

Прикладная биохимия и микробиология, 2020, T. 56, № 1, стр. 39-44
Очистка протеазы – активатора протеина с плазмы крови человека, продуцируемой микромицетом Aspergillus ochraceus ВКМ F-4104D
С. К. Комаревцев 1, Е. А. Попова 1, В. Г. Крейер 1, К. А. Мирошников 2, А. А. Осмоловский 1, *
1 Биологический факультет Московского государственного университета им. М.В. Ломоносова
119234 Москва, Россия
2 Институт биоорганической химии им. акад. М.М. Шемякина
и Ю.А. Овчинникова Российской академии наук
117997 Москва, Россия
* E-mail: aosmol@mail.ru
Поступила в редакцию 19.05.2019
После доработки 23.08.2019
Принята к публикации 30.08.2019
Аннотация
Разработан метод очистки протеазы − активатора протеина С плазмы крови человека из культуральной жидкости микромицета Aspergillus ochraceus ВКМ F-4104D. Свойства этого белка близки свойствам активатора протеина С из яда змеи Agkistrodon contortrix contortrix, который используется в современной лабораторной диагностике протеина С. Метод представляет собой комбинацию фракционирования сульфатом аммония и гидрофобной, ионообменной и гель-проникающей хроматографии. В результате был получен высокоочищенный препарат активатора протеина С, удельная активность которого в процессе очистки возросла более чем в 350 раз.
Протеин С – витамин-К-зависимый антикоагулянтный белок, образующийся главным образом в печени и эндотелии в форме неактивного одноцепочечного профермента. Активная форма протеина С образуется в результате ограниченного протеолиза профермента под действием комплекса тромбина с тромбомодулином и представляет собой сериновую протеазу. Функция активной формы протеина С заключается в ингибировании процесса свертывания крови за счет стимуляции фибринолиза и прерывания тромбиногенеза [1–3]. Вследствие важной физиологической роли протеина С в медицине существуют различные методы определения его функциональной активности, необходимые для диагностики ряда заболеваний системы гемостаза и их своевременной профилактики. В основе подавляющего большинства этих методов лежит использование активаторов протеина С из яда змей, чаще всего – южноамериканского медноголового щитомордника Agkistrodon contortrix contortrix [4, 5]. Ограниченная доступность ключевого компонента препятствует более широкому использованию диагностических методов в практике.
Ранее было показано, что ряд штаммов микромицета Aspergillus ochraceus секретирует протеазы, обладающие способностью активировать протеин С. Эти белки по ряду свойств сходны с активаторами, получаемыми из змеиного яда [6–8]. В частности, они обладают высокой тромбино- и плазминоподобной активностями и специфически расщепляют соответствующие хромогенные пептидные субстраты. Поэтому активаторы протеина С, продуцируемые микромицетами A. ochraceus, могут рассматриваться как потенциально более доступные аналоги активаторов из змеиного яда [9, 10].
В связи с этим становится актуальной проблема выделения и очистки активатора протеина С из культуральной жидкости A. ochraceus. Метод изоэлектрофокусирования, использованный для выделения активатора в ряде работ [8–10], позволяет получать небольшие количества высокоочищенного препарата, однако он трудно масштабируем и не может быть внедрен в промышленное производство. Разработанный ранее способ хроматографической очистки протеаз A. ochraceus основан на использовании труднодоступных носителей сочетанного действия с невысокой воспроизводимостью и не обеспечивает достаточной степени очистки конечного продукта [11].
Цель работы – разработка способа очистки активатора протеина С из культуральной жидкости микромицета A. ochraceus ВКМ F4104D с использованием современных хроматографических носителей.
МЕТОДИКА
Продуцент и условия культивирования. Использовали изученный ранее [8–10] штамм микромицета A. ochraceus ВКМ F-4104D, продуцирующий протеазу-активатор протеина С. Культивирование микромицета проводили в две последовательные стадии, выращивая его на посевной (состав в %: сусло – 6.7, глюкоза – 1, пептон – 0.1, рН 5.5–6.0) и ферментационной средах (состав в %: глюкоза – 3.5, гидролизат рыбной муки – 1, NaCl – 0.2, крахмал – 0.125, пептон – 0.1, KH2PO4 – 0.05, MgSO4 – 0.05, рН 5.5–6.0). Инокулят, получаемый смыванием посевной средой спор микромицета, выращенного в течение 7 сут на скошенном сусло-агаре 3°Б, вносили в посевную среду и культивировали в течение 2 сут, после чего часть биомассы переносили в ферментационную среду и культивировали ещё 2 сут. Культивирование проводили в качалочных колбах объемом 750 мл, содержащих 100 мл питательной среды, на орбитальной качалке при 200 об./мин при 28°С [12].
Фракционирование культуральной жидкости. После окончания культивирования мицелий отделяли от культуральной жидкости фильтрованием через фильтровальную бумагу (“ФС”, Россия). Протеазу-активатор протеина С осаждали из полученного фильтрата сульфатом аммония, подбирая оптимальную для осаждения белка степень его насыщения [13, 14]. Для этого к аликвотам культуральной жидкости медленно добавляли кристаллический сульфат аммония, получая различные степени насыщения: 0.5, 0.55, 0.6, 0.65, 0.7, 0.75 и 0.8 (за насыщенный раствор принимали 4.1 М раствор сульфата аммония, содержащий 767 г соли на 1 л), инкубировали в течение 12 ч при 4°С, после чего осадок отделяли центрифугированием при 15000 g на Роторе JA-20 (“Beckman J2-21”, Германия) в течение 20 мин. Для определения оптимальной для осаждения белка степени насыщения сульфатом аммония в супернатантах измеряли остаточную активность целевого белка.
Гидрофобная хроматография. Полученный при осаждении из культуральной жидкости белок, содержащий активатор протеина С, растворяли в 50 мМ Трис-HCl-буфере, рН 8.0, содержавшем сульфат аммония 0.35 степени насыщения, центрифугировали для удаления не растворившихся белков при 15000 g в течение 20 мин, после чего супернатант наносили на колонку с фенил-сефарозой CL-4B (“GE Healthcare”, США), уравновешенной тем же буфером. Элюцию проводили градиентом концентрации сульфата аммония в 50 мМ Трис-HCl-буфере, рН 8.0, от степени насыщения 0.35 до 0 [15, 16].
Ионообменная хроматография. Полученные после гидрофобной хроматографии активные компоненты, содержащие активатор протеина С, объединяли и наносили для дальнейшей очистки на колонку с ДЭАЭ-сефарозой (“GE Healthcare”, Швеция), предварительно уравновешенной 50 мМ Трис-HCl-буфером, рН 8.0. Элюцию проводили линейным градиентом концентрации NaCl от 0 до 1.0 М в стартовом буфере [17, 18].
Гель-фильтрация. Обогащенные целевым белком компоненты, полученные после ионообменной хроматографии, объединяли и концентрировали центрифугированием на мембранном фильтре Millipore (“Merck”, Германия), после чего наносили для дальнейшей очистки на колонку с Сефадексом G-50 (“Pharmacia”, Швеция), уравновешенную 50 мМ Трис-HCl-буфером, рН 8.0. Элюцию проводили тем же буфером. Активные очищенные фракции, содержащие протеазу-активатор протеина С, объединяли для дальнейшего анализа [19, 20].
Определение протеолитической активности. Ферментативную активность активатора протеина С определяли по реакции специфического расщепления бесцветного хромогенного пептидного субстрата п-нитроанилида тозил-глицил-пролил-аргинина (Tos-Gly-Pro-Arg-pNA), сопровождающегося накоплением свободного п-нитроанилина, окрашенного в желтый цвет. Для качественного определения активности в хроматографических фракциях, соответствующих максимумам поглощения при 280 нм, использовали дот-метод на парафильме. Для этого к 20 мкл 50 мМ Трис-HCl-буфера, рН 8.0, содержавшего 5 мМ CaCl2, добавляли по 20 мкл фермента и субстрата и визуально оценивали интенсивность окраски смеси против контроля, в который вместо субстрата вносили буфер [9, 10].
Количественное измерение активности проводили с использованием планшетного спектрофотометра “Wallac 1420” (“PerkinElmer”, Финляндия) при 405 нм. Реакцию проводили в 50 мМ Трис-HCl-буфере, рН 8.0, содержавшем 5 мМ CaCl2, при 37°С. К 50 мкл раствора фермента добавляли 50 мкл раствора субстрата (0.5 мг/мл), инкубировали 1 мин в описанных выше условиях и останавливали реакцию добавлением 100 мкл 50%‑ной уксусной кислоты. За единицу ферментативной активности (ед.) принимали количество фермента, расщепляющее 1 мкмоль субстрата за 1 мин. Измерения проводили в условиях линейной зависимости скорости накопления продукта реакции от времени [21, 22].
Анализ белкового состава компонентов. Для определения белкового состава полученных компонентов проводили электрофорез в полиакриламидном геле в присутствии ДДС-Na (концентрация акриламида в концентрирующем геле – 6%, в разделяющем – 12.5%). В качестве маркеров молекулярных масс использовали набор “Unstained Protein Molecular Weight Marker” (“Thermo Fisher Scientific”, США). После окончания электрофореза гель окрашивали 0.1%-ным раствором Кумасси бриллиантового голубого R-250 [23, 24]. Концентрацию белка определяли спектрофотометрическим методом на приборе “NanoDrop One” (“Thermo Fisher Scientific”, США) с использованием коэффициента экстинкции 0.667 при 280 нм [25].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Культуральная жидкость микромицета A. ochraceus, полученная по окончании срока культивирования продуцента, после удаления биомассы представляла собой прозрачную жидкость коричневого цвета, содержащую остатки компонентов питательной среды и различные, в том числе окрашенные, метаболиты микромицета. Суммарная концентрация белка в культуральной жидкости составила 5.7 мг/мл, а удельная ферментативная активность активатора протеина С – 0.02 ед./мг (табл. 1). На первой стадии очистки использовали высаливание сульфатом аммония, поскольку оно является доступным способом концентрирования целевого белка из большого объема культуральной жидкости и позволяет одновременно отделить значительное количество примесей, остающихся в растворенном виде.
Таблица 1.
Результаты очистки активатора протеина С из культуральной жидкости A. ochraceus
| Стадия очистки | Объем, мл | Концент-рация белка, мг/мл | Суммарный белок, мг | Удельная активность, ед./мг | Степень очистки, раз | Суммарная активность, ед. | Выход по актив-ности, % |
|---|---|---|---|---|---|---|---|
| Культуральная жидкость | 740 | 5.7 | 4218 | 0.02 | 1 | 84 | 100 |
| Гидрофобная хроматография | 35 | 0.7 | 24.5 | 2 | 100 | 49 | 58 |
| Ионообменная хроматография (после концентрирования) | 1 | 5.6 | 5.6 | 5.5 | 275 | 31 | 37 |
| Гель-фильтрация | 7.5 | 0.4 | 3 | 7.5 | 375 | 23 | 27 |
Для определения оптимальной степени насыщения сульфата аммония, необходимой для осаждения активатора протеина С, измеряли ферментативную активность в культуральной жидкости до и после высаливания различными концентрациями сульфата аммония. На рис. 1 приведена зависимость между степенью насыщения сульфата аммония и остаточной ферментативной активностью культуральной жидкости после высаливания. Снижение остаточной активности до минимальных значений произошло в опытах со степенями насыщения сульфата аммония 0.7, 0.75 и 0.8. Следовательно, степень насыщения сульфата аммония 0.7 является оптимальной для осаждения активатора протеина С из культуральной жидкости в процессе его выделения и очистки. Использование более высоких степеней насыщения 0.75 и 0.8 было нецелесообразным, поскольку это не приводило к существенному увеличению выхода целевого белка, но повышало количество соосаждающихся с ним примесей.
Рис. 1.
Зависимость от степени насыщения сульфата аммония (%) остаточной ферментативной активности активатора протеина С в культуральной жидкости A. ochraceus (%) после высаливания.
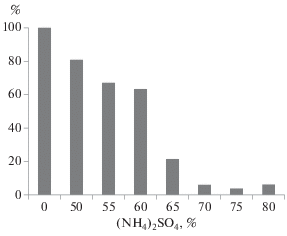
Электрофоретический анализ осадка, полученного в процессе высаливания культуральной жидкости сульфатом аммония при степени насыщения 0.7, показал, что вместе с целевым белком с молекулярной массой около 33 кДа содержалось значительное количество примесей, поэтому он нуждался в дальнейшей очистке (рис. 2, дорожка 2). Визуально осадок имел насыщенный коричневый цвет из-за содержания окрашенных продуктов метаболизма микромицета. Следует отметить, что основная часть примесей также начинала осаждаться при степенях насыщения сульфата аммония 0.65–0.7. В связи с этим было сделано заключение, что проведение предварительных стадий высаливания при степенях насыщения ниже 0.65 для осаждения посторонних компонентов культуральной жидкости не является обязательным, поскольку не приводит к существенному увеличению чистоты целевого белка, получаемого на данном этапе очистки.
Рис. 2.
Результаты электрофоретического анализа белкового состава осадка после высаливания культуральной жидкости A. ochraceus (2), компонентов, обогащенных активатором протеина С, после ионообменной хроматографии на ДЭАЭ-сефарозе (3), очищенной протеазы-активатора протеина С после гель-фильтрации (4). Белки-маркеры молекулярных масс (1): β-галактозидаза – 116.0, БСА – 66.2, овальбумин – 45.0, лактатдегидрогеназа – 35.0, РНКаза – 25.0, β-лактоглобулин – 18.4 и лизоцим – 14.4 кДа.

В дальнейшем было необходимо разработать способ очистки протеазы-активатора протеина С, образуемой A. ochraceus. В качестве следующей стадии очистки выбрали гидрофобную хроматографию на фенил-сефарозе. Ее результаты приведены на рис. 3а. Использованный хроматографический буфер, содержавший сульфат аммония 0.35 степени насыщения, обеспечил как растворение осадка протеазы-активатора протеина С, так и ее хорошую сорбцию на фенил-сефарозе. В то же время значительное количество примесей в этих условиях не связалось с носителем и элюировалось со свободным объемом колонки. Целевой белок элюировался в конце снижающегося градиента сульфата аммония, что обеспечило его эффективную грубую очистку. Раствор активатора протеина С, полученный после данного этапа очистки, имел желтый оттенок, удельная активность целевого белка в нем возросла в 100 раз по сравнению с первоначальной (табл. 1).
Рис. 3.
Результаты хроматографии компонентов, обогащенных активатором протеина С, на фенил-сефарозе (а), ДЭАЭ-сефарозе (б) и Сефадексе G-50 (в); величины оптической плотности элюата при 280 нм (1), градиенты (2) сульфата аммония (а) и (3) NaCl (б), компоненты, содержащие протеазу-активатор протеина С, отмечены “+".
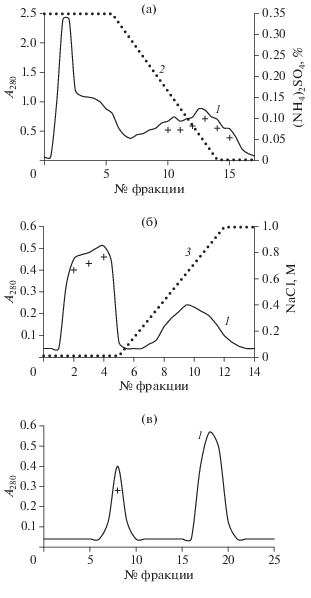
При дальнейшей очистке с использованием ДЭАЭ-сефарозы большое количество оставшихся примесей прочно связалось с хроматографическим носителем. Из результатов хроматографии на ДЭАЭ-сефарозе, приведенных на рис. 3б, видно хорошее разделение целевого белка, элюировавшегося в первых компонентах, и примесей, которые элюировались только при значительном увеличении концентрации NaCl в хроматографическом буфере. Компоненты, содержавшие активатор протеина С, после данного этапа очистки имели бледно-желтый цвет, удельная активность при этом возросла в 275 раз по отношению к исходной (табл. 1). Электрофоретический анализ полученных компонентов показал их значительное обогащение целевым белком и снижение количества примесей (рис. 2, дорожка 3).
На заключительном этапе очистки использовали гель-фильтрацию на Сефадексе G-50, предварительно сконцентрировав наносимый раствор белка на мембранном фильтре. Протеаза-активатор протеина С элюировалась с сорбента уже в начале процесса (рис. 3в), что обеспечило практически полное ее отделение от оставшихся примесей. Полученные после данного этапа очистки компоненты, содержавшие активатор протеина С, образуемого A. ochraceus, были бесцветны, их удельная активность возросла в 375 раз по сравнению с первоначальной (табл. 1). Электрофоретический анализ этих компонентов показал, что они содержали высокоочищенный целевой белок (рис. 2, дорожка 4).
Суммарный выход активатора протеина С после всех стадий очистки составил 27% (табл. 1), что незначительно ниже чем выход 35%, полученный при способе очистки протеазы другого штамма – A. ochraceus 513, и основанный на использовании бациллихин-силохрома, который был ранее опубликован в работе [11]. Однако возрастание итоговой степени очистки целевого белка в 375 раз, достигнутое при использовании предложенного в данной работе способа, значительно превышало ранее полученные результаты, в которых степень очистки возрастала только в 36 раз [11]. Это, а также труднодоступность бациллихин-силохрома в настоящее время, делают предложенный метод очистки более перспективным и легкодоступным для получения протеазы-активатора протеина С A. ochraceus.
Таким образом, с использованием современных хроматографических носителей была получено значительное количество высоко очищенной протеазы-активатора протеина С, образуемой A. ochraceus.
Список литературы
Bouwens E.A., Stavenuiter F., Mosnier L.O. // J. Thromb. Haemost. 2013. V. 11. № 1. P. 242–253.
Griffin J.H., Fernandez J.A., Gale A.J., Mosnier L.O. // J. Thromb. Haemost. 2007. V. 5. № 1. P. 73–80.
Струкова С.М. // Биохимия. 2004. Т. 69. № 10. С. 1314–1331.
Gempeler–Messina P.M., Müller C. // Toxin Rev. 2006. V. 25. № 4. P. 335–349.
Stoker K., Fisher H., Meier J., Brogli M., Svedsen L. // Toxicon. 1987. V. 25. № 3. P. 239–252.
Осмоловский А.А., Крейер В.Г., Кураков А.В., Баранова Н.А., Егоров Н.С. // Прикл. биохимия и микробиология. 2012. Т. 48. № 5. С. 537–542.
Пaтeнт PФ. 2011. № 2460772.
Осмоловский А.А., Крейер В.Г., Баранова Н.А., Кураков А.В., Егоров Н.С. // Прикл. биохимия и микробиология. 2013. Т. 49. № 6. С. 580–586.
Осмоловский А.А., Крейер В.Г., Баранова Н.А., Кураков А.В., Егоров Н.С. // Прикл. биохимия и микробиология. 2015. Т. 51. № 1. С. 86–92.
Осмоловский А.А., Крейер В.Г., Баранова Н.А., Егоров Н.С. // Прикл. биохимия и микробиология. 2017. Т. 53. № 4. С. 373–379.
Батомункуева Б.П., Егоров Н.С. // Микробиология. 2001. Т. 70. № 5. С. 602–606.
Пaтeнт PФ. 2011. № 2468081.
Green A.A., Hughes W.L. // Methods Enzymol. 1955. V. 1. № 10. P. 67–90.
Scopes R. Protein Purification: Principles and Practice / Ed. C.R. Cantor. N.Y.: Springer, 1993. 345 p.
Kennedy R.M. // Methods Enzymol. 1990. V. 182. № 27. P. 339–343.
McCue J.T. // Methods Enzymol. 2009. V. 463. № 25. P. 405–414.
Bollag D.M. // Methods Mol. Biol. 1994. V. 36. № 2. P. 11–22.
Cummins P.M., Rochfort K.D., O’Connor B.F. // Methods Mol. Biol. 2017. V. 1485. № 11. P. 209–223.
Stellwagen E. // Methods Enzymol. 1990. V. 182. № 25. P. 317–328.
Bollag D.M. // Methods Mol. Biol. 1994. V. 36. № 1. P. 1–9.
Lorsch J.R. // Methods Enzymol. 2014. V. 536. № 1. P. 3–15.
Harris T.K., Keshwani M.M. // Methods Enzymol. 2009. V. 463. № 7. P. 57–71.
Brunelle J.L., Green R. // Methods Enzymol. 2014. V. 541. № 12. P. 151–159.
Brunelle J.L., Green R. // Methods Enzymol. 2014. V. 541. № 13. P. 161–167.
Noble J.E., Bailey M.J. // Methods Enzymol. 2009. V. 463. № 8. P. 73–95.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология