

Прикладная биохимия и микробиология, 2022, T. 58, № 3, стр. 253-263
Альтернативные пути синтеза цефамандола с использованием биокаталитического ацилирования, катализируемого иммобилизованной синтетазой цефалоспоринов-кислот
А. В. Скляренко 1, *, И. А. Грошкова 1, И. Н. Крестьянова 1, С. В. Яроцкий 1
1 Национальный исследовательский центр “Курчатовский институт”
(НИЦ – “Курчатовский институт”)
123182 Москва, Россия
* E-mail: asklyarenko@yandex.ru
Поступила в редакцию 30.08.2021
После доработки 04.10.2021
Принята к публикации 10.01.2022
- EDN: TAPXIO
- DOI: 10.31857/S0555109922030126
Аннотация
Оптимизированы процессы биокаталитического ацилирования 1-метил-5-меркапто-1,2,3,4-тетразолил-7-аминоцефалоспорановой кислоты (7-TMCA) и 7-аминоцефалоспорановой кислоты (7-ACA) метиловым эфиром манделовой кислоты (MEMA) с использованием иммобилизованной синтетазы цефалоспоринов-кислот. В оптимизированных условиях в водно-органической среде, содержащей 43% (об./об.) этиленгликоля, при 30°С в спонтанно устанавливающемся градиенте рН в диапазоне 8.0–6.0 достигнуты выходы биокаталитической трансформации 80.8 ± 1.9% при ацилировании 7‑TMCA (концентрация 100–120 мМ) с образованием цефамандола (CFM) и 88.6 ± 2.0% при ацилировании 7-ACA (концентрация 140–170 мМ) с образованием полупродукта синтеза CFM (S-p CFM). Различия в исходных концентрациях 7‑TMCA и 7-ACA определяются различиями в растворимости этих субстратов. С учетом несомненных экологических преимуществ процесса химической трансформации полупродукта S-p CFM в CFM перед процессом получения 7‑TMCA из 7‑ACA сделан вывод о предпочтительности химико-биокаталитического синтеза CFM по второму пути.
Цефамандол (CFM) – парентеральный цефалоспориновый антибиотик второго поколения, обладающий широким спектром действия и характеризующийся высокой устойчивостью к действию плазмидных β-лактамаз, в том числе стафилококковых пенициллиназ [1]. К достоинствам CFM можно отнести его высокую эффективность при инфекциях, вызванных гемофильной палочкой, а также возможность сочетания данного цефалоспорина с пенициллинами и аминогликозидами.
Биокаталитический синтез полусинтетических β-лактамных антибиотиков – перспективное направление в области органического синтеза, альтернатива химическому синтезу в производстве лекарственных препаратов. Биокаталитический синтез β-лактамов является предметом многочисленных исследований [2–5], поскольку позволяет избежать использования токсичных реагентов, больших объемов органических растворителей, низких температур реакций, а также очистки промежуточных продуктов при многоступенчатом процессе химического синтеза β‑лактамных антибиотиков. Таким образом, применение биокаталитической трансформации для получения β-лактамных антибиотиков безусловно может снизить нагрузку на окружающую среду и уменьшить затраты на производство [6, 7].
Для биокаталитических процессов трансформации органических соединений используют ферменты не в растворенном виде, а в виде гетерогенных биокатализаторов (БК) – иммобилизованных (связанных с твердыми носителями) ферментов. Благодаря высокой специфичности фермента удается получать целевые продукты высокой степени чистоты [8–10].
Ранее в работе [11] по сопоставлению эффективности двух химико-биокаталитических путей синтеза антибиотика цефазолина (CEZ) был изучен и оптимизирован его биокаталитический синтез путем ацилирования 3-[(5-метил-1,3,4-тиадиазол-2-ил)-тиометил]-7-аминоцефалоспо-рановой кислоты (TDA), а также синтез полупродукта CEZ (S-p CEZ) путем ацилирования 7-аминоцефалоспорановой кислоты (7-ACA). На основании полученных результатов был сделан вывод о предпочтительности использования биокаталитического ацилирования 7-ACA для разработки химико-биокаталитической технологии получения CEZ.
Настоящая работа посвящена сопоставлению двух биокаталитических путей синтеза другого β‑лактамного антибиотика – цефамандола. CFM может быть синтезирован двумя химико-биокаталитическими путями, показанными на рис. 1. Традиционный химико-биокаталитический подход к синтезу CFM основан на том, что сначала химическим методом вводят необходимый заместитель в С3-положение 7-ACA (рис. 1, трансформация 1), а затем осуществляют биокаталитическое ацилирование аминогруппы полученного производного 1-метил-5-меркапто-1,2,3,4-тетразолил-7-аминоцефалоспорановой кислоты (7-TМСА) с образованием CFM (рис. 1, трансформация 2). Альтернативным химико-биокаталитическим подходом является использование биокатализа для ацилирования аминогруппы в С7-положении 7-ACA (рис. 1, трансформация 3) с последующим химическим превращением полупродукта CFM (S-p CFM) в целевой антибиотик (рис. 1, трансформация 4).
Рис. 1.
Пути химико-биокаталитического синтеза CFM. 1 – xимический синтез 7-TMCA из 7-ACA; 2 ‒ биокаталитический синтез CFM из 7-TMCA; 3 – биокаталитический синтез S-p CFM из 7-ACA; 4 – xимический синтез CFM из S-p CFM.

Биокаталитический синтез CFM и S-p CFM проводили методом ацильного переноса (кинетически-контролируемого синтеза) [6, 10, 11], используя 7-TMCA и 7-ACA в качестве ключевых аминокислот (КА) и метиловый эфир манделовой кислоты (МЕМА) в качестве ацилирующего агента (АА). В качестве биокатализатора использовали иммобилизованную синтетазу цефалоспоринов-кислот (IECASA). Фермент катализирует три конкурентные реакции: синтез CFM или S-p CFM и две побочные гидролитические реакции, а именно, гидролиз ацилирующего агента MEMA и расщепление ациламидной связи целевого продукта с образованием побочного продукта – манделовой кислоты (МА). Выход β-лактамного антибиотика кинетически контролируется, поскольку он зависит от скорости всех протекающих реакций.
Высокоспецифичным к цефалоспоринам-кислотам является фермент синтетаза цефалоспоринов-кислот (Cephalosporin-acid synthetase, CASA КФ 3.5.1.11) [6, 12]. Было установлено [12], что CASA кодируется геном, который является гомологом гена широко специфичной пенициллинацилазы (ПА) из Escherichia coli . В настоящей работе в качестве БК был использован фермент CASA, продуцируемый рекомбинантным штаммом E. coli ВКПМ В-12316 и иммобилизованный путем ковалентного связывания с эпокси-активированным макропористым носителем [5].
Цель работы – оптимизация ключевых стадий двух химико-биокаталитических путей получения CFM, а именно, процессов биокаталитического ацилирования 7-ТМСА с образованием CFM и 7-ACA с образованием полупродукта S-p CFM, катализируемых IECASA. В качестве критериев оптимизации использовали выход целевого продукта биокаталитической трансформации по отношению к КА и достигаемую концентрацию β-лактамного продукта в конечной реакционной смеси. Оптимизацию осуществляли по таким параметрам, как начальная концентрация КА и состав реакционной среды, а именно, концентрация в ней смешивающегося с водой двухатомного спирта этиленгликоля (ЭГ).
МЕТОДИКА
Материалы. В работе использованы коммерческие образцы 7-ACA фирмы “Anhui BBCA Pharmaceutical Co., LTD” (Китай) 93%-ной чистоты, определеной методом ВЭЖХ; 7‑ТМСА, “Shandong Sihuan Pharmaceutical Co., Ltd.”, (Китай) 96.5%-ной чистоты, также определенной методом ВЭЖХ; МЕМА (“Sigma-Aldrich”, США) – чистота 99%, определена методом ВЭЖХ; цефамандол натриевая соль (“Sigma-Aldrich”, США) – чистота 99% (по ВЭЖХ); МА (“Sigma-Aldrich”, США) – чистота 99%, (по ВЭЖХ); ЭГ (“РеаХимЛаб”, Россия) – ГОСТ 19770–83, 99.9%, концентрат. Лабораторный образец стандарта S-p CFM (97% чистоты, определено методом ВЭЖХ) любезно предоставлен Сычуаньским индустриальным институтом антибиотиков (Китай).
Получение биокатализатора IECASA. Биокатализатор IECASA получали согласно методике, описанной в работах [5, 11], путем иммобилизации фермента CASA из рекомбинантного штамма E. coli ВКПМ В-12316 на макропористом эпокси-активированном носителе Seplite LX-1000EP (“Sunresin New Materials”, Китай). Для работы был получен образец IECASA с синтетазной активностью 340 МЕ/г влажного БК при содержании сухих веществ 37%.
Синтетазную активность образца гетерогенного биокатализатора IECASA определяли по начальной скорости реакции синтеза CEZ из TDA и метилового эфира 1(H)-тетразолилуксусной кислоты (METzAA) с контролем содержания CEZ в реакционной смеси методом ВЭЖХ.
За 1 международную единицу (МЕ) синтетазной активности образца в реакции синтеза СEZ принимали такое количество препарата, которое катализирует образование 1 мкмоля CEZ за 1 мин в растворе, содержащем 60 мМ TDA и 240 мМ METzAA при температуре 30°С и начальном значении рН 7.5 [5, 11].
Анализ методом ВЭЖХ. Анализ реакционных смесей, получаемых при биокаталитическом синтезе CFM и S-p CFM, а также смесей, получаемых при изучении растворимости субстратов, проводили методом ВЭЖХ в изократическом режиме на хроматографе фирмы “Gilson” (США) с использованием хроматографической колонки “Spherisorb ODS”, 250 × 4 мм с диаметром частиц 7.5 мкм. В качестве мобильной фазы использовали смесь, состоящую из 0.05 М фосфатно-аммонийного буфера, рН 4.0, и метанола в различных соотношениях в зависимости от изучаемого процесса (табл. 1). Скорость потока мобильной фазы – 1.0 мл/мин. Детектирование пиков анализируемых веществ осуществляли спектрофотометрически при длине волны 218 или 254 нм. Условия проведения анализа методом ВЭЖХ и времена удерживания анализируемых веществ (RT) представлены в табл. 1.
Таблица 1.
Условия проведения анализа методом ВЭЖХ
| Процесс | Определяемый компонент | Содержание метанола в мобильной фазе, % (об./об.) | Детекция, нм | RT, мин |
|---|---|---|---|---|
| Биокаталитический синтез S-p CFM | 7-ACA | 1 | 218 | 7.8–8.0 |
| MA | 5.3–5.8 | |||
| S-p CFM | 35 | 4.1–4.5 | ||
| MEMA | 6.8–7.3 | |||
| Биокаталитический синтез CFM | 7-TMCA | 5 | 218 | 11.6–12.0 |
| MA | 4.5–4.7 | |||
| MEMA | 35 | 7.0–7.3 | ||
| CFM | 4.5–5.5 | |||
| Определение растворимости 7‑ACA | 7-ACA | 5 | 254 | 2.8–5.0 |
| Определение растворимости 7-TMCA | 7-TMCA | 8 | 254 | 8.0–8.3 |
| Определение растворимости MEMA | MEMA | 35 | 218 | 7.0–7.3 |
Проведение синтеза CFM и S-p CFM, катализируемого IECASA. Биокаталитический синтез CFM и S-p CFM осуществляли путем ацилирования соответствующей КА (7-ТМСА или 7‑ACA) метиловым эфиром манделовой кислоты. Для проведения процесса синтеза использовали стеклянный реактор емкостью 75 мл, оборудованный лопастной механической мешалкой и снабженный системами поддержания температуры и рН. Для приготовления раствора субстратов в реакторе при температуре 30 ± 1°С суспендировали навеску КА в 0.3 М фосфатно-натриевом буфере (ФБ), рН 8.3, с добавлением или без добавления ЭГ. Растворение КА осуществляли под контролем рН при постоянном перемешивании и порционном добавлении 2 М NaOH вплоть до полного растворения КА. При этом рН не превышал величину 8.0. После растворения КА вносили навеску МЕМА. Раствор перемешивали в течение 3–5 мин до полного или частичного растворения АА и начинали процесс биокаталитического синтеза, добавляя навеску IECASA, необходимую для создания в реакционной смеси требуемого содержания активного фермента (СЕ = 10–30 МЕ/мл). Значение рН снижалось по ходу реакции за счет образования МА. Процесс синтеза CFM или S-p CFM проводили при постоянном умеренном перемешивании реакционной смеси, температуре (30 ± 1)°С в спонтанно устанавливающемся градиенте рН. После достижения значения рН, равного 6.0, его поддерживали добавлением 2.0 М NaOH. Синтез останавливали, отделяя IECASA вакуумной фильтрацией на пористом стеклянном фильтре.
Для исследования динамики процесса синтеза каждые 5–10 мин отбирали пробу реакционной смеси и контролировали в ней методом ВЭЖХ содержание четырех компонентов: КА (7-TMCA или 7-ACA), ацилирующего агента MEMA, целевого β-лактамного продукта (CFM или S-p CFM) и побочного продукта МА. Синтез проводили до достижения стабильного плато на кривой зависимости относительной концентрации целевого продукта от времени (плато максимальной концентрации целевого продукта, ${\text{C}}_{{{\text{прод}}}}^{{{\text{макс}}}},$ мМ). Относительное содержание целевого продукта (CFM или S-p CFM) в текущий момент времени рассчитывали по отношению к содержанию в реакционной смеси КA (7‑ТМСА или 7‑ACA) в начальный момент времени (${\text{C}}_{{{\text{KA}}}}^{{\text{o}}},$ мМ). Максимальную степень трансформации КА в целевой продукт (максимальный выход целевого продукта, ${{{{\eta }}}^{{{\text{макс}}}}},$ %) рассчитывали по формуле:
(средние данные на плато).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Растворимость субстратов биокаталитического синтеза CFM и S-p CFM. Учет растворимости КА и АА является важным инструментом для выбора оптимальных условий процессов биокаталитического синтеза β-лактамов [14]. Растворимость 7-TMCA, 7-ACA и МЕМА изучали в условиях, моделирующих условия проведения биокаталитических процессов, а именно: 30°С, 0.3 М ФБ, начальная концентрация ЭГ (СЭГ, % об./об.) – 0 или 43%. Эксперименты проводили методом насыщения, описанным в работах [5, 14, 16]. Полученные результаты представлены в табл. 2.
Таблица 2.
Влияние этиленгликоля на кислотно-основные свойства и растворимость субстратов биокаталитического синтеза CFM и S-p CFM
| Соеди-нение | Структура | Электрохими-ческая природа | Константы ионизации | Характерис-тическая раствори-мость, мМ* | Концент-рация этилен-гликоля CЭГ, % (об./об.) |
|
|---|---|---|---|---|---|---|
| pK1 | pK2 | |||||
| MEMA |  |
Неэлектролит | _ | _ | 134 ± 4 | 0 |
| 360 ± 11 | 43 | |||||
| 7-TMCA | 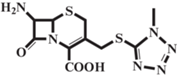 |
Электролит, аминокислота |
2.16 ± 0.03 | 4.71 ± 0.05 | 0.22 ± 0.03 | 0 |
| 2.33 ± 0.03 | 4.72 ± 0.05 | 0.15 ± 0.03 | 43 | |||
| 7-ACA | 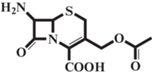 |
Электролит, аминокислота |
1.73 ± 0.02 | 4.70 ± 0.05 [11] | 2.78 ± 0.09 | 0 |
| 2.25 ± 0.03 | 4.58 ± 0.05 | 1.42 ± 0.14 | 43 | |||
Не зависящая от рН растворимость МЕМА, не являющегося электролитом, была изучена при рН 6.5, что обеспечило практическое отсутствие спонтанного гидролиза данного эфира в процессе проведения экспериментов. Из табл. 2 видно, что присутствие ЭГ существенно влияло на растворимость МЕМА. Так в водной буферной среде при СЭГ = 0 % (об./об.) растворимость MEMA составляла 130 мМ, а при СЭГ = 43% (об./об.) она достигала 360 мМ.
Для 7-TMCA и 7-ACA было изучено влияние рН на растворимость этих аминокислот при слабокислом, нейтральном и слабощелочном рН и определены константы ионизации карбоксильной (рK2) и аминогруппы (рK1), а также характеристическая растворимость (S±).
Для аминокислот 7-TMCA и 7‑ACA зависимость растворимости (S, мМ) от рН, описывается уравнением:
(1)
$S = {{S}^{ \pm }}\frac{{{{{[{{{\text{H}}}^{ + }}]}}^{2}} + {{K}_{1}}[{{{\text{H}}}^{ + }}] + {{K}_{1}}{{K}_{2}}}}{{{{K}_{1}}[{{{\text{H}}}^{ + }}]}}.$При нейтральных и щелочных рН в условиях полного депротонирования карбоксильной группы аминокислоты может быть использовано уравнение:
(1.1)
$S = {{S}^{ \pm }} + {{S}^{ \pm }}{{K}_{2}}\frac{{\text{1}}}{{{\text{[}}{{{\text{H}}}^{ + }}{\text{]}}}}.$При кислых рН в условиях полного протонирования аминогруппы аминокислоты применимо уравнение:
где [H+] – концентрация ионов водорода в растворе при заданном рН, мМ;K2 – константа ионизации карбоксильной группы аминокислоты, мM;
K1 – константа ионизации аминогруппы аминокислоты, мM;
S± – растворимость индивидуальной электронейтральной цвиттерионной формы аминокислоты (характеристическая растворимость электролита), мM.
На рис. 2а и 3а представлена линеаризация экспериментальных данных в координатах уравнения (1.1, S vs. 1/[H+]), полученных при изучении влияния pH на растворимость аминокислот 7-ТМСА и 7-ACA при нейтральном и слабощелочном pH при СЭГ = 0% (об./об.) и СЭГ = 43% (об./об.) соответственно. Линеаризация в координатах уравнения (1.2, S vs. [H+]) экспериментальных данных, полученных при изучении влияния pH на растворимость аминокислот при нейтральном и слабокислом pH при СЭГ = 0% (об./об.) и СЭГ = 43% (об./об.), представлена на рис. 2б и 3б соответственно.
Рис. 2.
Зависимости растворимости 7-ТМСА от рН в 0.3 M ФБ при 30°C: а – в координатах уравнения (1.1), б – в координатах уравнения (1.2). 1 – СЭГ = 0% (об./об.); 2 – СЭГ = 43% (об./об.).

Рассчитанные значения констант ионизации и характеристической растворимости для 7-ТМСА и 7-ACA приведены в табл. 2. На величину pK2 7‑ТМСА и 7-ACA этиленгликоль практически не влиял, однако увеличивал величину pK1, особенно в случае 7-ACA (на 0.5 ед. рН). Для обеих аминокислот в присутствии ЭГ характеристические растворимости снижались: в 1.5 раза для 7-ТМСА и в 2 раза для 7-ACA.
Рис. 3.
Зависимости растворимости 7-АСА от рН в 0.3 M ФБ при 30°C: а – в координатах уравнения (1.1), б – в координатах уравнения (1.2). 1 – СЭГ = 0% (об./об.); 2 – СЭГ = 43% (об./об.).

По уравнению (1) с использованием констант, представленных в табл. 2, были рассчитаны теоретические кривые зависимости растворимости 7-ТМСА и 7-ACA от pH при СЭГ = 0% (об./об.) и СЭГ = 43% (об./об.) (кривые 1–4 на рис. 4). На рис. 4 представлена также растворимость МЕМА в водной среде (кривая 5) и в присутствии ЭГ (кривая 6).
Рис. 4.
Теоретические кривые зависимости растворимости субстратов от рН, рассчитанные с использованием констант, представленных в табл. 2 (30°C, 0.3 M ФБ). Кривые 1–4 рассчитаны по уравнению (1). 1 – 7-TМСA при СЭГ = 0%; 2 – 7-TМСA при СЭГ = 43%; 3 – 7-АСA при СЭГ = 0%; 4 – 7-АСA при СЭГ = 43%; 5 – МЕМА при СЭГ = 0%; 6 – МЕМА при СЭГ = 43%.

Влияние ЭГ на растворимость КА различно: растворимость 7-TMCA в присутствии ЭГ снижалась (кривые 1 и 2, рис. 4), а растворимость 7-АСА существенно увеличивалась (кривые 3 и 4, рис. 4). При этом во всем изученном диапазоне рН растворимость 7-АСА значительно превышала растворимость 7-TMCA как в водной среде (кривые 3 и 1, рис. 4), так и в присутствии ЭГ (кривые 4 и 2, рис. 4). В рабочем диапазоне pH 6.0–7.0, где CASA обладает высокой активностью и стабильностью [5], растворимость 7‑ТМСА в водной среде изменялась от 4.5 до 43 мМ, а растворимость 7‑АСА превышала ее более чем в 10 раз, изменяясь от 55 до 570 мМ (расчет по уравнению (1)). Присутствие в среде ЭГ (СЭГ = 43%, (об./об.)) увеличивало разрыв в растворимости КА в диапазоне pH 6.0–7.0 до 80 раз: растворимость 7-ТМСА изменялась от 3.0 до 30 мМ, а растворимость 7-АСА от 250 до 2500 мМ (расчет по уравнению (1)). В присутствии ЭГ (СЭГ = 43%, (об./об.)) растворимость МЕМА увеличивалась в 2.8 раза по сравнению с водной средой (СЭГ = 0%, (об./об)) (кривые 6 и 5 на рис. 4, табл. 2). При изучении синтеза CFM для достижения в реакционной смеси исходной концентрации 7-ТМСА, превышающей ее растворимость в рабочем диапазоне pH 6.0–7.0, раствор субстратов готовили при рН около 8.0. В ходе ацилирования 7-ТМСА метиловым эфиром манделовой кислоты рН снижался за счет образования МА, однако 7-ТМСА не выпадала в осадок за счет эффекта перенасыщения, как это описано ранее для TDA при синтезе CEZ в работе [5].
Таким образом, с точки зрения возможности создания в реакционной среде высоких исходных концентраций КА $\left( {{\text{C}}_{{{\text{KA}}}}^{{\text{o}}}} \right)$ и АА $\left( {{\text{C}}_{{{\text{АA}}}}^{{\text{o}}}} \right),$ предпочтительным является биокаталитический процесс ацилирования 7-АСА с образованием S-p CFM, протекающий в среде, содержащей ЭГ.
Оптимизация процессов биокаталитического синтеза CFM и S-p CFM, катализируемого IECASA. Для повышения выхода целевого β-лактама в процессах синтеза с ацильным переносом может быть использована водно-органическая среда, содержащая смешивающийся с водой двухатомный спирт ЭГ. Наблюдаемый эффект является следствием снижения активности воды в присутствии органических растворителей, влекущей за собой уменьшение скорости непродуктивных гидролитических процессов. При этом замедляется гидролиз как ацилирующего агента, так и целевых продуктов [16, 17]. В ряде работ было продемонстрировано существенное увеличение выхода амино-β-лактамных антибиотиков ампициллина [18–20] и цефалексина [21–23] в процессах их кинетически-контролируемого синтеза, катализируемого иммобилизованной различными способами ПА из E. coli, за счет присутствия ЭГ в реакционной среде. Было показано, что растворитель не оказывал инактивирующего действия на ПА из E. coli [18, 19]. В настоящей работе ЭГ был использован для увеличения выхода CFM и S-p CFM, относящихся к классу β-лактамов-кислот, в процессах их биокаталитического синтеза, катализируемого БК на основе рекомбинантного фермента CASA, гомологичного ПА из E. coli [12].
Процессы биокаталитического ацилирования 7-TMCA и 7-ACA метиловым эфиром манделовой кислоты проводили в следующих условиях: 30°С, 0.3 М ФБ, Хо = 3.3 M/М (мольный избыток АА над КА, ${{{\text{X}}}^{{\text{o}}}} = \frac{{{\text{С}}_{{{\text{AA}}}}^{{\text{o}}}}}{{{\text{С}}_{{{\text{КА}}}}^{{\text{о}}}}}$), спонтанный градиент pH (спонтанное снижение рН от значения, при котором растворяют субстраты, до рН 6.0 и поддержание этого значения рН до завершения процесса). Эти условия были выбраны для биокатализатора IECASA при оптимизации процессов синтеза CEZ и S-p CEZ [5, 11]. В настоящей работе при оптимизации процессов синтеза CFM и S-p CFM варьировали два параметра: начальную концентрацию КА (${\text{C}}_{{{\text{7}} - {\text{TMCA}}}}^{{\text{о}}}$ = 60–120 мМ или ${\text{C}}_{{{\text{7}} - {\text{ACA}}}}^{{\text{о}}}$ = = 60–170 мМ) и концентрацию ЭГ в реакционной смеси (СЭГ = 0–43%, (об./об.)). Биокатализатор IECASA вносили в количестве, обеспечивающем содержание активного фермента в реакционной смеси СЕ = 10–30 ME/мл, так, чтобы длительность процесса, включая плато максимальной концентрации продукта, составляла 70–90 мин.
Отметим, что ЭГ тормозит не только побочные процессы гидролиза, протекающие при синтезе с ацильным переносом, но и (в меньшей степени) целевой процесс синтеза [17]. Было показано, что начальная скорость накопления CFM в процессе его биокаталитического синтеза снижалась в два раза при СЭГ = 43% (об./об.) по сравнению с начальной скоростью процесса в водной среде. Процессы биокаталитического синтеза CFM и S-p CFM при концентрациях ЭГ более 43% (об./об.) не изучались, так как их осуществление за время, не превышающее 90 мин, требует содержания IECASA в реакционной смеси более 100 мг/мл, что затрудняет массообменные процессы.
В каждом эксперименте по синтезу β-лактама осуществляли динамический контроль состава реакционной смеси по всем компонентам (КА, АА, целевой продукт, МА) и рассчитывали балансы по β-лактаму и МА. Результаты двух экспериментов, проведенных при СЭГ = 43% (об./об.), показаны на рис. 5 и 6 для биокаталитического ацилирования 7-ТМСА и 7-ACA соответственно, при максимальной использованной концентрации КА для каждого процесса: ${\text{C}}_{{{\text{7}} - {\text{TMCA}}}}^{{\text{о}}}$ = 120 мМ (рис. 5) и ${\text{C}}_{{{\text{7}} - {\text{ACA}}}}^{{\text{о}}}$ = 170 мМ (рис. 6).
Рис. 5.
Изменение состава реакционной смеси (относительные концентрации, %) от времени при синтезе CFM, катализируемом IECASA (30°C, 0.3 M ФБ, CE = = 30 МЕ/мл, ${\text{C}}_{{{\text{KA}}}}^{{\text{o}}}$ = 120 мМ, ${\text{C}}_{{{\text{AA}}}}^{{\text{o}}}$ = 400 мМ, Xo = = 3.3 М/М, СЭГ = 43% (об./об.), спонтанный градиент рН в диапазоне рН 8.0–6.0). 1 – 7-ТМСА; 2 – CFM, рассчитаны по отношению к ${\text{C}}_{{{\text{KA}}}}^{{\text{o}}};$ 3 – баланс (%) по β-лактаму, сумма относительных концентраций 7‑ТМСА и CFM; 4 – MEMA, 5 – MА, рассчитаны по отношению к ${\text{C}}_{{{\text{AA}}}}^{{\text{o}}};$ 6 – баланс (%) по MA – сумма относительных концентраций CFM, MEMA и MA.

Рис. 6.
Изменение состава реакционной смеси (относительные концентрации, %) от времени при синтезе S-p CFM, катализируемом IECASA (30°C, 0.3 M ФБ, CE = 30 МЕ/мл, ${\text{C}}_{{{\text{KA}}}}^{{\text{o}}}$ = 170 мМ, ${\text{C}}_{{{\text{AA}}}}^{{\text{o}}}$ = 560 мМ, Xo = = 3.3 М/М, СЭГ = 43% (об./об.), спонтанный градиент рН в диапазоне рН 8.0–6.0): 1 – 7-ACA; 2 – S-p CFM, рассчитаны по отношению к ${\text{C}}_{{{\text{KA}}}}^{{\text{o}}};$ 3 – баланс (%) по β‑лактаму, сумма относительных концентраций 7-ACA и S-p CFM; 4 – МЕMА; 5 – MA, рассчитаны по отношению к ${\text{C}}_{{{\text{AA}}}}^{{\text{o}}};$ 6 – баланс (%) по MA – сумма относительных концентраций S-p CFM, MEMA и MA.

В обоих процессах наблюдалось продолжительное плато на кривых накопления целевого продукта (рис. 5 и 6, кривые 2). Максимальные выходы CFM и S-p CFM, рассчитанные как среднее значение на плато, составили в этих экспериментах 81.3 ± 2.2 и 88.8 ± 2.3% соответственно. Особенностью процесса синтеза S‑p CFM, результаты которого представлены на рис. 6, было использование АА при начальной концентрации 560 мМ, превышающей растворимость MEMA. Из-за неполного растворения MEMA в течение первых 15 мин наблюдалось отсутствие баланса не только по МА (рис. 6, кривая 6), но и по β-лактаму (рис. 6, кривая 3). Это можно объяснить тем, что часть 7-AСА также выпадала в осадок в присутствии нерастворенных кристаллов АА. Далее оба баланса достигали 100%, т.е. оба субстрата полностью растворялись, а кроме того, в системе отсутствовали побочные процессы, в том числе с участием β-лактамного ядра.
На основании результатов всех проведенных экспериментов по синтезу CFM и S-p CFM, катализируемому IECASA, были построены зависимости максимального выхода продукта реакции (${{{{\eta }}}^{{{\text{макс}}}}},$ %) от концентрации ЭГ (СЭГ, % (об./об.)) и от начальной концентрации КА (${\text{C}}_{{{\text{КA}}}}^{{\text{о}}},$ мМ), представленные на рис. 7 и 8 соответственно.
Рис. 7.
Зависимость максимального выхода β-лактамного продукта (ηмакс, %) от концентрации этиленгликоля (СЭГ, % (об./об.)) в реакционной смеси при синтезе с ацильным переносом, катализируемом IECASA (30°C, 0.3 M ФБ, Xo = 3.3 M/M, спонтанный градиент рН): 1 – синтез CFM из 7-TMCA и МЕМА при CE = 10–20 МЕ/мл; ${\text{C}}_{{{\text{KA}}}}^{{\text{o}}}$ = 60 мМ; 2 – синтез S-p CFM из 7-АCA и МЕМА при CE = 30 МЕ/мл; ${\text{C}}_{{{\text{KA}}}}^{{\text{o}}}$ = = 150 мМ.

Рис. 8.
Зависимость максимального выхода β-лактамного продукта (ηмакс, %) от начальной концентрации КА (${\text{C}}_{{{\text{KA}}}}^{{\text{o}}},$ мМ) при синтезе с ацильным переносом, катализируемом IECASA (30°C, 0.3 M ФБ, Xo =3.3 M/M, CE = 20–30 МЕ/мл, спонтанный градиент рН, СЭГ = 43% (об./об.)): 1 – синтез CFM из 7‑TMCA и МЕМА; 2 – синтез S-p CFM из 7-ACA и МЕМА.
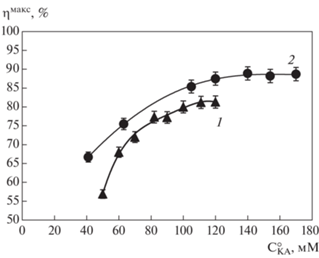
Использование водно-органической среды, содержащей ЭГ, позволило повысить выход CFM (рис. 7, кривая 1) с 60% (СЭГ = 0% (об./об.)) до 70% (СЭГ = 43%, (об./об.)). С увеличением концентрации ЭГ от 0 до 43% (об./об.) выход S-p СFM возрос с 80% до 88% (рис. 7, кривая 2).
Для дальнейших исследований процессов биокаталитического синтеза СFM и S-p СFM, катализируемых IECASA, была выбрана водно-органическая среда, содержащая ЭГ в концентрации 43% (об./об.), поскольку она обеспечивала наиболее высокий выход целевых продуктов. Зависимости максимального выхода каждого из процессов (ηмакс, %) от начальной концентрации КА (${\text{C}}_{{{\text{КА}}}}^{{\text{о}}},$ мМ) представлены на рис. 8. Зависимость выхода CFM от ${\text{C}}_{{{\text{КА}}}}^{{\text{о}}}$ была исследована в диапазоне 50–120 мМ (рис. 8, кривая 1), поскольку растворимость 7-ТМСА в условиях проведения эксперимента не позволяла достичь в исходной реакционной смеси более высокой концентрации КА, чем 120 мМ. Зависимость выхода S-p CFM от начальной концентрации 7-ACA была исследована в диапазоне ${\text{C}}_{{{\text{КА}}}}^{{\text{о}}}$ = 40–170 мМ (рис. 8, кривая 2). Растворимость 7-ACA в условиях проведения эксперимента при СЭГ = 43% (об./об.), рН 6.0–8.0 очень высока (рис. 4, кривая 4), однако диапазон используемой ${\text{C}}_{{{\text{КА}}}}^{{\text{о}}}$ при Хо = 3.3 М/М ограничен сверху растворимостью МЕМА.
Зависимости выхода продукта от ${\text{C}}_{{{\text{КА}}}}^{{\text{о}}}$ имеют вид кривых с насыщением (рис. 8). Выход CFM монотонно возрастал с ростом концентрации 7-ТМСА вплоть до ${\text{C}}_{{{\text{КА}}}}^{{\text{о}}}$ = 100 мМ и в диапазоне ${\text{C}}_{{{\text{КА}}}}^{{\text{о}}}$ от 100–120 мМ достигал среднего значения 80.8 ± 1.9%. Наибольший выход продукта процесса синтеза S-p CFM достигался в диапазоне ${\text{C}}_{{{\text{КА}}}}^{{\text{о}}}$ от 140–170 мМ и его среднее значение в данном диапазоне составляло 88.6 ± 2.0%.
На основании полученных результатов были выбраны следующие оптимальные условия процессов синтеза CFM и S-p CFM, катализируемых IECASA: 30°С, 0.3 М ФБ, СЭГ = 43% (об./об.), СЕ = 30 МЕ/мл, спонтанный градиент рН в диапазоне рН 8.0–6.0, Хо = 3.3 M/М, ${\text{C}}_{{{\text{КА}}}}^{{\text{о}}}$ = 100–120 мМ для процесса ацилирования 7-TMCA и ${\text{C}}_{{{\text{КА}}}}^{{\text{о}}}$ = = 140–170 мМ для процесса ацилирования 7-ACA. В таких оптимальных условиях продолжительность процессов ацилирования (время выхода на плато максимальной концентрации продукта) составляла 50 и 60 мин для синтеза CFM и S-p CFM соответственно.
В табл. 3 представлены результаты, достигнутые при синтезе CFM и S-p CFM, катализируемом IECASA, в выбранных оптимальных условиях, обеспечивающих высокую степень трансформации КА в β-лактам (ηмакс, %) и высокую концентрацию целевого продукта в конечной реакционной смеси (${\text{C}}_{{{\text{прод}}}}^{{{\text{макс}}}},$ мМ), что необходимо для разработки эффективного процесса выделения и очистки антибиотика. Полученные в настоящей работе результаты по кинетически-контролируемому синтезу CFM и S-p CFM, катализируемому IECASA, сопоставлены в табл. 3 с лучшими из имеющихся в литературе результатами по биокаталитическому синтезу этих соединений [24, 25].
Таблица 3.
Параметры эффективности процессов биокаталитического синтеза CFM и S-p CFM
| Продукт | Биокатализатор | Условия процесса | Конечная реакционная смесь | |||||
|---|---|---|---|---|---|---|---|---|
| ${\text{C}}_{{{\text{KA}}}}^{{\text{o}}}$, мМ | $X_{{}}^{o}$, М/М | Среда | Темпе-ратура, °С | Время, мин | ηмакс, % | ${\text{C}}_{{{\text{прод}}}}^{{{\text{макс}}}}$, мМ | ||
| CFM | IECASA | 100–120 | 3.3 | Однофазная система: 0.3 М ФБ, ЭГ (43%) | 30 | 50 | 80.8 ± 1.9 | 80–100 |
| ПА из E. сoli, на глиоксил- агарозе [21] |
50 | 1.5 | Водная двухфазная система: ПЭГ 600 (100%) и 3 М (NH4)2SO4 | 15 | 90 | 60.0 | 30 | |
| S‑p CFM | IECASA | 140–170 | 3.3 | Однофазная система: 0.3 М ФБ, ЭГ (43%) | 30 | 60 | 88.6 ± 2.0 | 125–150 |
| ПА из E. сoli, на Sepharose [22] | 50 | 3.0 | Водная двухфазная система: ПЭГ 600 (80%) и 4 М (NH4)2SO4 | 4 | 120 | 88.0 | 44 | |
Сравнение катализируемых IECASA процессов синтеза CFM из 7-TMCA, S-p CFM и 7-ACA, (рис. 1, трансформации 2 и 3) свидетельствовало о таких преимуществах второго процесса, как увеличение выхода продукта процесса ацилирования с 80.8 ± 1.9 до 88.6 ± 2.0% и увеличение концентрации целевого β-лактама в конце процесса в реакционной смеси в 1.5 раза. Следует отметить, что увеличение концентрации продукта S-p CFM в конечной реакционной смеси по сравнению с концентрацией CFM достигалось как за счет повышения выхода продукта процесса трансформации, так и в результате достижения более высокой начальной концентрации 7-ACA по сравнению с начальной концентрацией 7-TMCA.
Таким образом, процесс биокаталитического ацилирования 7-ACA метиловым эфиром манделовой кислоты (рис. 1, трансформация 3) протекал более эффективно, чем ацилирование 7-TMCA (рис. 1, трансформация 2). Существенно, что химическая трансформация S-p CFM в CFM (рис. 1, трансформация 4) может быть осуществлена без выделения S-p CFM из реакционной смеси [11, 25] и с экологической точки зрения предпочтительнее, так как протекает в более мягких условиях, чем химическая трансформация 7-ACA в 7-ТМСА (рис. 1, трансформация 1). В связи с этим химико-биокаталитический синтез CFM через полупродукт S-p CFM представляется более перспективным для разработки конкурентоспособной технологии получения данного антибиотика. Ранее аналогичный вывод был сделан нами при сопоставлении химико-биокаталитических путей синтеза другого цефалоспоринового антибиотика – CEZ [11].
Работа выполнена в рамках Государственного задания № 593-00003-19 ПР “Фундаментальные и прикладные научные работы в области биотехнологии”.
Список литературы
Sharon S.C. // xPharm: The Comprehensive Pharmacology Reference. Cefamandole. / Ed. S.J. Enna, D.B. Bylund. Elsevier Inc., 2008. P. 1–5.
Schmidt F.-R. The Mycota X. Industrial Applications. / Ed.K. Esser, M. Hofrichter. Berlin, Heidelberg: Springer-Verlag, 2010. V. 5. P. 101–121.
Srirangan K., Orr V., Akawi L., Westbrook A., Moo-Young M., Chou C.P. // Biotechnol Adv. 2013. V. 31. № 8. P. 1319–1332.
Volpato G., Rodrigues R.C., Fernandez-Lafuente R. // Curr. Med. Chem. 2010. V. 17. № 32. P. 3855–3873.
Wang Lu, Sklyarenko A.V., Li Duanhua, Sidorenko A.I., Zhao Chen, Li Jinjun, Yarotsky S.V. // Bioprocess Biosyst. Eng. 2018. V. 41. № 12. P. 1851–1867.
Kurochkina V.B., Sklyarenko A.V. // Biotechnology: State of the Art and Prospects for Development. / Ed. G.E. Zaikov. N.Y.: Nova Science Publishers, 2008. Ch. 20. P. 175–204.
Sheldon R.A., Woodley J.M. // Chem. Reviews. 2018. V. 118. № 2. P. 801–838.
Rajasekar V.W. // Enz. Eng. 2016. V. 5. № 1. P. 138–139.
Rodriguez–Herrera R., Puc L.E.C., Sobrevilla J.M.V., Luque D., Cardona-Felix C.S., Aguilar-González C.N., Flores-Gallegos A.C. Enzymes in the Pharmaceutical Industry for β-Lactam Antibiotic Production. / Ed. M. Kuddus. Acad. Press. 2019. Ch. 36. P. 627–643.
Sklyarenko A.V., Eldarov M. A., Kurochkina V.B., Yarotsky S.V. // Appl. Biochem. Microbiol. 2015. V. 51. № 6. P. 627–640.
Sklyarenko A.V., Groshkova I.A., Sidorenko A.I., Yarotsky S.V. // Appl. Biochem. Microbiol. 2020. V. 56. № 5. P. 452–464.
Эльдаров М.А., Скляренко А.В., Думина М.В., Медведева Н.В., Жгун А.А., Сатарова Д.Э., Сидоренко А.И., Епремян А.С., Яроцкий С.В. // Биомедицинская химия. 2015. Т. 61. № 5. С. 646–651.
Eldarov M.A., Sklyarenko A.V., Mardanov A.V., Belet-sky A.V., Zhgun A.A., Dumina M.V., Medvedeva N.V., Satarova D.E., Ravin N.V., Yarockii S.V. // Appl. Biochem. Microbiol. 2015. V. 51. № 5. P. 505–510.
Kurochkina V.B., Sklyarenko A.V., Satarova D.E., Yarosky S.V. // Bioprocess Biosyst. Eng. 2011. V. 34. № 9. P. 1103–1117.
McDonald M.A., Bommarius A.S., Rousseau R.W. // Chem. Eng. Sci. 2017. V. 165. P. 81–88.
Santana M., Ribeiro M.P.A., Leite G.A., Giordano R.L.C., Giordano R.C., Mattedi S. // AIChE J. 2010. V. 56. № 6. P. 1578–1583.
Курочкина В.Б., Скляренко А.В. // Антибиотики и химиотерапия. 2005. № 5–6. С. 39–58.
Illanes A., Fajardo A. // Molecul. Catalysis B: Enzymatic. 2001. № 11. C. 587–595.
Illanes A., Anjari S., Arrieta R., Aguirre C. // Appl. Biochem. Biotechnol. 2002. V. 97. № 3. P. 165–179.
Wei D.Z., Yang L. // Chem. Technol. Biotechnol. 2003. V. 78. № 4. P. 431–436.
Aguirre C., Toledo M., Medina V., Illanes A. // Process Biochem. 2002. V. 38. № 3. P. 351–360.
Illanes A., Cabrera Z., Wilson L., Aguirre C. // Process Biochem. 2003. V. 39. № 1. P. 111–117.
Illanes A., Anjari M.S., Altamirano C., Aguirre C. // J. Molecul. Catal. B: Enzymatic. 2004. V. 30. № 4. P. 95–103.
Hernández-Jústiz O., Terrenib M., Pagani G., J.L. Garcí A., Guisán J.M., Fernández–Lafuente R. // Enzyme Microb. Technol. 1999. V. 25. № 3–5. P. 336–343.
Terreni M., Ubiali D., Pagani G., Hernández-Jústiz O., Fernández–Lafuente R., Guisán J.M. // Enzyme Microb. Technol. 2005. V. 36. № 5–6. P. 672–679.
Дополнительные материалы отсутствуют.
Инструменты
Прикладная биохимия и микробиология