

Прикладная биохимия и микробиология, 2020, T. 56, № 1, стр. 52-59
Скрининг условий рефолдинга и получение рекомбинантного антистафилококкового эндолизина LysKCA в активной форме из телец включения E. coli
А. В. Жидецкий 1, *, С. Г. Голенченко 1, В. А. Прокулевич 1, М. В. Шолух 1
1 Белорусский государственный университет, биологический факультет
220030 Минск, Беларусь
* E-mail: Zhydzetski@gmail.com
Поступила в редакцию 17.04.2019
После доработки 29.07.2019
Принята к публикации 30.08.2019
Аннотация
Предложен пошаговый скрининг основных характеристик рефолдинг-буфера, с использованием которого из телец включения E. coli получен в активной форме рекомбинантный антистафилококковый эндолизин LysK, содержащий два каталитических домена – CHAP и амидазу-2. Определены оптимальные значения рН, температуры, окислительно-восстановительного потенциала рефолдинг-буфера и установлены оптимальные конечные концентрации белка, мочевины и тип антиагрегационного соединения. Выяснен состав системы ренатурации антистафилококкового эндолизина, который при 10°C и разведении целевого белка до конечной концентрации 150 мкг/мл содержал 20 мМ натрий-фосфатного буфера, рН 7.4, содержащего 2.5 мМ ДТТ и 0.4 М сахарозы. Выход рефолдинга после масштабирования составил 29.5 ± 6.7%, что позволило получить 16 ± 2.3 мг целевого белка из 2.25 г отмытых телец включения c ферментативной активностью 1.8 ± 0.73 × 103 МЕ/мг.
Технология рекомбинантных молекул ДНК открыла новую эру получения различного рода белков как для структурных и биохимических исследований, так и для индустриальных и коммерческих целей [1]. Экспрессия в трансформированных клетках позволяет получить редкие, ценные, дорогостоящие и трудно получаемые традиционными методами белки [2, 3]. В частности, с помощью данного метода можно получить ряд терапевтических белков, которые обладают большим потенциалом для применения в медицине и ветеринарии, и которые приходят на замену стандартным методам лечения [4, 5].
Одним из таких белков является эндолизин фага К (LysK) – пептидогликангидролаза, которая участвует в лизисе грамм-положительных бактериальных клеток рода Staphylococcus и экспрессируется на терминальном цикле выхода зрелых фаговых частиц из клеток-хозяев [6–8]. Эндолизин LysK состоит из трех доменов – двух каталитических (CHAP и амидаза-2), и одного структурного (SH3b), причем активностью обладают и укороченные варианты данного белка, содержащие CHAP-домен [9, 10]. Высокая антистафилококковая эффективность LysK и его производных при действии на клетки рода Staphylococcus делает его перспективным кандидатом на роль нового антимикробного агента в медицине и ветеринарии, особенно по отношению к штаммам, устойчивым к антибиотикам. Следует отметить, что данный белок обладает рядом преимуществ перед стандартными антистафилококковыми препаратами, главными из которых являются отсутствие развития к нему резистентности, проявление высокой видовой специфичности, отсутствие воздействия на нормальную микрофлору, а также возможность безопасного применения в пищевой индустрии [8, 11].
Вместе с тем определенные затруднения в изучении и дальнейшем применении LysK и его форм связаны с их выделением в ограниченных количествах. Работы, посвященные данному белку, основаны в большинстве случаев на его получении в штаммах-продуцентах в растворимой форме с дальнейшей очисткой на аффинных сорбентах [7, 8, 12, 13]. Однако при использовании такой технологии удается получить небольшое количество белка для исследовательских целей. В частности, в недавних работах удалось получить лишь 2–10 мг CHAPK и около 12 мг LysKCA с 1 л ферментируемой культуры [13–15]. В то же время потенциальное применение в ветеринарии, медицине и других отраслях народного хозяйства будет требовать больших количеств и соответственно более дешевых и эффективных методов получения данного белка [11].
Проблему достаточного количества целевого белка можно решить путем его получения из телец включения (ТВ) − плотных, сильно гидратированных образований, состоящих по большей части из неактивных агрегированных молекул целевого белка [16]. Хотя в данном случае возникает ряд дополнительных трудностей, главной из которых является активация молекул целевого белка, связанная с проведением процесса рефолдинга. Эффективность ренатурации рекомбинантных белков зависит от конкуренции между правильным фолдингом и агрегацией [17, 18]. Какой процесс будет доминировать, определяется составом и свойствами рефолдинг-буфера, правильный подбор которого является ключевым фактором в достижении эффективной ренатурации. К физическим параметрам буфера относятся рН, ионная сила, температура, окислительно-восстановительный потенциал среды и время, требуемое для полного принятия молекулой белка нативной конформации [18]. Помимо этого существенный эффект на процесс ренатурации оказывают конечная концентрация белка и денатуранта, а также его химический состав – присутствие антиагрегационных соединений [19–21].
Цель работы – разработать методический подход на основе скрининга условий рефолдинга LysKCA из ТВ Eschirichia coli, позволяющий получить данный белок в активной форме.
МЕТОДИКА
В работе использовали ТВ, содержащие рекомбинантный эндолизин К, состоявший из CHAP и амидазного домена (LysKCA), выделенные из клеток E. coli BL21-CodonPlus (DE3)-RIPL pET24b LysK528+ по методике, описанной ранее [22].
Получение LysKCA для скрининга условий рефолдинга. Полученные ТВ (1 г) отмывали в 20 мл в раствора 50 мМ трис-буфера, содержащего 50 мМ NaCl, до полного выхода примесных соединений. Отмытые ТВ солюбилизировали в растворе 50 мМ трис-HCl буфера, рН 10, содержащего 6 М мочевину и 10 мМ β-меркаптоэтанол, при постоянном перемешивании на магнитной мешалке в течение 2–3 ч. Для удаления не растворившихся частиц полученный солюбилизат центрифугировали при 8000 g в течение 30 мин при 20°С. Осветленный солюбилизат наносили на колонку с DE-сефарозой (35 мл) (“GE Healthcare”, Швеция). Не связавшуюся фракцию (проскок), содержащую целевой белок, наносили на колонку с SP-сефарозой (35 мл). Для уравновешивания и промывки колонок использовали солюбилизирующий буфер. Изократическую элюцию с SP-сефарозы проводили тем же буфером, содержащим 1.0 М NaCl. Полученный элюат использовали для скрининга условий ренатурации.
Финальную очистку и сбор целевого белка после масштабирования рефолдинга проводили на колонке с SP-сефарозой (35 мл) (“GE Healthcare”, Швеция). Для этого ее уравновешивали буфером А1 (20 мМ натрий-фосфатный буфер, рН 7.4, NaФБ) и промывали тем же буфером после нанесения белка. Примесные белки удаляли буфером Б1, который содержал 20 мМ NaФБ, рН 7.4, с 0.27 М NaCl. Целевой продукт элюировали градиентом 0 → 100% буферов Б1 → Б2 (объемом 350 мл), Б2 состоял из буфера А1 с 1.0 М NaCl. Фракции, содержащие целевой белок, объединяли, фильтровали через фильтр (0.2 мкм) и хранили при 4°С. Рассчитывали выход и специфическую ферментативную активность полученного препарата белка.
Скрининг условий рефолдинга методом разведения. Для определения условий рефолдинга LysKCA использовали фракцию, элюированную с SP-сефарозы, содержавшую 1.8 мг белка/мл. Полученную фракцию разводили в 12 раз до конечной концентрации белка 150 мкг/мл в различных рефолдинг системах. Конечный объем этих систем − 3 мл.
На первом этапе ренатурации применяли при 10 или 20°С системы с различными значениями рН, включая рН 5.0 и 6.0 (20 мМ MES) и 7.0–9.0 (20 мМ трис-HCl), содержащие 0.4 M сахарозы, 4 мМ L-цистеина и 0.4 мМ L-цистина.
Затем оптимизировали окислительно-восстановительный потенциал ренатурирующего буфера. Сравнительную эффективность восстанавливающих агентов определяли в 20 мМ NaФБ, pH 7.4, содержащего 0.4 М сахарозу и дитиотреитол (ДТТ) с различной концентрацией (0.1, 1, и 5 мМ) или пары L-цистеин/L-цистин в соотношении 0.1/0.1, 1/0.1, 5/0.1, 0.1/1, 1/1, 5/1, 0.1/5, 1/5, и 5/5 мМ.
Для установления оптимальной конечной концентрации белка и мочевины проводили рефолдинг в 20 мМ NaФБ, pH 7.4, содержащем 0.4 М сахарозу, 2.5 мМ ДТТ и белок в конечной концентрации 150, 200, 300 и 600 мкг/мл. Концентрация мочевины при этом составляла 0.5, 0.67, 1.0 и 2.0 М соответственно.
На заключительном этапе был проведен скрининг соединений, обладающих антиагрегационной активностью. Протестированы: 0.1, 0.5, и 1.0%-ные растворы Kolliphor EL, 0.1, 0.5 и 1%-ный полиэтиленгликоль 3000 (ПЭГ с Mr 3000), 0.1, 0.2, и 0.4 М глицерол, 0.1, 0.2, и 0.4 М сахароза. Конечная концентрация белка – 150 мкг/мл. Все соединения готовили на основе 20 мМ NaФБ, pH 7.4, с 2.5 мМ ДТТ.
Лучшей системой на каждом этапе рефолдинга считалась та, в которой после 12–14 ч инициации ренатурации детектировалась наиболее высокая ферментативная активность и мономерная форма целевого белка, а также наблюдалась наименьшая степень агрегации.
Масштабирование процесса ренатурации. Определив оптимальный состав и параметры рефолдинг-буфера – 20 мМ NaФБ, pH 7.4, содержащий 0.4 М сахарозы и 2.5 мМ ДTT, при 10°C было проведено масштабирование процесса ренатурации в 100 раз. Для этого 25 мл фракции, полученной после элюции с SP-сефарозы, как описано выше, разводили в 12 раз в этом рефолдинг буфере в один этап при постоянном перемешивании на магнитной мешалке до конечной концентрации белка 150 мкг/мл. Сосуд с разведенным белком переносили в холодильник на 18 ч. Окончательную очистку и сбор целевого белка проводили, как описано выше для очистки на SP-сефарозе, после чего рассчитывали выход и активность полученного препарата белка.
Измерение специфической активности. Антистафилококковую активность измеряли по модифицированному методу, описанному в работе [9]. Его принцип заключался в регистрации снижения оптической плотности при 600 нм стационарной культуры Staphilococcus aureus subsp. aureus Rosenbach (ATCC 25923) после лизиса бактериальных клеток при добавлении фракции, содержащей ренатурированный LysKCA. За единицу активности (МЕ) принимали такое количество белка, которое приводило к снижению оптической плотности культуры S. aureus при 600 нм с 0.4 до 0.2 ед. за 15 мин после добавления исследуемого белка. Измерение проводили на термостатируемом планшетном ридере SpectraMax M2 (“Molecular Devices”, США).
ДДС-электрофорез в полиакриламидном геле. Чистоту белковых фракций и содержание мономерной формы рекомбинантного LysKCA после ренатурации оценивали методом электрофореза с Na-ДДС в 12.5%-ном полиакриламидном геле (ПААГ) на приборе Mini-PROTEAN II (“BioRad”, США) [23]. Окраску гелей проводили красителем кумасси ярко-синим R-250 (CBB R-250) или серебром по методу, описанному в работе [24].
УФ-видимая спектроскопия. Для определения концентрации общего белка использовали модифицированный метод Лоури [25]. Агрегацию во время подбора условий рефолдинга регистрировали при 340 нм через 30 мин и 14 ч после инициации ренатурации на спектрофотометре Cary-50 Bio (“Varian”, Австралия). Концентрацию очищенного препарата эндолизина К определяли, используя коэффициент молярной экстинкции LysKCA ε280 = 89395 M–1 cm–1 [26].
Обработка и визуализация результатов. Обработка и графическое отображение результатов выполнено по программе SigmaPlot 13.1, определение чистоты (денситометрический анализ) целевого белка – по TotalLab 2.0, построение 3D-модели LysKCA осуществлено с применением онлайн платформы Phyre2, а визуализация полученной модели выполнена по программе Chimera 1.8.1.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
К настоящему времени описано большое количество методов получения рекомбинантных белков из ТВ. Но, несмотря на общность алгоритмов получения таких белков в активной форме, которые включают этапы солюбилизации ТВ, очистки целевого белка от примесных соединений и его рефолдинг, на данный момент не существует универсального. Это в первую очередь связано с индивидуальными характеристиками каждого отдельного белка [16, 21]. Как уже отмечалось, этап ренатурации является лимитирующим и определяет эффективность выхода целевого продукта. Он зависит, как от физико-химических условий среды (рефолдинг буфера), так и от конечной концентрация белка [16–19].
Параметры рефолдинг буфера (рН, температура, редокс потенциал, конечная концентрация белка и химический состав среды) были протестированы в данной работе на этапе определения условий ренатурации LysKCA методом разведения.
На первом этапе изучали зависимость рефолдинга LysKCA от рН (5–10) и температуры (10 и 20°C) ренатурирующего буфера, который содержал 0.4 М сахарозу в качестве стабилизатора и редокс систему цистеин/цистин (4 мМ /0.4 мМ). Установлено, что при повышенной температуре (20°C) и приближении значения рН к pI целевого белка (9.58) возрастала его агрегация в этих системах, хотя специфическая активность была обнаружена во всех исследуемых растворах. При 20°C в системах с рН от 7 до 9 шло формирование молекулярных изоформ целевого белка, которые были видны на электрофореграмме в виде двойных полос, чего не наблюдалось при 10°C (результаты не представлены).
Можно предположить, что увеличение степени агрегации при повышенных значениях рН связано, во-первых, с повышенной реакционной способностью цистеинов и образованием межмолекулярных и некорректных внутримолекулярных дисульфидных связей. А, во-вторых, щелочные значения рН среды, близкие к pI молекулы LysKCA, могли приводить к элиминации электростатического отталкивания промежуточных форм молекулы, что вызывало их взаимодействие, и как следствие, дальнейшую агрегацию [4, 20].
На следующем этапе работы была подобрана оптимальная редокс система для рефолдинга L-ysKCA. Для этого изучена зависимость активности и агрегации целевого продукта от концентрации и соотношения цистина, цистеина и дитиотреитола в ренатурирующем буфере (рис. 1). Установлено, что литическая активность LysKCA восстанавливалась, достигала максимально возможных значений и оставалась неизменной вплоть до соотношения цистеин : цистин 5 : 1. Увеличение концентрации L-цистина до 5 мМ приводило к снижению активности фермента в 4 раза (рис. 1a). Это, вероятно, было обусловлено окислением SH-групп цистеина и образованием не нужной дисульфидной связи в этой среде, в том числе возможно и межмолекулярных, так как степень агрегации после 1 ч ренатурации в данных условиях оказывалась выше, чем в среде с более низкой концентрацией цистина.
Рис. 1.
Влияние окислительно-восстановительного потенциала рефолдинг буфера на эффективность ренатурации рекомбинантного LysKCA, активность LysKCA (III) и степень агрегации образцов через 1(I) и 14 (II) ч (а), и электрофореза в 12.5%-ном ПААГ в не редуцирующих условиях (б) в присутствии: 1 – буфер без низкомолекулярных тиолов, 2 – 0.1 мМ ДДТ, 3 – 1 мМ ДДТ, 4 – 5 мМ ДДТ, 5 – 0.1 мМ L-цистеина и L-цистина, 6 – 1 мМ L-цистеина и 0.1 мМ L-цистина, 7 – 5 мМ L-цистина и 0.1 мМ L-цистина, 8 – 0.1 мМ L-цистеина и 1 мМ L-цистина, 9 – 1 мМ L-цистеина и L-цистина, 10 – 5 мМ L-цистеина и 1 мМ L-цистина, 11 – 0.1 мМ L-цистеина и 5 мМ L-цистина, 12 – 1 мМ L-цистеина и 5 мМ L-цистина и 13 – 5 мМ L-цистеина и 5 мМ L-цистина; M – белки-маркеры молекулярных масс (250, 130, 100, 70, 55, 35, 25, 15 и 10 кДа); окраска CBB R-250. Все системы готовили на основе 20 мМ Na-ФБ, рН 7.4, с 0.4 М сахарозой.
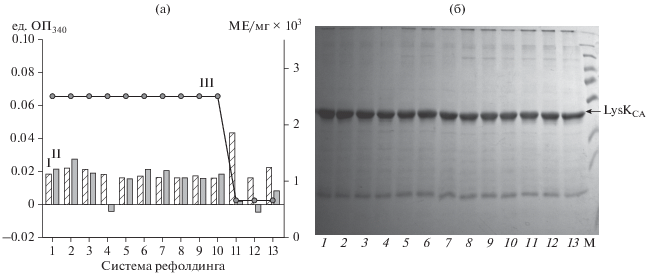
Было выбрано 2.5 мМ ДТТ, которое составило промежуточное значение между концентрациями 1 и 5 мМ, поскольку в этих условия ренатурация целевого белка проходила с образованием мономерной и активной формы фермента, а также сопровождалась низкой степенью агрегации. Это обусловлено тем, что использованное соединение позволяло поддержать в восстановленном состоянии три сульфгидрильные группы остатков цистеина в структуре белка (рис. 2а).
Рис. 2.
Трехмерная структура эндолизина LysKCA, включающая элементы вторичной структуры с тремя остатками цистеина (белые стрелки, а), остатки цистеина не присутствовали на поверхности молекулы (б), располагались в углублениях − карманах активных центров (черные стрелки, в и г).
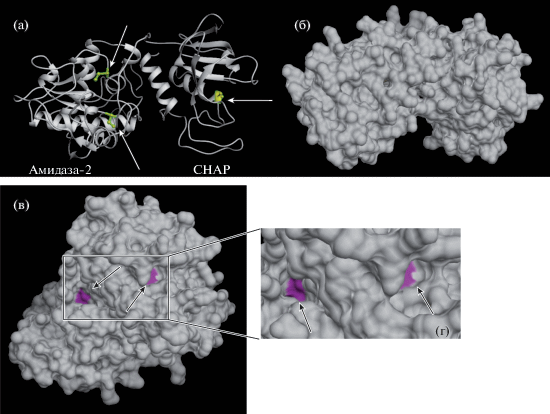
Анализ аминокислотной последовательности показал, что в структуре LysKCA имелось три аминокислотных остатка цистеина. Моделирование пространственной структуры на базе сервиса Phyre2, которое выявило 99 и 100%-ную гомологию CHAP и амидаза-2 домена, соответственно, с соответствующими доменами эндолизина LysGH15, показало, что все три остатка находятся в свободном состоянии (рис. 2а). Однако они не участвовали в формировании поверхности молекулы, а располагались в углублениях активных центров доменов (рис. 2б, 2в). Одна сульфгидрильная группа находилась в каталитическом центре CHAP-домена [27]. Две другие SH-группы, как и в родственном эндолизине LysGH15, располагались в амидазном домене, одна из которых также в активном центре [28, 29]. Таким образом, в активном состоянии LysKCA должен содержать все три остатка Цис в восстановленном состоянии.
В связи с этим можно объяснить наличие активности данного белка в среде с ДТТ и цистеином, присутствующих в рефолдинг буфере. Они обеспечивали поддержание свободных сульфгидрильных групп в восстановленном состоянии именно во время ренатурации, так как данные группы не участвовали как в формировании поверхности молекулы, так и соответственно в каких либо межмолекулярных взаимодействиях после принятия нативной конформации (рис. 2б). Об этом также свидетельствовало отсутствие влияния низкомолекулярных тиолов (ДТТ и β-меркаптоэтанола) на структуру и активность полноразмерной нативной молекулы LysK, что было установлено ранее и для LysKCA [30].
В то же время противоположный эффект наблюдался при наличии в среде высоких концентраций окисленного тиолата (рис. 1а). Так, при содержании в буфере 5 мМ цистина ферментативная активность LysKCA практически полностью отсутствовала. Можно предположить, что при окислении SH-групп во время рефолдинга формировалась одна из трех возможных дисульфидных связей, образуя неправильную конформацию белка, или формировался смешанный дисульфид с цистином сульфгидрильной группы в каталитическом центре CHAP домена уже после ренатурации, поскольку именно этот домен и этот аминокислотный остаток из триады Цис-Гис-Глу в активном центре ответственны за проявление специфической активности [8, 9, 27].
После установления физических параметров ренатурирующего буфера была изучена зависимость эффективности рефолдинга от конечной концентрации целевого продукта и мочевины. Наилучший эффект был обнаружен при разведении исходного белка до его конечной концентрации в рефолдинг буфере 0.15 и 0.2 мг/мл, а мочевины 0.5 и 0.67 М соответственно. Такая же активность (2.5 × 103 МЕ/мг) наблюдалась при тех же разведениях, но с конечной концентрацией мочевины 2 М, хотя на основании турбидиметрического исследования это хаотропное соединение снижало степень агрегации (результаты не представлены). При разведении белка до конечной концентрации 0.3 мг/мл, а мочевины до 1 М активность LysKCA падала в 4 раза, а при конечной концентрации белка 0.6 мг/мл и мочевины 2 М активность полностью исчезала. Это связано с тем, что при повышении в растворе концентрации белка возрастала и степень неправильного фолдинга и агрегации, напрямую зависящие от нее. Образование нативной конформации протекало в соответствии с кинетикой реакций первого порядка, в то время как агрегация должна подчиняться процессам второго (или высших) порядков, соответственно скорость агрегации выше скорости ренатурации при высоких исходных концентрациях денатурированного белка в растворе [16, 19].
В результате было выбрано разведение в 12 раз с конечной концентрацией белка 0.15 мг/мл, при котором наблюдалась наименьшая степень агрегации, а LysKCA находился в мономерной и активной форме.
Как уже было отмечено, остатки цистеина не участвовали в формировании поверхности, что могло бы стать причиной неспецифического взаимодействия между молекулами белка и дальнейшей агрегации. В переходном состоянии “расплавленной” глобулы эти остатки поддерживались в восстановленном состоянии присутствием ДТТ или L-цистеина. Можно предположить, что за агрегацию отвечали неспецифические гидрофобные взаимодействия переходных форм молекулы LysKCA, особенно при его высоких концентрациях. В связи с этим при рефолдинге методом разведения часто применяют добавки различной химической природы, обладающие антиагрегационным действием и ингибирующие взаимодействие переходных состояний между собой [19, 21].
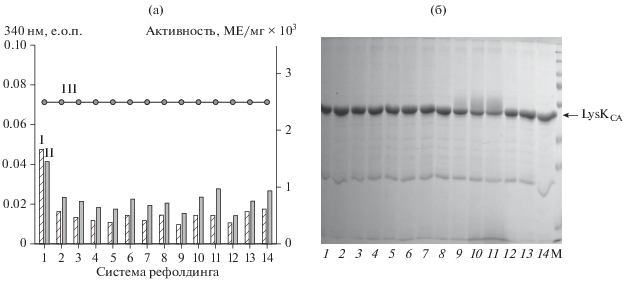
На наличие такого эффекта на заключительном этапе данной работы были протестированы сахароза и глицерол, мочевина, Kolliphor EL и ПЭГ 3000. Как видно из полученных результатов, сравнение с системой без добавок показало, что все соединения проявили примерно одинаковый антиагрегационный эффект (рис. 3а). Однако из результатов по стабилизации переходных состояний молекулы LysKCA, и антиагрегационного действия изученных соединений, видно, что полного подавления агрегации не наблюдалось. Практически для всех соединений удалось добиться снижения мутности растворов в 2 раза по сравнению с контролем без добавок. Это значение светорассеяния в скрининг растворах наблюдалось и на первых его трех этапах, так как первоначально во всех системах присутствовала сахароза. Этот эффект наблюдали и в присутствии 2 М мочевины на этапе определения ее конечной концентрации. В итоге в качестве антиагреганта была выбрана 0.4 М сахароза, как одно из наиболее эффективных, доступных и дешевых соединений.
Рис. 3.
Сравнительная эффективность действия антиагрегационных соединений на рефолдинг рекомбинантного Lys-KCA (III) по результатам турбидиметрического анализа (а) активность (III) и степень агрегации образцов через 1(II) и 14 (II) ч) и электрофореза в 12.5%-ном ПААГ в не редуцирующих условиях (б) в присутствии: 1 – буфера без добавок, 2 – 1.5 М мочевины, 3 – 0.1 М сахарозы, 4 – 0.2 М сахарозы, 5 – 0.4 М сахарозы, 6 – 0.1 М глицерола, 7 – 0.2 М глицерола, 8 – 0.4 М глицерола, 9 – 0.1% Kolliphor EL, 10 – 0.5% Kolliphor EL, 11 – 1.0% Kolliphor EL, 12 – 0.1% ПЭГ 3000, 13 – 0.5% ПЭГ 3000, 14 – 1% ПЭГ 3000; M – белки-маркеры молекулярных масс; окраска CBB R-250; все системы содержали 20 мМ NaФБ, рН 7.4, содержащем 2.5 мМ ДТТ.
Условия рефолдинга, определенные на этапе скрининга для каждого белка, были отправной точкой при масштабировании процесса ренатурации. Однако увеличение масштабов данного процесса могло привести к нежелательным последствиям, связанным с увеличением агрегации. Это могло быть вызвано увеличением общего количества внесенного белка, а также повышенной локальной его концентрацией и денатуранта, что возникало вследствие сложности перемешивания больших объемов, и приводило к увеличению вероятности встречи несвернутых или переходных состояний молекул белка друг с другом [16, 20]. Стократное масштабирование рефолдинга методом разведения в выбранном рефолдинг буфере увеличивало степень агрегации и в данном случае.
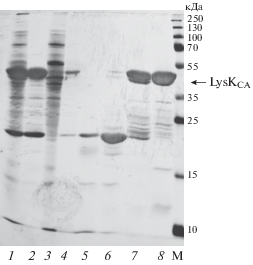
Тем не менее после масштабирования процесса ренатурации и сбора целевого белка на SP-сефарозе выход рефолдинга целевого белка составил 29.5 ± 6.7% с чистотой целевого продукта около 75%, рассчитанной на основании анализа результатов ЭФ (рис. 4). На рис. 4 видна гетерогенность LysKCA на всех этапах его получения – от солюбилизации ТВ до сбора целевого продукта после ренатурации. Введение дополнительного этапа очистки на катионообменнике позволило получить LysKCA с чистотой около 90% в конечном препарате белка, обладающего ферментативной активностью 1.8 ± 0.73 × 103 МЕ/мг, что почти в три раза выше активности, проявляемой полноразмерной молекулой LysK (0.621 × 103 МЕ/мг) [9, 13].
Рис. 4.
Электрофорез в 12.5%-ном ПААГ с Na-ДДС продуктов, получаемых на различных этапах масштабирования очистки и рефолдинга методом разведения рекомбинантного LysKCA: 1 – солюбилизат ТВ, 2 – свободный объем с DE-сефарозы, 3 – элюция с DE-сефарозы, 4 – фракция в рефолдинг буфере, 5 – фракция 1, элюирующаяся буфером Б1 с SP-сефарозы, 6 – фракция 2, элюирующаяся буфером Б1 с SP-сефарозы, 7 – фракция 1, элюирующаяся буфером Б2 с SP-сефарозы, 8 – фракция 2, элюирующаяся буфером Б2 с SP-сефарозы; M – белки-маркеры молекулярных масс (окраска серебром).
Можно заключить, что наибольший эффект на ренатурацию данного белка оказывали физические параметры буфера, правильно подобрав которые можно добиться увеличения выхода ренатурированного белка, если такой скрининг не проводить. Следует добавить, что при подборе условий рефолдинга может быть весьма полезной информация анализа третичной структуры белка, если не известно количество дисульфидных связей при наличии в его молекуле остатков цистеина. Зная их расположение, можно определить тип окислительно-восстановительного соединения.
Масштабирование процесса рефолдинга позволило получить рекомбинантный LysKCA с выходом 29.5 ± 6.7% и активностью 1.8 ± 0.73 × 103 МЕ/мг. В перерасчете на массу ТВ и объем культуры можно из 2.25 г отмытых ТВ (0.16 л культуры) выделить около 16 ± 2.3 мг целевого белка, то есть около 100 мг с 1 л ферментируемой культуры.
Список литературы
Rosano G.L., Ceccarelli E.A. // Front. Microbiol. 2014. V. 5. P. 172.
Palomares L.A., Kuri–Breña F., Ramírez O.T. // Encyclopedia of Life Support Systems. 2002. V. 6. № 3. P. 8.
Birch J.R., Onakunle Y. Therapeutic Proteins. Totowa, N.J.: Humana Press, 2005. P. 1–16.
Li Y. // Protein Expr. Purif. 2011. V. 80. № 2. P. 260–267.
Czaplewski L., Bax R., Clokie M., Dawson M., Fairhead H., Fischetti V.A., Henderson I.R. // Lancet Infect. Dis. 2016. V. 16. № 2. P. 239–251.
O’flaherty S., Coffey A., Meaney W., Fitzgerald G.F., Ross R.P. // J. Bacteriol. 2005. V. 187. № 20. P. 7161–7164.
Becker S.C., Foster–Frey J., Donovan D.M. // FEMS Microbiol. Lett. 2008. V. 287. №. 2. P. 185–191.
Becker S.C., Dong S., Baker J.R., Foster–Frey J., Pritchard D.G., Donovan D.M. // FEMS Microbiol. Lett. 2009. V. 294. № 1. P. 52–60.
Horgan M., O’Flynn G., Garry J., Cooney J., Coffey A., Fitzgerald G.F., McAuliffe O. // Appl. Environ. Microbiol. 2009. V. 75. № 3. P. 872–874.
Fenton M., Casey P.G., Hill C., Gahan C.G., McAuliffe O., O’Mahony J., Coffey A. // Bioengineered Bugs. 2010. V. 1. № 6. P. 404–407.
Schmelcher M., Loessner M.J. // Curr. Op. Biotechnol. 2016. V. 37. P. 76–87.
Schmelcher M., Shen Y., Nelson D.C. Eugster M.R., Eichenseher F., Hanke D.C., Becker S.C. // J. Antimicrob. Chemother. 2015. V. 70. № 5. P. 1453–1465.
Haddad Kashani H., Fahimi H., Dasteh Goli Y., Moniri R. // Front. Cell. Infect. Microbiol. 2017. V. 7. P. 290–301.
Fenton M., Ross R.P., McAuliffe O., O’Mahony J., Coffey A. // Appl. Microbiol. 2011. V. 111. № 4. P. 1025–1035.
Keary R., Sanz–Gaitero M., J. van Raaij M., Mahony J., Fenton M., McAuliffe O., Coffey A. // Curr. Prot. Pept. Sci. 2016. V. 17. № 2. P. 183–190.
Jungbauer A., Kaar W. // J. Biotechnol. 2007. V. 128. № 3. P. 587–596.
Lilie H., Schwarz E., Rudolph R. // Curr. Op. Biotechnol. 1998. V. 9. № 5. P. 497–501.
Shin H. C. // Biotechnol. Bioprocess Eng. 2001. V. 6. № 4. P. 237–243.
Rudolph R., Lilie H. // FASEB J. 1996. V. 10. № 1. P. 49–56.
Eiberle M.K., Jungbauer A. // Biotech. J. 2010. V. 5. № 6. P. 547–559.
Qoronfleh M.W., Hesterberg L.K., Seefeldt M.B. // Protein Expr. Purif. 2007. V. 55. № 2. P. 209–224.
Жидецкий А.В., Шолух М.В., Ханнеман Ю.Р., Хандрик Р., Мотылевич Ж.В., Химанен Ю.П. // Журн. Белорус. гос. ун-та. Биология. 2017. № 2. С. 58–65.
Laemmli U.K. // Nature. 1970. V. 227. № 5259. P. 680–685.
Fairbanks G., Steck T.L., Wallach D.F.H. // Biochemistry. 1971. V. 10. № 13. P. 2606–2617.
Peterson G.L. // Method. Enzymol. 1983. V. 91. P. 95–119.
Pace N., Vajdos F., Fee L., Grimsley G., Gray T. // Protein Sci. 1995. V. 4. № 11. P. 2411–2423.
Sanz–Gaitero M., Keary R., Garcia–Doval C., Coffey A., van Raaij M.J. // Virol. J. 2014. V. 11. № 1. P. 133–144.
Firczuk M., Bochtler M. // FEMS Microbiol. Rev. 2007. V. 31. № 6. P. 676–691.
Gu J., Feng Y., Feng X., Sun C., Lei L., Ding W., Liu X // PLoS Pathog. 2014. V. 10. № 5. P. e1004109.
Filatova L.Y., Becker S.C., Donovan D.M., Gladilin A.K., Klyachko N.L. // Biochimie. 2010. V. 92. № 5. P. 507–513.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология