

Теплофизика высоких температур, 2019, T. 57, № 6, стр. 870-881
Включение кулоновского взаимодействия в схему модели погруженного атома. Система литий–свинец
Д. К. Белащенко *
Московский институт стали и сплавов
Москва, Россия
* E-mail: dkbel@mail.ru
Поступила в редакцию 22.06.2018
После доработки 05.05.2019
Принята к публикации 16.05.2019
Аннотация
Предложена схема включения экранированного кулоновского взаимодействия в модель погруженного атома, позволяющая описывать методом молекулярной динамики двух- и многокомпонентные растворы с сильным взаимодействием компонентов. Эффективные заряды частиц удовлетворяют условию электронейтральности и определяются путем минимизации полной энергии. При расчетах используются потенциалы чистых компонентов и подгоночные перекрестные парные потенциалы с учетом электронных вкладов в энергию и давление. Для пар 1–2 в растворах Li–Pb (1 – Li, 2 – Pb) предложен парный потенциал вида “8–4”. Выполнены расчеты для нескольких расплавов Li–Pb при нулевом давлении и температурах до 1000 К, а также для раствора Li17Pb83 в условиях ударного сжатия при температурах до 25 000 К и давлениях до 470 МПа. Термодинамические свойства раствора Li17Pb83 представлены в виде таблиц. Рассчитаны также диффузионные и структурные свойства этого и других растворов, коэффициенты Грюнайзена и адиабата Гюгонио.
ВВЕДЕНИЕ
В настоящее время расплавы системы Li–Pb привлекают интерес благодаря их использованию в ядерной энергетике. Свойства этих расплавов изучены довольно подробно [1–3]. Исследованы плотность этих расплавов [4–7], термодинамические свойства [4, 5, 7–12], структура [13–17], диффузионная подвижность [17–19] и широкий спектр физических свойств. Считается, что из-за большой разницы электроотрицательностей в расплавах происходит перенос электронного заряда с атомов лития на атомы свинца, так что связь становится частично ионной. Наиболее сильно этот эффект наблюдается при составе Li4Pb [16, 20–23].
Опубликовано значительное число работ по моделированию расплавов Li–Pb, в том числе методом ab initio [12, 22]. В первых работах применялись преимущественно методы теории жидкостей, использующие концепцию парного взаимодействия, а также методы молекулярной динамики и Монте-Карло с эффективными парными потенциалами [24], в том числе с учетом кулоновского взаимодействия [16, 25]. Этот подход практически непригоден для описания термодинамических свойств растворов. Более близкие к реальным результаты получены с применением потенциалов погруженного атома (Embedded atom model, ЕАМ) [26–28].
Однако все еще остается много неясного в поведении расплавов Li–Pb. Методами компьютерного моделирования недостаточно исследованы особенности, связанные с наличием вклада ионной связи. Концепция трансферабельности потенциала ЕАМ переносит все особенности взаимодействия компонентов на парный потенциал для пар 1–2 (1 – Li, 2 – Pb). Такая схема может хорошо работать в системах, близких к идеальности, но для систем с сильным взаимодействием компонентов (кулоновским или ковалентным) она может быть недостаточной.
В настоящей работе рассмотрены два варианта описания расплавов литий–свинец: 1) “трансферабельный” вариант и 2) вариант с явным включением кулоновского взаимодействия в схему модели погруженного атома. Для проверки трансферабельного варианта выполнено моделирование растворов Li–Pb с иным выбором потенциалов ЕАМ. Использовались (без изменения) потенциалы ЕАМ для лития [29–31] и свинца [31–33], разработанные специально для описания жидких металлов.
ТРАНСФЕРАБЕЛЬНЫЙ ВАРИАНТ
В этом варианте моделирования методом молекулярной динамики (МД) парные вклады в потенциал (для пар 1–1 и 2–2) и потенциалы погружения ЕАМ взяты такими же, как в случае соответствующего чистого компонента (так называемое свойство транферабельности), и дополнительно вводится только парный потенциал взаимодействия 1–2. Форма и параметры потенциала пар 1–2 подбираются с учетом термодинамических свойств двойной системы – обычно теплоты образования (смешения) и данных по плотности растворов. Концепция транферабельности представляется адекватной в случае систем, близких к идеальным, когда состояние частиц в растворе мало отличается от их состояния в однокомпонентной жидкости. Однако в случае систем с сильным взаимодействием компонентов (например, в растворах металлов IV–V групп с щелочными металлами и в растворах Al в 3d-металлах, когда связь перестает быть чисто металлической) такая трансферабельность может нарушаться.
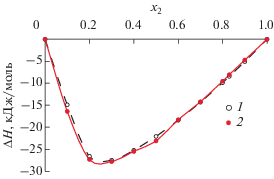
Растворы Li–Pb являются примером такой системы. Теплота смешения ΔHf расплавов Li–Pb измерена в [8, 9] (рис. 1). Довольно глубокий минимум изотермы ΔH1000 (около –27.5 кДж/моль) находится вблизи состава Li4Pb. “Трансферабельный” вариант ЕАМ был применен ранее для моделирования расплавов системы Li–Pb в работах [26–28]. В [27] удалось получить неплохое согласие значений ΔHf МД-моделей с экспериментальными данными по теплоте смешения [8, 9], но со сдвигом минимума от 80 до 60 ат. % лития (рис. 1). Неплохое согласие получено в [27] также для изотермы мольного объема (рис. 2), причем здесь минимум находится на правильном месте (80% лития [6, 7]). Соответствует данным [6, 7] и коэффициент теплового расширения в области граничных растворов. В МД-работе [28] согласуется с экспериментом теплоемкость раствора Li17Pb83, но минимум изотермы теплоты смешения ΔHf расплавов сдвинут к 50% лития. Что касается структуры расплавов, то неплохое согласие с дифракционными данными [13, 14] по суммарному структурному фактору расплава Li17Pb83 получено в МД-работе [27] и умеренное согласие с экспериментом [15] в [28].


Для пар 1–2 (т.е. Li–Pb) в настоящей работе применяется потенциал 8–4:
Параметры потенциала ε и r0 подобраны по экспериментальным данным для теплоты образования растворов Li–Pb при 1000 К [8, 9] (рис. 1). Оптимальные значения параметров для пар 1–2 составляют: r0 = 2.553 Å и ε = 0.0880 эВ. Совокупность этих потенциалов для системы Li–Pb далее обозначена как ЕАМ-1. Близкие результаты получаются с потенциалом Морзе:
В трансферабельном варианте модели растворов Li–Pb имели размер 2000 или 16 000 атомов в основном кубе и релаксировались с потенциалом ЕАМ-1. Применялся алгоритм Верле в режимах NpT или NVT. Радиус обрыва взаимодействия равен 7.50 Å для пар 1–1 и 9.01 Å для пар 1–2 и 2–2. Радиус сферы Верле равен 10.5 или 17.5 Å.
Важное значение в методе молекулярной динамики имеет выбор шага по времени. В компьютерной системе расчетов используются обычно три основных единицы: длины L = 1 Å, энергии E = 1 эВ и массы m (масса атома, в данном случае – лития). Единица времени является здесь производной величиной и рассчитывается по выражению t0 = (mL2/E)1/2. При расчете t0 по средней массе атома лития (6.94) получается t0 = 2.682 × 10–14 с. В случае растворов Li–Pb массы атомов компонентов отличаются более чем в 29 раз, что усложняет расчеты, поскольку обычная длина шага в алгоритме Верле (Δt ≈ 0.01t0) может оказаться неподходящей для одного из компонентов. Поэтому здесь длина шага выбрана из условия, по которому при прогоне длиной 1000 шагов в режиме NVE (т.е. в режиме изолированной системы) энергия модели менялась не более, чем в пятом–шестом знаке (в пределах 0.004% за 1 пс). При расчете t0 по средней массе атома лития оптимальная величина Δt для растворов Li–Pb равна 0.05t0 = 1.341 фс.
Результаты расчетов энергии и теплоты образования ΔUMD моделей растворов Li–Pb с потенциалом ЕАМ-1 показаны на рис. 1 и в табл. 1. В таблице приведены экспериментальные значения теплоты образования ΔHf [9] (вторая колонка) и мольного объема растворов Li–Pb [6] (третья колонка). В четвертой колонке приведены значения энергии моделей UMD с потенциалом ЕАМ-1 при 1000 К и нулевом давлении, в пятой – теплота образования ΔH1000 = ΔU1000 моделей раствора (разность между энергией раствора и аддитивной величиной, рассчитанной по энергии чистых компонентов). При 1000 К электронные поправки к энергии и давлению (см. ниже) невелики и ими можно пренебречь. На рис. 2 показаны экспериментальные и расчетные мольные объемы сплавов Li–Pb при 1000 К. Настоящие данные с потенциалом ЕАМ-1 (линия 3) в целом близки к результатам МД-расчетов [27] и согласуются с экспериментальными данными [6] при концентрациях X1 ≤ 0.6. При X1 > 0.6 расхождения с экспериментом увеличиваются. Расчетные объемы чистых компонентов практически совпадают с экспериментальными данными (14.96 см3/моль для Li [34] и 20.39 для Pb [35]).
Таблица 1.
Термодинамические данные моделей растворов Li–Pb при 1000 К (λ = 1.1, ε = 2.553, r00 = 0.0880)
| X1 | ΔHf, кДж/моль [9] | V, см3/моль [6] |
UMD, кДж/моль | ΔHMD, кДж/моль | V, см3/моль | $q_{{\text{1}}}^{*}$ | UMD, кДж/моль | UCoul, кДж/моль | UTrans, кДж/моль | ΔHMD, кДж/моль | d, г/см3 | V, см3/моль |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| без учета кулоновского взаимодействия | с учетом кулоновского взаимодействия | |||||||||||
| 1.0 | 0 | 15.52 | –131.15 | 0 | 14.96 | – | –131.15 | – | – | 0.00 | 0.4639 | 14.96 |
| 0.9 | –15.47 | 13.97 | –143.18 | –8.66 | 14.64 | 0.11 | –150.94 | –1.21 | –5.52 | –16.4 | 1.913 | 14.10 |
| 0.8 | –27.59 | 13.96 | –153.65 | –15.75 | 14.54 | 0.13 | –165.16 | –1.24 | –8.27 | –27.3 | 3.402 | 13.81 |
| 0.7 | –28.35 | 13.96 | –161.60 | –20.32 | 14.72 | 0.141 | –169.12 | –1.05 | –3.99 | –27.8 | 4.796 | 13.96 |
| 0.6 | –25.85 | 14.75 | –166.80 | –22.15 | 15.15 | 0.0926 | –170.19 | –0.30 | –1.62 | –25.5 | 5.906 | 14.74 |
| 0.5 | –22.48 | 15.43 | –169.33 | –21.30 | 15.80 | 0.10 | –171.14 | –0.13 | –0.74 | –23.1 | 6.895 | 15.53 |
| 0.4 | –18.66 | 16.74 | –169.67 | –18.27 | 16.60 | 0.00 | –169.70 | 0 | 0 | –18.3 | 7.660 | 16.59 |
| 0.3 | –14.49 | 17.62 | –168.81 | –14.04 | 17.50 | 0.00 | –168.94 | 0 | 0 | –14.2 | 8.419 | 17.47 |
| 0.2 | –9.99 | 18.60 | –167.67 | –9.52 | 18.42 | 0.00 | –167.67 | 0 | 0 | –9.52 | 9.074 | 18.42 |
| 0.17 | –8.43a | 18.74a | –167.16 | –8.00 | 18.72 | 0.00 | –167.21 | 0 | 0 | –8.05 | 9.250 | 18.72 |
| 0.1 | –5.16 | 19.45 | –166.20 | –4.68 | 19.39 | 0.00 | –166.24 | 0 | 0 | –4.71 | 9.641 | 19.41 |
| 0 | 0 | 20.45 | –164.90 | 0 | 20.37 | 0.00 | –164.90 | 0 | 0 | 0.00 | 10.16 | 20.37 |
Расхождение с экспериментом при больших концентрациях лития обусловлено недостаточной точностью приближения трансферабельности. Ни один из упомянутых выше потенциалов ЕАМ не дает реально наблюдаемого минимума изотермы теплоты смешения при составе Li4Pb [8, 9].
На рис. 3 показана зависимость плотности d расплава Li17Pb83 от температуры по разным данным. Расхождения результатов расчета плотности модели с потенциалом ЕАМ-1 и экспериментальных данных [5] невелики и составляют 0.6–2.6%. Модели расплава Li17Pb83 устойчивы при давлении p ≈ 0 до температуры немного выше 2000 К. Плотность модели с потенциалом работы [28] занижена.

При высокотемпературных расчетах следует учитывать электронный вклад EeT в энергию и давление, рассчитываемый по методу [30, 33]. Для расплава Li17Pb83 он увеличивается от нуля при 298 К до 3.129 кДж/моль при 2000 К. На рис. 4 показана зависимость энергии этой модели от температуры, которая хорошо описывается выражением
(1)
$\begin{gathered} {{U}_{{{\text{MD}}}}} + {{E}_{{eT}}} = 1.1398 \times {{10}^{{--10}}}{{T}^{3}} + \\ + \,\,5.9003 \times {{10}^{{--7}}}{{T}^{2}} + 0.027274T--194.30. \\ \end{gathered} $Рис. 4.
Зависимость энергии раствора Li17Pb83 от температуры: 1 – данные [28], 2 – авторские данные с потенциалом ЕАМ-1 (метод МД).

Данные [28] располагаются немного ниже и почти параллельно кривой, полученной по (1). Дифференцированием (1) получается теплоемкость Cp = 3.4194 × 10–7T 2 + 1.1801 × 10–3T + 27.274. Она немного возрастает с температурой (от 27.95 Дж/(моль К) при 500 К до 31.00 при 2000 К). Эксперимент показывает, что теплоемкость расплава Li17Pb83 слабо убывает от 32.9 Дж/(моль К) при 513 К до 32.8 при 623 К [1, 2]. Эти цифры выше примерно на 2–4 Дж/(моль К), чем полученные МД-данные. В МД-работе [27] теплоемкость монотонно убывает при нагревании от 37 Дж/(моль К) при 600 К до 30 при 1200 К (электронный вклад не учитывался). В [28] значения теплоемкости монотонно убывают от 34.1 Дж/(моль К) при 600 К до 30.2 при 1000 К.
В целом данные МД-моделирования с потенциалами ЕАМ [27, 28] и ЕАМ-1 показывают, что трансферабельный вариант приводит к значительным расхождениям с экспериментом для расплавов, богатых литием, в частности к смещению минимума изотермы теплоты смешения от состава Li80Pb20 к эквиатомному составу. В обычном потенциале ЕАМ не заложено инструментов, позволяющих варьировать положение минимума теплоты смешения двойных систем.
ЕАМ С УЧЕТОМ КУЛОНОВСКОГО ВЗАИМОДЕЙСТВИЯ
В работах [36, 37] структура растворов литий–свинец рассмотрена в различных вариантах теории жидкостей в приближении парного взаимодействия с межчастичными экранированными кулоновскими потенциалами (потенциалами Юкавы) вида
(2)
${{\varphi }_{{ij}}}(r) = ({{{{a}_{{ij}}}} \mathord{\left/ {\vphantom {{{{a}_{{ij}}}} r}} \right. \kern-0em} r}){\text{exp}}(--\lambda r),$Концепцию включения кулоновского взаимодействия в схему ЕАМ можно сформулировать следующим образом.
1) В результате частичного перетекания электронного заряда из ячеек Вигнера–Зейтца (полиэдров Вороного) менее электроотрицательного компонента (лития) в ячейки более электроотрицательного компонента (свинца) возникают избыточные эффективные заряды этих ячеек qi, которые экранируются электронами проводимости.
2) Избыточные заряды ионов компонентов q1 и q2 удовлетворяют условию электронейтральности: q1X1 + q2X2 = 0 (Xi – атомные доли компонентов).
3) Экранированный электрический потенциал i-го иона имеет вид (qi/r) exp(–λr), является короткодействующим, и поэтому при расчетах кулоновского взаимодействия не требуется применять процедуру Эвальда.
4) При переходе части электронов данного атома в состояние экранированного заряда qi соответствующая доля электронов изымается из эффективной электронной плотности схемы ЕАМ ψi(r) = p1i exp(–p2ir) или добавляется к ней, т.е. p1i = $p_{{{\text{1}}i}}^{0}$(1 – ${{{{q}_{i}}} \mathord{\left/ {\vphantom {{{{q}_{i}}} {q_{i}^{0}}}} \right. \kern-0em} {q_{i}^{0}}}$), где $q_{i}^{0}$ = 1 для лития и 4 для свинца. Впрочем, этот эффект очень слаб и его можно не учитывать.
5) Равновесные значения зарядов ионов определяются конкуренцией между кулоновской энергией раствора ECoul (ECoul < 0) и энергией перезарядки ионов ETrans (ETrans > 0). Изменение энергии при перезарядке i-го иона ΔEi может быть задано функцией, которая при qi = 1 принимает значение первого потенциала ионизации Ii, а при qi = –1 равна энергии сродства к электрону со знаком минус (–Qi). Кроме того, ΔEi = 0 при qi = 0. Можно, например, выбрать функцию ΔEi в виде
6) Эффективные заряды ионов qi рассчитываются как величины, минимизирующие полную энергию модели при заданных давлении и температуре. Оптимальные заряды $q_{i}^{*}$ получаются путем коррекции минимизирующих зарядов для лучшего согласия плотности модели с экспериментальными данными.
7) Экранированный кулоновский потенциал и кулоновские межчастичные силы добавляются к потенциалам и силам, рассчитываемым обычным образом на основе ЕАМ-1. Добавка ETrans на межчастичные силы не влияет.
Далее такой гибридный потенциал обозначен как ЕАМ-2.
Расчет энергии перезарядки ETrans выходит за рамки метода МД и является предметом теории металлов. Чтобы оставаться в пределах метода компьютерного моделирования, следует параметризовать вклад ETrans, выразив его через значения зарядов ионов. Оптимальное выражение для ETrans получено (с учетом п. 2) сравнением энергии моделей и реальных растворов в виде
В варианте с включением кулоновского взаимодействия модели растворов Li–Pb имели размер 2000 или 16000 атомов в основном кубе и релаксировались с гибридным потенциалом ЕАМ-2, равным сумме потенциала ЕАМ-1 и экранированного кулоновского потенциала (2), в котором aij = 14.399$q_{i}^{*}q_{j}^{*}$ (14.399 – коэффициент пересчета из компьютерной системы единиц в СГС). Оптимальный параметр экранирования оказался равным 1.10, т.е. таким же, как значение, найденное в [16]. В режимах NpT или NVT применялся алгоритм Верле. Радиус обрыва взаимодействия равен 7.50 Å для пар 1–1 и 9.01 Å для пар 1–2 и 2–2. Радиус сферы Верле составляет 17.5 Å.
Для каждой исследованной концентрации растворов Li–Pb выполнена релаксация моделей с различными допустимыми эффективными зарядами 0 < $q_{{\text{1}}}^{*}$ < 1 и –1 < $q_{{\text{2}}}^{*}$ < 0 (с учетом п. 2). Минимизирующие заряды найдены из условия минимума энергии модели при температуре 1000 К и нулевом давлении, а оптимальные заряды определены с учетом реального значения плотности раствора [6]. Минимизирующие и оптимальные значения зарядов приведены в табл. 2. Они значительно меньше найденных в [16] (0.5 и –2.0), однако значения в [16] приводят к неприемлемым расхождениям с экспериментом по плотности расплавов. При концентрациях лития менее 50% учет кулоновского взаимодействия не требуется, так как минимизирующие заряды близки к нулю.
Таблица 2.
Минимизирующие qi и оптимальные $q_{i}^{*}$ заряды ионов в расплавах Li–Pb при 1000 К
| X1 | q1 | $q_{1}^{*}$ | $ - q_{2}^{*}$ |
|---|---|---|---|
| 0.9 | 0.11 | 0.11 | 0.99 |
| 0.8 | 0.15 | 0.13 | 0.52 |
| 0.7 | 0.141 | 0.141 | 0.329 |
| 0.6 | 0.10 | 0.0926 | 0.139 |
| 0.5 | 0.12 | 0.075 | 0.075 |
| 0.4 | 0.00 | 0.00 | 0.00 |
| 0.3 | 0.00 | 0.00 | 0.00 |
| 0.2 | 0.00 | 0.00 | 0.00 |
| 0.1 | 0.00 | 0.00 | 0.00 |
Релаксация моделей проведена с включением зарядов $q_{i}^{*}.$ Полученные значения расчетной теплоты смешения показаны на рис. 5 и в табл. 1. Включение кулоновского взаимодействия позволяет значительно улучшить согласие расчетной теплоты смешения с экспериментальными данными (колонки 2 и 11). На рис. 2 и в табл. 1 (колонки 3 и 13) показаны значения мольного объема растворов при 1000 К. Отсюда также видно, что включение кулоновского взаимодействия значительно улучшает согласие с экспериментом [6].
Рис. 5.
Изотерма теплоты смешения расплавов Li–Pb при 1000 К: 1 – данные [9], 2 –авторские данные (МД) с учетом кулоновского взаимодействия.
Несколько хуже выглядят результаты для производных свойств. С использованием потенциала ЕАМ-2 рассчитаны теплоемкости CV и Cp, а также изотермический модуль всестороннего сжатия KT моделей при 1000 К. Результаты приведены в табл. 3. Прямые измерения теплоемкостей и модуля, видимо, не проводились. Расчетные значения Cp в [27] (метод МД) располагаются выше настоящих данных примерно на 5–6 Дж/(моль К). Полученная в [28] методом МД величина 30.3 Дж/(моль К) для раствора Li17Pb83 при 1000 К заметно выше полученного здесь значения 25.8.
Таблица 3.
Свойства моделей Li–Pb с потенциалом ЕАМ-2 при 1000 К
| X1 | CV, Дж/(моль К) EAM |
Cp, Дж/(моль К) | KT, ГПа, ЕАМ | Ks, ГПа [6]b | us, м/с | α × 105, К–1 | (∂p/∂T)V, МПа | γ | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ЕАМ | [27] | ЕАМ | [6]b | 723 К [38] | ЕАМ | [6]b | ||||||
| 1.0 | 21.9 | 27.16 | 28.8 [34] | 8.28 | – | 4694 | 4195 [34] | – | 21.2 | 20.1 [34] | 1.848 | 1.26 |
| 0.9 | 22.3 | 27.0 | 29.1 | 9.58 | 10.8 | 2465 | 2278 | – | 17.4 | 14.5 | 1.863 | 1.18 |
| 0.8 | 22.5 | 29.2 | 38.2 | 12.45 | – | 2177 | – | – | 17.9 | 21.5 | 2.080 | 1.28 |
| 0.7 | 22.6 | 30.3 | 40.0 | 18.03 | – | 2130 | – | – | 16.6 | 15.0 | 2.583 | 1.60 |
| 0.6 | 22.5 | 28.2 | 42.0 | 18.71 | 20.0 | 2002 | – | 1820 | 13.3 | 11.9 | 2.717 | 1.89 |
| 0.5 | 21.2 | 28.5 | 39.4 | 21.62 | 21.7 | 2053 | 1704 | 1770 | 13.3 | 10.4 | 2.744 | 2.00 |
| 0.4 | 21.0 | 27.7 | 38.0 | 23.89 | 24.7 | 2002 | 1676 | 1740 | 12.5 | 10.0 | 2.793 | 2.21 |
| 0.3 | 20.1 | 26.6 | 35.2 | 22.96 | 26.6 | 1902 | 1664 | 1740 | 11.6 | 9.3 | 2.603 | 2.26 |
| 0.2 | 20.3 | 26.3 | 33.6 | 22.61 | – | 1798 | – | – | 12.3 | 7.8 | 2.805 | 2.54 |
| 0.17 | 20.4 | 25.8 | 32.2 [1, 2, 39]b | 23.51 | – | 1793 | – | 1730 | 11.2 | 16.1 [40] | 2.860 | 2.62 |
| 0.1 | 20.2 | 26.5 | 31.2 | 21.70 | – | 1720 | – | 1740 | 12.3 | – | 2.664 | 2.56 |
| 0.0 | 20.5 | 29.1 | 30.0 | 19.5 | 27.0 | 1652 | 1708 1658a | 1775 | 12.8 | 13.3 | 2.936 | 2.93 |
a [41]. b Экстраполяция.
В табл. 3 приведены расчетные значения скорости звука в моделях при 1000 К в сравнении с экспериментальными данными [6, 38]. МД-данные выше экспериментальных при 30–60% Li в среднем на ∼10%, но убывают с приближением к чистым компонентам. Расчеты с потенциалом Морзе дают примерно то же самое, а потенциал Леннард-Джонса приводит к еще более высоким скоростям звука. Данные [6], возможно, занижены. Неплохо согласуются с экспериментом [6] значения коэффициента теплового расширения α. Производная давления (∂p/∂T)V зависит от концентрации с положительными отклонениями от аддитивности, а коэффициент Грюнайзена γ = = (V/CV)(∂p/∂T)V меняется почти аддитивно.
Структура. Структура расплавов исследована в [13, 14] методом рассеяния нейтронов в интервале температур 995–1225 К с использованием изотопа 7Li. На рис. 6 показаны парциальные парные корреляционные функции (ППКФ) модели расплава Li17Pb83 при 600 К. Координаты пиков ППКФ равны 5.05, 2.90 и 3.23 Å для пар 1–1, 1–2 и 2–2 соответственно. Первый пик для пар 1–2 с высотой 2.63 ниже пика для пар 2–2, но расположен левее. При температуре 1000 К координаты пиков ППКФ равны 4.92, 2.87 и 3.19 Å для пар 1–1, 1–2 и 2–2 при таких же соотношениях высот пиков (1.26, 2.24 и 2.59). Это указывает на слабую тенденцию к преимущественному окружению атомов частицами противоположного сорта.

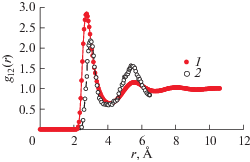
Расплавы Li–Pb моделировались также в [27, 28]. В [28] высота пика ППКФ для пар 1–2 модели Li17Pb83 при 600 К больше, чем в настоящей работе: 3.38 вместо 2.63, хотя координаты пиков в основном совпадают (2.93 Å) (рис. 6). Высоты пиков ППКФ моделей Li–Pb в [27] почти не зависят от состава и равны 2.2–2.4 при 1000 К, т.е. близки к полученным в настоящей работе. На рис. 7 показаны ППКФ для пар 1–2 в модели расплава Li80Pb20 при 1000 К. Учет кулоновского вклада в ЕАМ-2 приводит к небольшому росту высоты первого пика ППКФ g12(r) при 1000 К: от 2.68 при q1 = 0 до 2.75 при q1 = 0.13. Высота пика ППКФ пар 1–2 при 1000 К в модели Li80Pb20, полученная в [27], равна 2.18 (рис. 7). Эти различия обусловлены различной жесткостью потенциала пар 1–2: в [28] он более жесткий, а в [27] менее жесткий, чем в ЕАМ-2.
Рис. 7.
ППКФ пар 1–2 в расплаве с 80 ат. % Li при 1000 К: 1 – данные [27], 2 – авторские данные с учетом кулоновского взаимодействия.
На рис. 8 показаны зависимости от концентрации координат r1 и высот g1(r) первых пиков ППКФ для пар 1–2 в рассматриваемых моделях. Координата растет с увеличением концентрации свинца, а высота пика проходит через максимум при 60–80 ат. % Li, т.е. в интервале концентраций с наибольшим кулоновским вкладом.
Рис. 8.
Зависимость формы первого пика ППКФ пар 1–2 от концентрации расплавов Li–Pb с потенциалом ЕАМ-2 с учетом кулоновского взаимодействия при 1000 К: 1 – координата r1(12), 2 – высота g1(12).

В работах [13, 14] рассчитана функция
На рис. 9 приведена функция 4πr2n0ρCC(r) (n0 – число частиц в единице объема) для модели расплава Li4Pb при 1073 К в сравнении с данными [13, 14]. Согласие можно считать удовлетворительным только для первого минимума. Осциллирующая форма зависимости означает гетерокоординацию частиц, слоистость ближнего порядка вокруг частиц расплава и может являться признаком наличия вклада ионной связи, при которой чередуются слои частиц с зарядами разного знака. В МД-модели данной работы гетерокоординация быстро затухает с расстоянием из-за сильного экранирования кулоновского взаимодействия. В [27] наличие гетерокоординации вообще не отмечено.

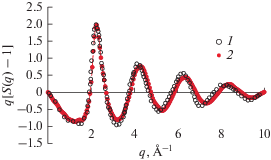
На рис. 10 показана функция q[S(q) – 1], где q и S(q) – вектор рассеяния и структурный фактор рассеяния нейтронов на расплаве 7Li17Pb83 при 773 К в сравнении с дифракционными данными [13]. В настоящих расчетах амплитуды рассеяния на частицах 7Li и Pb приняты равными –1.90 и 7.60. Согласие с данными [13] здесь очень хорошее, за исключением небольшого смещения 2–4 пиков вправо.
Рис. 10.
Функция q[S(q) – 1] расплава Li17Pb83 при 773 K: 1 – дифракционные (нейтронные) данные [13], 2 – МД-модель.
Диффузия. Коэффициенты самодиффузии $D_{i}^{0}$ в равновесных компьютерных моделях с потенциалом ЕАМ-2 рассчитывались по уравнению 〈Δr2〉 = 6Dt, где 〈Δr2〉 – средний квадрат смещения частиц за время t. В табл. 4 приведены коэффициенты самодиффузии компонентов 105D в см2/с при температурах до 2000 К. С увеличением концентрации лития скорость диффузии возрастает. На рис. 11, 12 показаны зависимости D(T) в логарифмических координатах для лития и свинца. Коэффициенты для прямых линий lg D = a1 lg T + a2 имеют следующие значения:
Таблица 4.
Коэффициенты самодиффузии 105D в моделях Li–Pb, см2/с
| T, К | Li17Pb83 | Li50Pb50 | Li80Pb20 | |||
|---|---|---|---|---|---|---|
| Li | Pb | Li | Pb | Li | Pb | |
| 600 | 4.12 | 3.02 | – | – | – | – |
| 800 | 7.32 | 4.39 | 6.88 | 5.42 | – | – |
| 1000 | 9.18 | 5.94 | 10.5 | 7.72 | 20.9 | 13.5 |
| 1200 | 17.3 | 8.62 | 15.3 | 10.6 | 29.5 | 17.8 |
| 1500 | 24.9 | 10.7 | 26.4 | 16.5 | 45.5 | 25.4 |
| 2000 | 47.2 | 18.0 | 49.7 | 23.8 | 76.4 | 39.3 |
Рис. 11.
Коэффициенты самодиффузии лития в моделях расплавов Li–Pb: 1 – Li17Pb83 и Li50Pb50, 2 – Li80Pb20.

Рис. 12.
Коэффициенты самодиффузии свинца в моделях расплавов Li–Pb: 1 – Li17Pb83, 2 – Li50Pb50, 3 – Li80Pb20.

Спрямление этих зависимостей означает, что температурная зависимость коэффициентов самодиффузии хорошо описывается выражением D = сT b. Такая форма зависимости выполняется вдоль изобар многих металлов [31]. Коэффициенты самодиффузии лития в расплаве Li17Pb83, полученные в [27], в 2–4 раза ниже приведенных в табл. 4. Коэффициенты, рассчитанные в [28], близки к полученным результатам при температурах выше 800 К, а при меньших температурах занижены.
Отметим, что графики расчетных коэффициентов самодиффузии из табл. 4 не спрямляются в координатах ln D–1/T и имеют вид парабол с положительной кривизной. Для приближенных оценок аппроксимация D = D0 exp(–Eact/RT) дает следующие величины:
| Расплав | D0(Li) × 104, см2/с | Eact(Li), Дж/моль | D0(Pb) × 104, см2/с | Eact(Pb), Дж/моль |
| Li17Pb83 | 9.88 | 16 880 | 3.07 | 12 300 |
| Li50Pb50 | 15.8 | 21 800 | 6.15 | 16 680 |
| Li80Pb20 | 26.8 | 21 560 | 11.0 | 17 760 |
Свойства расплавов Li–Pb в условиях ударного сжатия. Потенциал ЕАМ-2 позволяет рассчитывать свойства расплавов при высоких температурах и давлениях. Для чистых компонентов Li и Pb такие расчеты выполнены в [29–33] при температурах до 14 000 К (Li) и 25 000 К (Pb) и давлениях до 100 ГПа (Li) и 280 ГПа (Pb). Проведем аналогичные расчеты для раствора Li17Pb83. С этой целью следует найти зависимости энергии и давления раствора от объема и температуры до степеней сжатия Y = V/V0 = 0.4–0.5, где V – мольный объем в сжатом состоянии, а V0 – нормальный объем в исходном состоянии. При обычных условиях сплав со средним составом Li17Pb83 включает в себя две фазы: LiPb и почти чистый свинец. Для упрощения расчетов в качестве исходного состояния при 298 К принята аморфная фаза состава Li17Pb83. Эта модель имеет плотность 10.096 г/см3 (V0 = 17.152 см3/моль, аддитивный объем – 20.29) и энергию –186.79 кДж/моль (аддитивная энергия –181.5). Фактическая плотность сплава Li17Pb83 при 298 К равна 10.22 г/см3 [40]. При расчетах электронных вкладов в энергию Eel и давление pel применена методика из [29–31]. Электронные вклады в энергию рассчитаны при средней электронной плотности 3.49 эл/ат. Они приведены в табл. 5. Электронные вклады в давление рассчитаны по формуле pelV = (2/3)Eel.
Таблица 5.
Электронные вклады в энергию моделей Li17Pb83 (V0 = 17.152 см3/моль)
| T, К | Y = V/V0 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1.0 | 0.9 | 0.8 | 0.7 | 0.6 | 0.5 | 0.45 | 0.4 | 0.35 | 0.3 | |
| энергия, кДж/моль | ||||||||||
| 298 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 500 | 0.068 | 0.064 | 0.06 | 0.056 | 0.052 | 0.047 | 0.043 | 0.038 | 0.035 | 0.033 |
| 1000 | 0.17 | 0.160 | 0.150 | 0.140 | 0.130 | 0.118 | 0.107 | 0.095 | 0.088 | 0.082 |
| 2000 | 0.386 | 0.364 | 0.342 | 0.319 | 0.295 | 0.270 | 0.243 | 0.215 | 0.201 | 0.186 |
| 3000 | 0.915 | 0.864 | 0.811 | 0.756 | 0.699 | 0.639 | 0.577 | 0.511 | 0.477 | 0.440 |
| 5000 | 1.655 | 1.562 | 1.466 | 1.367 | 1.264 | 1.157 | 1.044 | 0.925 | 0.862 | 0.797 |
| 10 000 | 2.603 | 2.458 | 2.307 | 2.152 | 1.990 | 1.821 | 1.644 | 1.456 | 1.358 | 1.256 |
| 15 000 | 3.759 | 3.549 | 3.333 | 3.109 | 2.876 | 2.632 | 2.376 | 2.105 | 1.963 | 1.815 |
| 20 000 | 5.119 | 4.835 | 4.541 | 4.237 | 3.920 | 3.589 | 3.241 | 2.872 | 2.678 | 2.476 |
| 25 000 | 6.68 | 6.311 | 5.930 | 5.534 | 5.121 | 4.690 | 4.236 | 3.754 | 3.501 | 3.238 |
Далее построены модели некристаллического сплава Li17Pb83 при различных температурах и объемах с использованием потенциала ЕАМ-2. Заряды ионов считались не зависящими от температуры. Значения энергии и давления этих моделей с добавлением электронных поправок приведены в табл. 6 и 7, с помощью которых можно рассчитать ударную адиабату (рис. 13) методом, описанным в [29–33]. До давления 100 ГПа она почти совпадает с адиабатой чистого свинца, а при более высоких давлениях уходит вверх. В верхней точке адиабаты сплава температура достигает 21 400 К. На адиабате свинца при той же степени сжатия (Y = 0.55) температура составляет ∼14 200 К.
Таблица 6.
Энергия U моделей Li17Pb83 с учетом электронной поправки (V0 = 17.152 см3/моль)
| T, К | Y = V/V0 | ||||||
|---|---|---|---|---|---|---|---|
| 1.00 | 0.90 | 0.80 | 0.70 | 0.60 | 0.50 | 0.45 | |
| U, кДж/моль | |||||||
| 298 | –186.79 | –185.30 | –167.84 | –131.82 | –45.13 | 141.14 | 307.09 |
| 500 | –181.68 | –179.97 | –162.41 | –126.44 | –39.92 | 146.05 | 312.16 |
| 1000 | –169.71 | –166.10 | –148.25 | –112.38 | –26.64 | 158.60 | 326.40 |
| 2000 | –146.55 | –140.36 | –120.09 | –82.24 | 1.33 | 182.85 | 349.44 |
| 3000 | –124.05 | –116.25 | –94.50 | –55.41 | 29.20 | 209.63 | 377.07 |
| 5000 | –77.99 | –68.30 | –45.84 | –4.31 | 82.38 | 269.60 | 433.63 |
| 10 000 | 53.43 | 63.51 | 86.56 | 129.78 | 220.74 | 414.35 | 590.70 |
| 15 000 | 212.19 | 220.27 | 241.35 | 283.32 | 376.88 | 578.10 | 762.26 |
| 20 000 | 395.21 | 400.49 | 418.61 | 458.97 | 551.58 | 757.18 | 947.34 |
| 25 000 | 598.96 | 599.68 | 615.06 | 652.30 | 743.53 | 951.64 | 1146.55 |
Таблица 7.
Давление p моделей Li17Pb83 с учетом электронной поправки (V0 = 17.152 см3/моль)
| T, К | Y = V/V0 | ||||||
|---|---|---|---|---|---|---|---|
| 1.00 | 0.90 | 0.80 | 0.70 | 0.60 | 0.50 | 0.45 | |
| p, ГПа | |||||||
| 298 | 0.00 | 6.63 | 16.28 | 32.38 | 75.23 | 158.38 | 243.72 |
| 500 | 1.03 | 7.63 | 17.09 | 33.17 | 75.84 | 159.86 | 246.90 |
| 1000 | 3.02 | 10.03 | 19.16 | 35.29 | 77.64 | 163.90 | 254.41 |
| 2000 | 5.97 | 13.29 | 22.78 | 40.06 | 82.32 | 171.32 | 264.86 |
| 3000 | 8.37 | 15.88 | 25.88 | 44.31 | 87.69 | 179.77 | 277.01 |
| 5000 | 12.46 | 20.49 | 31.37 | 52.13 | 98.10 | 197.00 | 298.49 |
| 10 000 | 21.91 | 31.20 | 44.74 | 69.85 | 122.20 | 234.90 | 348.46 |
| 15 000 | 31.55 | 42.30 | 58.14 | 86.70 | 144.67 | 268.85 | 391.60 |
| 20 000 | 41.69 | 53.89 | 71.91 | 103.82 | 166.75 | 300.25 | 430.94 |
| 25 000 | 52.49 | 65.86 | 86.18 | 121.19 | 188.75 | 330.80 | 467.83 |

ОБСУЖДЕНИЕ
Выбором потенциалов ЕАМ для пар 1–1 и 2–2 и потенциала “8–4” для пар 1–2, а также включением экранированного кулоновского взаимодействия компонентов удалось получить хорошее согласие ряда термодинамических свойств моделей системы Li–Pb (плотности, энергии, коэффициента теплового расширения, модуля всестороннего сжатия, скорости звука) с опытными данными при температурах до 1000 К. Получено также согласие с дифракционными данными о структуре этих расплавов при 600–1073 К. Дополнительного улучшения согласия с экспериментом по упругим свойствам расплавов можно достичь, немного уменьшая степень жесткости парного потенциала для пар 1–2, но не допуская сближения частиц в этих парах ближе 1.9–2.0 Å. Применение потенциала Леннард-Джонса для пар 1–2 способствует росту модуля всестороннего сжатия моделей, а мягкий сплайновый потенциал пар 1–2 приводит к слишком тесному сближению частиц и к расхождению с дифракционными данными о структуре.
Структурные характеристики расплава Li4Pb рассчитаны в [16] на основе средне-сферического и гиперцепного приближений теории жидкостей, разработанных для систем с парным потенциалом. В расчетах [16] использованы дифракционные данные [13, 14]. Первый максимум ППКФ пар 1–2 в [16] при 1075 К с высотой около 2.6 расположен при r = 2.6 Å, а в случае 1073 К (рис. 7) он имеет высоту 2.75 и находится при r = 2.70 Å. Для пар 1–1 получено согласие и по положению, и по высоте пика. Однако для пар 2–2 расхождения гораздо больше: r = 4.8 Å в [16] и 5.00 Å (данная работа), а высоты этих пиков составляют соответственно 1.67 и 1.30. Эти расхождения указывают на недостаточную адекватность приближения парного взаимодействия при описании систем с сильным взаимодействием компонентов.
Значительные расхождения получены и с результатами МД-расчетов [27]. Так, при 1000 К первый пик ППКФ пар 1–2 расплава Li4Pb в [27] имеет высоту 2.00 и расположен при r = 3.00 Å, в данной работе: высота – 2.85 и r = 2.75 Å. Второй пик этой ППКФ в [27] расположен при r = 5.46 Å (высота – 1.45) и r = 5.57 Å (высота – 1.17) соответственно. Эта разница обусловлена различием потенциалов.
Ударная адиабата сплава Li17Pb83 очень близка к адиабате чистого свинца при давлениях до 100 ГПа, а при бóльших давлениях идет выше последней. Следовательно, нельзя рассчитать адиабату сплава, используя только адиабаты чистых компонентов, без расчетов межчастичных потенциалов. Для проверки адекватности потенциала ЕАМ-2 полезно построить экспериментально адиабату Гюгонио реального сплава Li17Pb83 или любого другого.
Предложенный в настоящей работе метод сочетания потенциалов ЕАМ и экранированного кулоновского потенциала позволяет распространить модель погруженного атома на системы с сильным взаимодействием компонентов, когда трансферабельность потенциалов ЕАМ сама по себе недостаточна.
ЗАКЛЮЧЕНИЕ
Построены две серии моделей жидких растворов Li–Pb: 1) в трансферабельном варианте с потенциалами ЕАМ и 2) с дополнительным включением экранированного кулоновского потенциала. Первый вариант хорошо описывает свойства растворов при содержании лития меньше 60 ат. %. Второй позволяет получить правильное положение минимумов на изотермах теплоты смешения и плотности, а также достичь согласия с опытом по совокупности термодинамических свойств и по структуре расплавов. Рассчитаны свойства раствора Li17Pb83 при температурах до 25 000 К и давлениях до 470 ГПа, и построена адиабата Гюгонио. Вариант с включением кулоновского взаимодействия расширяет набор используемых потенциалов и позволяет моделировать растворы с сильным взаимодействием компонентов. При использовании потенциалов ЕАМ без учета переноса заряда с менее электроотрицательного компонента на более электроотрицательный вряд ли можно добиться хорошего согласия расчетных свойств моделей растворов типа Li–Pb с экспериментальными данными.
Список литературы
Blanket, Shield Design, and Material Data Base. ITER Documentation Series. № 29. Vienna: IAEA, 1991.
Fusion Engineering and Design Journal. 1991. V. 14. № 3–4.
Nuclear Data for Science and Technology / Proc. Int. Conf. Ed. Bockhoff K.H. Antwerp. Belgium. 6–10 Sept. 1982.
Stankus S.V., Khairulin R.A., Mozgovoy A.G. et al. The Density and Thermal Expansion of Eutectic Alloys of Lead with Bismuth and Lithium in Condensed State // J. Phys.: Conf. Ser. 2008. V. 98. P. 062017.
Tiwari A., Allison B., Hohorst J.K. et al. Insertion of Lead Lithium Eutectic Mixture in RELAP/SCDAPSIM Mod 4.0 for Fusion Reactor Systems // Fusion Eng. Design. 2012. V. 87. P. 156.
Ruppersberg H., Speicher W. Density and Compressibility of Liquid Li–Pb Alloys // Z. Naturforsch. 1976. Bd. 31a. S. 47.
Saar J., Ruppersberg H. Calculation of Cp(T) for Liquid Lithium/Lead Alloys from Experimental ρ(T) and (dp/dT)s Data // J. Phys. F: Met. Phys. 1987. V. 17. P. 305.
Predel B., Oehme Z. Calorimetric Investigation of Liquid Li–Pb–alloys // Z. Metallkunde. 1979. Bd. 70. S. 450.
Terlicka S., Dębski A., Gąsior W. Thermodynamic Properties of Li–Pb System // J. Mol. Liquids. 2018. V. 249. P. 66.
Gąsior W., Moser Z. Thermodynamic Study of Liquid Lithium-lead Alloys Using the EMF Method // J. Nucl. Mater. 2001. V. 294. № 1–2. P. 77.
Becker W., Schwitzgebel G., Ruppersberg H. Thermodynamic Investigations of Liquid Lithium–Lead and Lithium–Silver Alloys – A Comparative Study // Z. Metallkunde. 1981. Bd. 72. № 3. S. 186.
Zhou Ch., Guo C., Li Ch., Du Zh. Thermodynamic Optimization of the Li–Pb System Aided by First-principles Calculations // J. Nucl. Mater. 2016. V. 477. P. 95.
Ruppersberg H., Egger H. Short-range Order in Liquid Li–Pb Alloys // J. Chem. Phys. 1975. V. 63. P. 4095.
Ruppersberg H., Reiter H. Chemical Short-range Order in Liquid LiPb Alloys // J. Phys. F. 1982. V. 12. P. 1311.
Mudry S., Shtablavyi I., Sklyarchuk V., Plevachuk Yu. Structure and Electrophysical Properties of Liquid Pb83Mg17 and Pb83Li17 Eutectics // J. Nucl. Mater. 2008. V. 376. № 3. P. 371.
Copestake A.P., Evans R., Ruppersberg H., Schirmacher W. A Model for the Structure of Liquid Li4Pb // J. Phys. F: Metal Phys. 1983. V. 13. № 10. P. 1993.
Soltwisch M., Quitmann D., Ruppersberg H., Suck J.B. Dynamical Concentration Fluctuations in Liquid Lithium–Lead (Li4Pb) // J. Phys. Colloq. 1980. V. C8. P. 167.
Schwitzgebel G., Langen G. Application of the Hard Sphere Theory to the Diffusion of Binary Liquid Alloy Systems // Z. Naturforsch. 1981. Bd. 36a. S. 1225.
Wang B., Xiao S., Gan X. et al. Diffusion Properties of Liquid Lithium–Lead Alloys from Atomistic Simulation // Comput. Mater. Sci. 2014. V. 93. P. 74.
Van der Marel C., Geertsma W., van der Lugt W. Lithium-7 Knight Shift of Liquid Lithium–Lead and Tin Alloys // J. Phys. F: Metal Phys. 1980. V. 10. № 10. P. 2305.
Holzhey Ch., Brouers F., Franz J.R., Schirmacher W. Self-consistent Study of Chemical Short-range Order and Charge Transfer in Liquid Alloys as a Function of Temperature // J. Non-Crystal. Solids. 1984. V. 61–62(1). P. 65.
Senda Y., Shimojo F., Hoshino K. The Ionic Structure and the Electronic States of Liquid Li–Pb Alloys Obtained from ab initio Molecular Dynamics Simulations // J. Phys.: Condens. Matter. 2000. V. 12. № 28. P. 6101.
Ruppersberg H., Schirmacher W. Ordering Potential in Liquid Lithium–Lead (Li4Pb) and Lithium–Silver (Li7Ag3) Calculated from Neutron Diffraction Data // J. Phys. F: Metal Phys. 1984. V. 14. № 12. P. 2787.
Jacucci G., Ronchetti M., Schirmacher W. Computer Simulation of the Liquid Lithium–Lead (Li4Pb) Alloy // J. Phys. Colloq. 1985. V. 46. P. 385.
Aniya M., Ginoza M. Screened Coulomb Model in a Uniform Background Charge for Temperature Dependence of the Structure of Liquid Lithium–Lead (Li4Pb) // J. Phys. Soc. Japan. 1987. V. 56. № 6. P. 2046.
Fraile A., Cuesta-López S., Iglesias R. et al. Atomistic Molecular Point of View for Liquid Lead and Lithium in Nuclear Fusion Technology // J. Nucl. Mater. 2013. V. 440. № 1–3. P. 98.
Fraile A., Cuesta-López S., Caro A. et al. Interatomic Potential for the Compound-forming Li–Pb Liquid Alloy // J. Nucl. Mater. 2014. V. 448. P. 103.
Gan X., Xiao Sh., Deng H. et al. Thermodynamic Properties of Li, Pb, and Li17Pb83 with Molecular Dynamics Simulations // Fusion Eng. Design. 2014. V. 89. P. 2946.
Белащенко Д.К., Островский О.И. Применение модели погруженного атома к жидким металлам. Жидкий литий // ТВТ. 2009. Т. 47. № 2. С. 231.
Белащенко Д.К. Гибридный потенциал межчастичного взаимодействия и расчет линии плавления лития методом молекулярной динамики // ТВТ. 2015. Т. 53. № 5. С. 683.
Belashchenko D.K. Liquid Metals. From Atomistic Potentials to Properties, Shock Compression, Earth’s Core, and Nanoclusters. Nova Science Publishers, 2018.
Belashchenko D.K. Computer Simulation of the Properties of Liquid Metals: Gallium, Lead, and Bismuth // Russ. J. Phys. Chem. 2012. V. 86. № 5. P. 779.
Белащенко Д.К. Расчет свойств жидких свинца и висмута в условиях ударного сжатия методом молекулярной динамики // ТВТ. 2017. Т. 55. № 3. С. 386.
Быстров П.И., Каган Д.Н., Кречетова Г.А., Шпильрайн Э.Э. Жидкометаллические теплоносители тепловых труб и энергетических установок. М.: Наука, 1988. 264 с.
Assael M.J., Kalyva A.E., Antoniadis K.D. et al. Reference Data for the Density and Viscosity of Liquid Antimony, Bismuth, Lead, Nickel, and Silver // High Temp.–High Pres. 2012. V. 41. P. 161.
Gallego L.J., Somoza J.A., Alonso J.A. On the Concentration Dependence of the Ordering Potential in Liquid Lithium–Lead Alloys // Phys. Chem. Liquids. 1987. V. 16. № 4. P. 249.
Gonzalez D.J., Silbert M. Ordering Potential and the Structural Properties of Binary Yukawa Mixtures // J. Phys. F: Metal Phys. 1988. V. 18. № 11. P. 2353.
Hoffman N.J., Darnell A., Blink J.A. Properties of Lead–Lithium Solutions // NLLL. Univ. California. Livermore, California. 1980. Preprint 9455L.
Brandt R., Schulz B. Specific Heat of Some Li–Compounds // J. Nucl. Mater. 1988. V. 152. № 2–3. P. 178.
Thermophysical Properties in the System Li–Pb // Kernforschungszentrum. Karlsruhe, 1986.
Филиппов С.И., Казаков Н.Б., Пронин Л.А. Скорость ультразвука, сжимаемость жидких металлов и их связь с различными физическими свойствами // Изв. вузов. Черная металлургия. 1966. № 3. С. 8.
Дополнительные материалы отсутствуют.
Инструменты
Теплофизика высоких температур