной и четвертичной структур, а также специфичностью по отношению к субстратам (нуклеозидам), что позволило их разделить на два семейства: NP-I и NP-II. Семейство NP-I обладает широкой субстратной специфичностью по отношению к пурин- и пиримидиннуклеозидам, в то время как NP-II проявляют избирательную активность по отношению 2'-дезоксирибонуклеозидам (например, тимидинфосфорилаза) [34, 40–42].
Четвертичные формы новых рекомбинантных пурин- и пиримидиннуклеозидфосфорилаз из H.chromatireducens AGD 8-3 исследовали с применением аналитической гель-фильтрации на колонке Tricorn 10/300 c сорбентом Superdex 200. Полученные результаты показали, что рекомбинантные тимидин- и пуриннуклеозидфосфорилазы формируют димерную и гексамерную четвертичные структуры, соответственно (табл. 2.). Показано, что тимидинфосфорилаза из H. chromatireducens AGD 8-3 проявляла активность только по отношению к 2'-дезоксирибонуклеозидам, что позволяет отнести ее к NP-II семейству нуклеозидфосфорилаз.
Таблица 2.
Сравнительные характеристики тимидин- и пуриннуклеозидфосфорилаз из E. coli и H. chromatireducens AGD 8-3
| Фермент | Удельная активность, ед./мг |
Топт, оС | рНопт | Четвертичная структура | Ссылка |
|---|---|---|---|---|---|
| ETPP | 105.3 ± 5.1 | 51.3 ± 1.6 | 6.6–7.3 | Димер | Данная работа |
| HrTPP | 235.2 ± 7.6 | 60.4 ± 1.3 | 7.2–8.9 | Димер | Данная работа |
| EPNP | 66.0 | 60.0 | 7.1–7.5 | Гексамер | [18] |
| HrPNP | 28.6 ± 3.8 | 55.2 ± 1.7 | 7.0–9.1 | Гексамер | Данная работа |
Сравнительное исследование полученных рекомбинантных нуклеозидфосфорилаз из H. chromatireducens AGD 8-3 и E. coli показало (табл. 2), что тимидинфосфорилаза проявляла более высокую, по сравнению с ее аналогом из E. coli, ферментативную активность, в то время как пуриннуклеозидфосфорилаза уступала по этому показателю референс-белку. Обращает на себя внимание и широкий диапазон значений pH, при которых оба выделенных рекомбинантных белка сохраняли свою ферментативную активность (табл. 2). Этот факт, вероятно, является отражением структурных изменений этих белков, как ответ на условия обитания самого микроорганизма (рН 9.5–10.1).
Однако сравнительное исследование влияния концентрации NaCl на активность тимидинфосфорилаз из H. chromatireducens AGD 8-3 и E. coli показало неожиданный результат. Несмотря на адаптацию клеток H. chromatireducens AGD 8-3 к экстремальным солевым условиям обитания, толерантность HrTPP из этого организма к данному показателю оказалась значительно ниже по сравнению с аналогичным ферментом из E. coli (табл. 3). Инактивация HrTPP при 2.0 М концентрации NaCl достигала 50%, а тимидинфосфорилаза из E. coli сохраняла 88% своей активности.
Таблица 3.
Влияние концентрации NaCl на активность рекомбинантной тимидинфосфорилазы
| № | NaCl, M | Активность тимидинфосфорилазы,% |
|
|---|---|---|---|
| H. chromati- reducens AGD 8-3 |
E. coli | ||
| 1 | 0 | 100.0 | 100.0 |
| 2 | 0.5 | 96.2 ± 2.1 | 98.3 ± 1.6 |
| 3 | 1.0 | 82.6 ± 1.6 | 95.4 ± 1.9 |
| 4 | 1.5 | 66.9 ± 2.1 | 94.3 ± 1.1 |
| 5 | 2.0 | 48.9 ± 1.7 | 88.1 ± 1.6 |
| 6 | 2.5 | 35.1 ± 1.5 | 82.5 ± 1.7 |
Полученные результаты выявили необходимость дополнительного сравнительного анализа первичных структур тимидинфосфорилаз из E. coli и H. chromatireducens AGD 8-3 (рис. 3).
Рис. 3.
Сравнение первичных структур тимидинфосфорилазы (TPP) из E. coli (EC) и H. chromatireducens AGD 8-3 (HR). Выделение элементов вторичных структур белков: жирный шрифт – α-спиральные структуры, а жирный курсив – β‑структуры, согласно работе [43]. Подчеркнуты аминокислотные остатки, входящие в состав фермент-субстратного комплекса в тимидинфосфорилазе из E. coli [34].

Анализ проводили с использованием алгоритма, заложенного в работе [44], и определяющего вероятность замены аминокислотных остатков в составе родственных белков в ходе эволюции. Обращает на себя внимание тот факт, что все аминокислотные остатки, принимающие участие при формировании фермент-субстратного комплекса для этих двух белков, остаются неизменными (рис. 3), хотя уровень гомологии исследуемых белковых структур невысок и составляет только 58.9%. Кроме того, в составе тимидинфосфорилазы из H. chromatireducens AGD 8-3 выявлялся дополнительный гидрофильный участок полипептидной цепи, расположенный между двумя α-спиралями (251–256 и 270–287 аминокислотные остатки, нумерация по ферменту из E. сoli). Дальнейшее выяснение особенностей строения полученных ферментов, а также роли отмеченных различий в первичной структуре и возможного их влияния на ферментативные и физико-химические свойства полученных тимидин- и пуриннуклеозидфосфорилаз из H. chromatireducens AGD 8-3 в настоящее время проводится с использованием рентгеноструктурного анализа.
Таким образом, в результате проведенного исследования получены рекомбинантные штаммы новых нуклеозидфосфорилаз из экстремофильной бактерии H. chromatireducens AGD 8-3. Исследованы основные свойства этих белков и показано, что они могут быть использованы при ферментативном синтезе производных нуклеозидов, а также для дальнейшего изучения молекулярных основ функционирования этого класса ферментов.
При проведении исследований использовали оборудование Центра коллективного пользования “Промышленные биотехнологии” Федерального исследовательского центра “Фундаментальные основы биотехнологии” Российской академии наук.
Исследование выполнено при частичной поддержке РФФИ (Грант № 18-04-00784 А).
Список литературы
Pugmire M.J., Cook W.J., Jasanoff A., Walter M.R., Ealick S.E. // J. Mol. Biol. 1998. V. 281. № 2. P. 285–299.
Caradoc–Davies T.T., Cutfield S.M., Lamont I.L., Cutfield J.F. // J. Mol. Biol. 2004. V. 337. № 2. P. 337–354.
Safonova T.N., Mordkovich N.N., Veiko V.P., Okorokova N.A., Manuvera V.A., Dorovatovskii P.V., Popov V.O., Polyakov K.M. // Acta Crystallogr. D. Struct. Biol. 2016. V. 72. № 2. P. 203–210.
Mordkovich N.N., Safonova T.N., Antipov A.N., Manuvera V.A., Polyakov K.M., Okorokova N.A., Veiko V.P. // Appl. Biochem. Microbiol. 2018. V. 54. № 1. P. 12–20.
Константинова И.Д., Леонтьева Н.А., Галегов Г.А., Рыжова О.И., Чувиковский Д.В., Антонов К.В., Есипов Р.С., Таран С.А., Веревкина К.Н., Феофанов С.А., Мирошников А.И. // Биоорг. химия. 2004. Т. 30. № 6. С. 613–620.
Utagawa T. // J. Molec. Cat. B: Enzymatic. 1999. V. 6. P. 215–222.
Komatsu H., Araki T. // Nucleosides, Nucleotides & Nucleic Acids. 2005. V. 24. № 5–7. P. 1127–1130.
Roivainen J., Elizarova T., Lapinjoki S., Mikhailopulo I.A., Esipov R.S., Miroshnikov A.I. // Nucleosides, Nucleotides & Nucleic Acids. 2007. V. 26. № 8–9. P. 905–909.
Михайлопуло И.А., Мирошников А.И. // Acta Naturae. 2010. Т. 2. № 2. С. 38–61.
Mahmoud S., Hasabelnaby S., Hammad S.F., Sakr T.M. // J. Advanced Pharmacy Reseach. 2018. V. 2. № 2. P. 73–88.
Liekens S, De Clercq E, Neyts J. // Biochem. Pharmacol. 2001. V. 61. № 3. P. 253–270.
Carmeliet P. // Nature. 2005. V. 438. № 7070. P. 932–936.
Furukawa T., Tabata S., Yamamoto M., Kawahara K., Shinsato Y., Minami K., Shimokawa M., Akiyama S. // Pharmacological Research. 2018. V. 132. P. 15–20.
Ashour O.M., Naguib F.N.M., Khalifa M.M.A., Abdel-Raheem M.H., Panzica R.P., el Kouni M.H. // Cancer Res. 1995. V. 55. № 5. P. 1092–1098
Iigo M., Nishikata K., Nakajima Y., Szinai I., Veres Z., Szabolcs A., De Clercq E. // Biochem. Pharmacol. 1990. V. 39. № 7. P. 1247–1253
Погосян Л.Г., Акопян Ж.И. // Биомедицинская химия. 2013. Т. 59. № 5. С. 483–497.
Погосян Л.Г., Нерсесова Л.С., Газарянц М.Г., Мкртчян З.С., Меликсетян Г.О., Акопян Ж.И. // Укр. биохи-м. журн. 2008. Т. 80. № 5. С. 95–104.
Jensen K.F., Nygaard P. // Eur. J. Biochem. 1975. V. 51. № 1. P. 253–265.
Xie X., Xia J. He K., Lu L., Xu Q., Chen N. // Biotechnol. Lett. 2011. V. 33. № 6. P. 1107–1112.
Visser D.F., Hennessy F., Rashamuse K., Louw M.E., Brady D. // Extremophiles. 2010. V. 14. № 2. P. 185–192.
Xie X., Huo W., Xia J., Xu Q., Chen N. // Enzyme Micr-ob. Technol. 2012. V. 51. № 1. P. 59–65.
Liu K., Zhou Y., Zhang J., Chu J., Zhang Y., He B. // Biotechnol Lett. 2017. V. 39. № 12. P. 1903–1910.
Zhou X., Szeker K., Janocha B., Böhme T., Albrecht D., Mikhailopulo I.A., Neubauer P. // FEBS J. 2013. V. 280. № 6. P. 1475–1490.
Almendros M., Gago J.V., Carlos J.B. // Molecules. 2009. V. 14. № 3. P. 1279–1287.
Szeker K., Zhou X., Schwab T., Casanueva A., Cowan D., Mikhailopulo I.A., Neubauer P. // J. Mol. Catal. B. Enzy-m. 2012. V. 84. P. 27–34.
Sambrook J., Fritsch E.F., Maniatis T. Molecular Cloning: a Laboratory Manual. N.Y.: Cold Spring Harbor Laboratory Press, 1989. 545 p.
Вейко В.П., Сипрашвили З.З., Ратманова К.И., Гулько Л.Б., Андрюхина Р.В., Дебабов В.Г. // Биотехнология. 1994. № 4. С. 2–4.
Вейко В.П., Сипрашвили З.З., Ратманова К.И., Гулько Л.Б., Миронов А.А., Андрюхина Р.В., Дебабов В.Г. // Докл. РАН. 1994. Т. 339. № 6. С. 819–821
Вейко В.П., Осипов А.С., Шехтер И.И., Букленков М.Т., Ратманова К.И., Гулько Л.Б., Чибискова Н.А., Эррайс Л.Л., Деревщикова Е.Б., Дебабов В.Г. // Биоорг. химия. 1995. Т. 21. № 5. С. 354–358.
Шаповалова А.А, Хижняк Т.В., Турова Т.П., Сорокин Д.Ю. // Микробиология. 2009. Т. 78. № 1. С. 117–127.
Вейко В.П., Ратманова К.И., Осипов А.С., Буленков М.Т., Пугачев В.В. // Биоорг. химия. 1991. Т. 17. № 5. С. 685–689.
Bradford M.M. // Anal. Biochem. 1976. V. 2. P. 248–254.
Laemmli U.K. // Nature. 1970. V. 227. № 5259. P. 680–685.
Панова Н.Г., Алексеев К.С., Кузьмичев А.С., Щевелёва Е.В., Гаврюшов С.А., Поляков К.М., Крицын А.М., Михайлов С.Н., Есипов Р.С., Мирошников А.И. // Биохимия. 2007. Т. 72. № 1. С. 27 – 35.
Xie X., Xia J., He K., Lu L., Xu Q., Chen N. // Biotechnol. Lett. 2011. V. 33. № 6. P. 1107–1112.
Мордкович Н.Н., Манувера В.А., Вейко В.П., Дебабов В.Г. // Биотехнология. 2012. № 1. С. 21–30.
Shapovalova A.A., Khijniak T.V., Tourova T.P., G. Muyzer G., Sorokin D.Y. // Extremophiles. 2008. V. 12. № 5. P. 619–625.
Sharko F.S., Shapovalova A.A., Tsygankova S.V., Komova A.V., Boulygina E.S., Teslyuk A.B., Gotovtsev P.M., Namsaraev Z.B., Khijniak T.V., Nedoluzhko A.V., Vasilov R.G. // Genome Announcements. 2016. V. 4. № 2. P. 1–2.
Овчарова И.В., Еремина С.Ю., Миронов А.С. // Генетика. 2004. Т. 40. № 1. С. 15–25.
Pugmire M.J., Ealick S.E. // Biochem J. 2002. V. 361. № 1. P. 1–25.
Mushegian A.R., Koonin E.V. // Protein Sci. 1994. V. 3. № 7. P. 1081–1088.
Mahor D., Priyanka A., Prasad G.S., Thakur K.G. // PLOS One. 2016. V. 11. № 10. e0164279.
Walter M.R., Cook W.J., Cole L.B., Short S.A., Koszalka G.W., Krenitsky T.A., Ealick S.E. // J. Biol. Chem. 1990. V. 265. № 23. P. 14016–14022.
Dayhoff, M.O. Atlas of Protein Sequence and Structure: National Biomedical Research Foundation. / Ed. M.O. Dayhoff. MD.: Silver Spring, 1972.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология