

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 4, стр. 253-268
Радикальная полимеризация стирола в присутствии динитронов различного строения
Е. В. Колякина a, А. Б. Алыева a, Е. В. Сазонова a, Е. А. Захарычев a, Д. Ф. Гришин a, *
a Национальный исследовательский Нижегородский государственный университет им. Н.И. Лобачевского
603950 Нижний Новгород, пр. Гагарина, 23, Россия
* E-mail: grishin@ichem.unn.ru
Поступила в редакцию 18.02.2020
После доработки 05.03.2020
Принята к публикации 19.03.2020
Аннотация
Изучено влияние сопряженных динитронов N,N–диметилглиоксальдинитрона, N,N–ди-трет-бутилглиоксальдинитрона и N,N–дифенилглиоксальдинитрона на радикальную полимеризацию стирола, инициированную динитрилом азоизомасляной кислоты. Установлено, что при значениях температуры ниже 90°С в процессе полимеризации стирола происходит обрыв цепей на исследуемых нитронах с образованием высокомолекулярных нитроксильных радикалов. Закономерности синтеза полистирола в присутствии сопряженных нитронов при 130°С (первый порядок реакции по мономеру во всем интервале превращений, линейный рост молекулярной массы с конверсией и осуществление постполимеризации) свидетельствуют о возможности полимеризации стирола в условиях обратимого ингибирования в высокотемпературном режиме. Выявлено, что акцептирование и особенности полимеризации стирола в присутствии нитроксильных радикалов, образующихся in situ, напрямую связаны с исходной структурой динитронов. Так, наличие сопряжения между фенильной и нитронильной группами в дифенилдинитроне приводит к уменьшению его акцептирующей способности, понижая эффективность высокомолекулярных нитроксилов как регуляторов полимеризации в высокотемпературном режиме. Методами статического и динамического светорассеяния исследованы гидродинамические размеры макромолекул, рассчитан форм-фактор Rg/Rh для линейного полимера и полимеров, синтезированных с участием динитронов.
ВВЕДЕНИЕ
Контролируемая радикальная полимеризация или полимеризация в режиме “живых” цепей занимает одну из ведущих позиций в современной синтетической химии полимеров [1], поскольку существенно расширяет возможности традиционной радикальной полимеризации в плане синтеза узкодисперсных гомо- и сополимеров с заданными молекулярно-массовыми характеристиками и четкой топологией [2]. Известны три основных направления контролируемой радикальной полимеризации: Stable Free-Radical Polymerization (SFRP) – полимеризация с участием стабильных радикалов, в том числе нитроксильных (данный метод в зарубежной литературе называется Nitroxide Mediated Radical Polymerization, NMP); Atom Transfer Radical Polymerization (ATRP) – полимеризация с переносом атома; Degenerative Transfer Processes (DTP) – полимеризация с вырожденной передачей цепи [1, 2]. Среди указанных методов синтез полимеров с участием стабильных нитроксильных радикалов в режиме обратимого ингибирования является одним из наиболее известных и хорошо изученных [3–6]. Полученные в условиях обратимого ингибирования полимеры, как правило, не нуждаются в дополнительной очистке, кроме традиционного переосаждения для удаления непрореагировавшего мономера. Неслучайно, метод NMP достаточно широко применяют для синтеза материалов биомедицинского назначения, сенсоров, органических материалов для электроники, наноразмерных полимерных структур и ряда других высокотехнологичных полимеров [3, 7–10].
Существенным недостатком метода NMP можно назвать ограниченный круг мономеров, способных к полимеризации по данному механизму. На начальном этапе становления этого направления круг мономеров, которые могут быть заполимеризованы в режиме обратимого ингибирования в присутствии нитроксилов, ограничивался стиролом и его аналогами [4, 5]. В последнее десятилетие для проведения контролируемой радикальной полимеризации акрилатов, акриламидов и диенов были предложены достаточно эффективные регуляторы на основе так называемых нитроксильных радикалов и алкоксиаминов второго поколения [11]. Гомополимеризация сложных эфиров метакриловой кислоты до некоторого времени оставалась труднореализуемой. Так, стерически затрудненный алкоксиамин на основе имидазолина, содержащий в своем составе спироциклический фрагмент, был получен авторами работы [12] и апробирован в NMP метакрилатов, однако хороший контроль наблюдался лишь до 55%-ной конверсии. С разработкой алкоксиаминов нового поколения, в частности Dispolreg-007 и его аналогов, появилась возможность осуществления NMP метилметакрилата, стирола и бутилакрилата с высокой степенью контроля молекулярно-массовых характеристик, вплоть до глубоких степеней превращения мономера [13, 14]. Следует отметить, что синтез указанных выше регуляторов весьма сложен и трудоемок.
В этой связи заслуживает внимания несколько иной подход к осуществлению NMP, заключающийся в образовании нитроксильных радикалов и алкоксиаминов in situ, – непосредственно в процессе полимеризации. При этом источниками нитроксильных радикалов могут выступать нитроны и нитрозосоединения, амины и гидроксиламины, окситриазены и другие соединения [15–26]. Способы получения перечисленных агентов вполне доступны, многие из них являются коммерческими соединениями и несомненно представляют интерес в плане применения их в качестве прекурсоров нитроксильных радикалов и алкоксиаминов. Использование прекурсоров, генерирующих нитроксильные радикалы непосредственно в ходе полимеризации (in situ NMP), позволило, с одной стороны, осуществить процесс контролируемого синтеза в более комфортных с практической точки зрения температурных условиях (70–90°С), а с другой – возможность расширить круг мономеров, полимеризацию которых можно вести в режиме обратимого ингибирования [15–26].
В настоящей работе впервые предложено применять в качестве агентов контролируемой радикальной полимеризации стирола сопряженные динитроны на основе глиоксаля различного строения, способные генерировать пространственно затрудненные нитроксилы при взаимодействии с радикалами (схема 1).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы
Стирол (“Aldrich”) очищали от ингибитора 10%-ным водным раствором гидроксида натрия, отмывали щелочь водой до нейтральной реакции и сушили прокаленным хлоридом кальция. Затем перегоняли, собирая фракцию с температурой кипения 48°С при 20 мм рт.ст. [27]. ДАК (“Aldrich”) перекристаллизовывали из этанола по стандартной методике [28]. Хлористый метилен, изопропиловый спирт и ацетон (“Компонент-реактив”, Россия) сушили прокаленным хлоридом кальция и перегоняли. Толуол (“Компонент-реактив”) первоначально сушили хлоридом кальция, после чего перегоняли над металлическим натрием. ДМСО сушили гидроксидом натрия и перегоняли при пониженном давлении. Физико-химические константы всех растворителей соответствовали литературным данным [29].
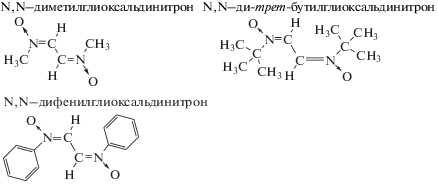
N,N–диметилглиоксальдинитрон (МДН), N,N–ди-трет-бутилглиоксальдинитрон (БДН), N,N–дифенилглиоксальдинитрон (ФДН) получали в соответствии с известной методикой [30]. Данные продукты характеризовали методами жидкостной хроматографии, ИК- и ЯМР-спектроскопии.
N,N–диметилглиоксальдинитрон – спектр ЯМР 1H (CDCl3, 400 MГц, δ, м.д.): 3.8 (s, 6H, СH3), 7.8 (s, 2H, CH=N(O)); ИК-спектр (KBr, ν/см–1): 3109 с (валентные колебания связей С–Н), 1547 (валентные колебания связей С=N), 1176 ср. (валентные колебания связей N–О).
N,N–ди-трет-бутилглиоксальдинитрон – спектр ЯМР 1H (CDCl3, 400 MГц, δ, м.д.): 1.57 (s, 18H, C(CH3)3), 8.27 (s, 2Н, CH=N(O)); ИК-спектр (KBr, ν/см–1): 3163 с (валентные колебания связей С–Н), 1504 с (валентные колебания связей С=N) 1127 ср. (валентные колебания связей N–О).
N,N–дифенилглиоксальдинитрон – спектр ЯМР 1H (CDCl3, 400 MГц, δ, м.д.): 7.85 м.д. (m, 4Н, м-C6H5), 7.52 м.д.(m, 6H, о- и п-C6H5), 8.67 (s, 2H, CH=N(O)); ИК-спектр (KBr, ν/см–1): 3134 с (валентные колебания связей С–Н), 1500 с (валентные колебания связей С=N), 1477 ср. (скелетные колебания связей С–С) 1092 ср. (валентные колебания связей N–О), 779 ср. (деформационные колебания связей С–Н).
Полимеризация стирола с участием динитронов
В ампулу помещали 0.8 мл раствора стирола с соответствующими динитронами (0.5, 1.5 или 3.0 мол. %) и ДАК (1.0 мол. %). При пониженном давлении ампулу трижды дегазировали путем повторения циклов замораживания–размораживания до остаточного давления 2 × 10–2 мм рт.ст. и отпаивали. Контроль за кинетикой полимеризации осуществляли гравиметрическим методом. По истечении определенного времени ампулу вынимали и замораживали в жидком азоте для прерывания полимеризации. Полученный полимер высаживали в изопропиловый спирт. С целью очищения от остатков мономера образцы переосаждали из раствора хлористого метилена, затем при пониженном давлении и 50°С сушили до постоянной массы. Степень превращения мономера в полимер Р рассчитывали по формуле:
Анализ молекулярно-массовых характеристик полимеров
Молекулярно-массовые характеристики полимеров определяли методом ГПХ на жидкостном хроматографе “Knauer”, снабженном каскадом колонок “Phenomenex” (300 × 7.8 мм, феногель, 10 мкм) с диаметром пор 105 и 103 Å и двумя детекторами (рефрактометрическим и ультрафиолетовым). Элюентом служил ТГФ, температура колонки составляла 25.0 ± 0.1°С. Для калибровки применяли узкодисперсные стандарты ПС [31].
ЯМР-спектроскопия
Исследование структуры динитронов осуществляли на спектрометре “Agilent DD2-400” с рабочей частотой 400 МГц. Спектры ЯМР 1H снимали в CDCl3 при комнатной температуре относительно сигнала остаточных протонов дейтерированного растворителя.
Регистрация ИК-спектров
Регистрацию ИК-спектров проводили на приборе “Infralum FT-801”. Спектры динитронов снимали в прессованных таблетках KBr. Диапазон волновых чисел лежал в пределах 5500–550 см–1. Погрешность определения составляла ±0.05 см–1.
Молекулярно-массовые и гидродинамические характеристики полимеров
Для определения ММ, статических и гидродинамических размеров макромолекул использовали методы статического и динамического светорассеяния. Гидродинамические характеристики макромолекул синтезированных полимеров измеряли на установке “NanoBrook Omni” (“Brookhaven Instruments”, США). В качестве источника света использовали лазер с длиной волны λ0 = 640 нм и мощностью 35 мВт. Корреляционную функцию интенсивности рассеянного света получали с помощью коррелятора исследовательского уровня “TurboCorr” с числом каналов 510. Время накопления корреляционной функции 180–300 с, при этом число фотонов, попадающих на детектор в 1 с, находится в диапазоне 50–450 килоимпульсов в секунду. Средний гидродинамический диаметр рассчитывали по значению из 5–10 параллельных измерений. Кварцевую кювету перед работой “обеспыливали” двух–трехкратным ополаскиванием растворителем, отфильтрованным через 0.2 мкм нейлоновым фильтром (“Chromafil Xtra”). Измерения проводили при угле рассеивания θ = = 173°. Значения гидродинамических радиусов Rh определяли по уравнению Стокса–Эйнштейна. В качестве растворителя использовали толуол (плотность ρ = 0.862 г/см3, динамическая вязкость η0 = 0.560 сП и показатель преломления n0 = 1.494). Все эксперименты вели при температуре 25°С. Исследуемые растворы фильтровали через нейлоновые фильтры (“Chromafil Xtra”) с диаметром пор 0.2 мкм.
Величину Mw и радиус инерции Rg измеряли на спектрометре статического светорассеяния на приборе “BI-MwA molecular Weight Analyzer” (“Brookhaven Instruments”, США). Данные величины находили по методу Дебая [32] и методом двойной экстраполяции Зимма. В качестве источника света использовали лазер с длиной волны λ0 = 660 нм и мощностью 30 мВт с вертикальной поляризацией.
Инкремент показателя преломления измеряли на дифференциальном рефрактометре “BI-DNDC” (“Brookhaven Instruments”, США). Величину dn/dc определяли по тангенсу угла наклона зависимости показателя преломления исследуемого раствора от концентрации полимера.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Как известно [33], нитроны являются эффективными акцепторами свободных радикалов. Они могут присоединять реакционноспособные радикальные центры, в том числе радикалы роста и инициатора, с образованием достаточно стабильных спин-аддуктов, и традиционно применяются в качестве спиновых ловушек при исследовании свободно радикальных процессов. Особенность полимеризации в присутствии нитронов заключается в том, что нитроксилы не вводятся в систему в “готовом виде”, а образуются непосредственно в ходе полимеризации (in situ). Таким образом, создается возможность синтеза полимеров на основе виниловых и (мет)акриловых мономеров различного строения с заданными молекулярно-массовыми характеристиками и архитектурой [15, 16]. В плане поиска новых медиаторов полимеризации на основе нитроксильных радикалов, формирующихся in situ, в данной работе была изучена радикальная полимеризация стирола в присутствии ряда сопряженных динитронов на основе глиоксаля, содержащих в своем составе при атоме азота различные пространственно-затрудненные радикалы (схема 1).
Особенности полимеризации стирола в присутствии динитронов различного строения
На первоначальном этапе был проведен скрининг полимеризации стирола в присутствии динитронов различного строения в широком температурном интервале. Из данных, представленных в табл. 1, видно, что введение динитронов независимо от их структуры влечет существенное увеличение времени полимеризации до высоких значений конверсии.
Таблица 1.
Молекулярно-массовые характеристики образцов ПС, синтезированных в присутствии динитронов и 1.0 мол. % ДАК при разных значениях температуры
| Динитрон | Концентрация, мол. % | Т, °С | Время, ч | Конверсия, % | Mn × 10–3 | Mw × 10–3 | Ð |
|---|---|---|---|---|---|---|---|
| – | – | 70 | 8.0 | 100 | 45.2 | 225.0 | 4.98 |
| 90 | 1.3 | 87 | 17.6 | 64.5 | 3.67 | ||
| 110 | 4.3 | 85 | 15.9 | 81.4 | 5.12 | ||
| 130 | 2.2 | 89 | 12.2 | 84.3 | 6.90 | ||
| МДН | 0.5 | 70 | 14 | 88 | 32.9 | 215.8 | 6.56 |
| 90 | 25 | 85 | 11.6 | 64.4 | 5.57 | ||
| 110 | 100 | 89 | 14.2 | 59.9 | 4.21 | ||
| 130 | 9 | 74 | 16.9 | 48.1 | 2.84 | ||
| МДН | 1.5 | 70 | 100 | 68 | 12.6 | 77.1 | 6.12 |
| 90 | 120 | 50 | 12.5 | 45.5 | 3.64 | ||
| 110 | 100 | 76 | 12.6 | 42.1 | 3.35 | ||
| 130 | 15 | 73 | 12.5 | 23.9 | 1.91 | ||
| МДН* | 1.5 | 70 | 100 | 41 | 8.1 | 26.1 | 3.21 |
| 90 | 120 | 50 | 8.1 | 20.2 | 2.49 | ||
| 110 | 100 | 86 | 6.7 | 15.7 | 2.35 | ||
| 130 | 15 | 89 | 6.4 | 11.1 | 1.75 | ||
| МДН | 3.0 | 70 | 140 | 24 | 6.8 | 47.5 | 6.94 |
| 90 | 140 | 46 | 10.6 | 36.1 | 3.41 | ||
| 110 | 100 | 77 | 19.3 | 34.4 | 1.79 | ||
| 130 | 24 | 87 | 13.5 | 22.3 | 1.65 | ||
| БДН | 0.5 | 70 | 100 | 90 | 17.1 | 40.4 | 2.35 |
| 90 | 100 | 79 | 12.1 | 21.7 | 1.79 | ||
| 110 | 100 | 84 | 10.7 | 24.3 | 2.26 | ||
| 130 | 32 | 87 | 9.7 | 26.8 | 2.75 | ||
| БДН | 1.5 | 70 | 100 | 28 | 4.8 | 7.4 | 1.54 |
| 90 | 100 | 55 | 7.0 | 11.8 | 1.68 | ||
| 110 | 100 | 72 | 7.1 | 20.6 | 2.90 | ||
| 130 | 60 | 79 | 7.4 | 21.1 | 2.83 | ||
| БДН | 3.0 | 70 | 100 | 26 | 5.8 | 8.2 | 1.42 |
| 90 | 100 | 44 | 5.9 | 9.3 | 1.58 | ||
| 110 | 100 | 76 | 7.5 | 15.4 | 2.06 | ||
| 130 | 73 | 76 | 7.4 | 17.8 | 2.41 | ||
| ФДН | 0.5 | 70 | 11 | 98 | 48.3 | 160.8 | 3.33 |
| 90 | 7 | 97 | 18.2 | 60.1 | 3.31 | ||
| 110 | 55 | 89 | 14.4 | 59.1 | 4.09 | ||
| 130 | 6 | 77 | 15.4 | 66.0 | 4.29 | ||
| ФДН | 1.5 | 70 | 28 | 100 | 46.0 | 126.3 | 2.75 |
| 90 | 80 | 80 | 14.2 | 31.5 | 2.22 | ||
| 110 | 100 | 81 | 9.3 | 19.2 | 2.06 | ||
| 130 | 32 | 82 | 17.5 | 56.4 | 3.21 | ||
| ФДН | 3.0 | 70 | 63 | 99 | 35.4 | 82.1 | 2.32 |
| 90 | 80 | 80 | 14.0 | 29.0 | 2.06 | ||
| 110 | 100 | 75 | 12.1 | 28.5 | 2.35 | ||
| 130 | 32 | 81 | 13.7 | 40.9 | 2.99 |
При этом с увеличением концентрации динитронов наблюдается понижение ММ синтезированных полимеров, что может быть связано с акцептированием радикалов роста динитронами и образованием стабильных нитроксильных радикалов:
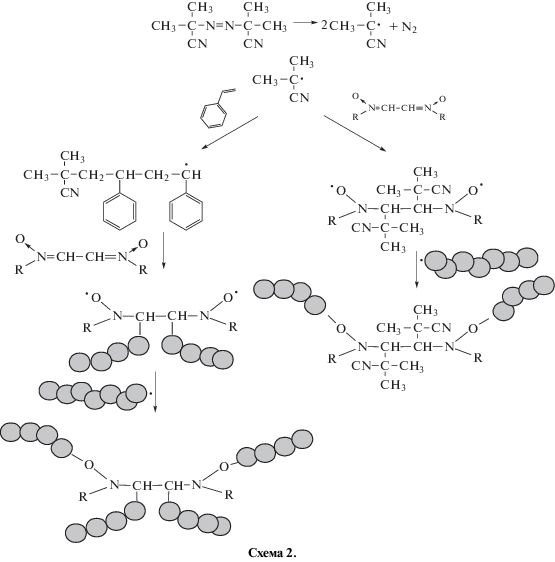
Теоретически данные динитронов способны акцептировать два радикала с образованием бирадикалов нитроксильного типа, однако в литературе имеются лишь единичные примеры участия динитронов в реакциях такого типа [34]. В частности, в указанной работе установлено, что БДН, имеющий в своем составе объемные трет-бутильные радикалы, эффективно акцептирует различные углерод-, кислород-, серо- и азотцентрированные активные радикалы. При этом образования бинитроксилов не обнаружено. Сведений в отношении использования МДН и ФДН для фиксации реакционноспособных радикалов в литературе не найдено.
Следует отметить некоторые особенности полимеризации стирола, связанные с различным строением использованных динитронов. Так, на основании данных табл. 1 можно заключить, что МДН при концентрации 0.5 и 1.5 мол. % не оказывает значительного влияния на коэффициенты полидисперсности полимеров, синтезированных в температурном интервале 70–90°С. При более высоких значениях температуры происходит уменьшение полидисперсности по сравнению с образцами, полученными на ДАК. Вероятно, это обусловлено увеличением вклада обратимого ингибирования по сравнению с традиционными способами обрыва цепи при радикальной полимеризации путем рекомбинации и диспропорционирования. Причем предпочтительнее в плане синтеза ПС с относительно низкой полидисперсностью – использование высоких значений концентрации МДН (1.5 и 3.0 мол. %) и повышенной температуры (130°С). Следует отметить эффективное влияние МДН на процесс полимеризации стирола в ДМСО, что обусловлено хорошей растворимостью динитронов в полярных растворителях, а значит, наиболее высокой акцептирующей способностью по отношению к активным радикалам роста.
При использовании в качестве акцептора свободных радикалов БДН установлено, что он оказывает существенное влияние на процесс полимеризации стирола при более низких значениях температуры (70–90°С), фактически приближенных к условиям промышленного синтеза (табл. 1). При этом синтезированные образцы имеют меньшее значение коэффициентов полидисперсности, чем ПС, полученный в условиях инициирования ДАК без добавок динитронов. Полидисперсность синтезированных образцов увеличивается с повышением температуры. Как показано в работе [34], нитроксильные радикалы на основе углеродцентрированных радикалов и БДН способны распадаться под действием УФ-излучения и высокой температуры с образованием 2-метил-2-нитрозопропана и трет-бутилвинилнитроксилов. Вероятно, именно такие побочные процессы происходят и в системах, исследуемых в настоящей работе. В частности, о вероятном распаде нитроксильных радикалов при полимеризации стирола в присутствии БДН в условиях высокой температуры (110°С и выше) косвенно свидетельствует изменение окраски полимеризационной системы, а также повышение полидисперсности ПС, синтезированного в данных условиях.
В отличие от МДН и БДН, процесс полимеризации в присутствии ФДН, содержащего в своем составе два фенильных кольца, находящихся в сопряжении с нитронильной группой, сопровождается менее выраженным понижением ММ полимеров и их коэффициентов полидисперсности, независимо от концентрации динитрона и температурного режима. Хотя во всех случаях синтеза ПС в присутствии ФДН, значения Ð ниже соответствующих значений полидисперсности полимеров, синтезированных в присутствии ДАК без добавок динитронов (табл. 1).
Особенности влияния динитронов на процесс полимеризации стирола при температуре 90°С
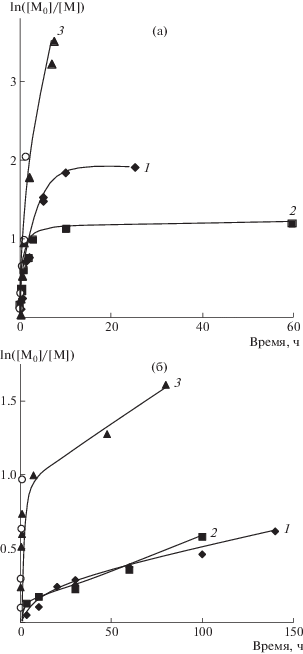
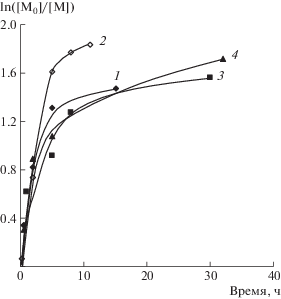
Детальное исследование кинетических особенностей полимеризации стирола с участием динитронов было проведено при 90°С, поскольку данный температурный показатель достаточно близок к условиям синтеза полимеров в промышленности. На рис. 1 представлены зависимости ln([M0]/[M]) от времени полимеризации стирола в присутствии динитронов различного строения при концентрации 0.5 и 3.0 мол. %. Так, в присутствии 0.5 мол. % динитронов на кривых полимеризации можно выделить два участка. На первом из них независимо от природы нитрона процесс реализуется практически с одинаковой скоростью (рис. 1а). Вероятно, это связано с тем, что на начальном этапе происходит постепенный распад инициатора (десять периодов полураспада ДАК при указанной температуре ~0.5 ч) и акцептирование активных радикалов инициатора или радикалов роста динитронами с образованием нитроксильных радикалов и алкоксиаминов (схема 2). Второй этап сопровождается понижением скорости полимеризации стирола, связанным с практически полным распадом инициатора, а также участием образовавшихся нитроксильных радикалов и алкоксиаминов в полимеризации в качестве ингибиторов. Данные закономерности наиболее четко проявляются при увеличении концентрации динитронов в среде мономера (рис. 1б). Как видно на рисунке, структура динитронов особым образом отражается на кинетических закономерностях полимеризации стирола. Так, ФДН обладает меньшей акцептирующей способностью по отношению к радикалам роста, и, неслучайно, что в его присутствии скорость полимеризации всегда выше по сравнению МДН и БДН. В случае последних метильные и трет-бутильные заместители при атоме азота в структуре динитронов не способны к сопряжению в отличие от фенильных колец и создают меньше препятствий к подходу активных радикалов к нитронильной группе. Следовательно, процесс акцептирования с их участием более эффективен. Отметим, что в случае использования МДН и БДН с увеличением их концентрации понижается предельная степень превращения мономера в полимер и при 3.0 мол. % конверсия стирола составляет лишь ~50%.
Рис. 1.
Полулогарифмические кинетические зависимости полимеризации стирола при Т = 90°С в присутствии 1.0 мол. % ДАК, а также 0.5 (а) и 3.0 мол. % динитронов (б): 1 – МДН, 2 – БДН, 3 – ФДН.
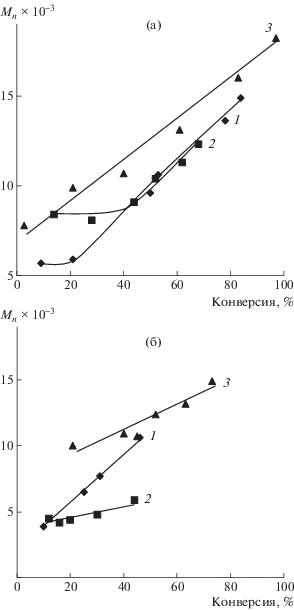
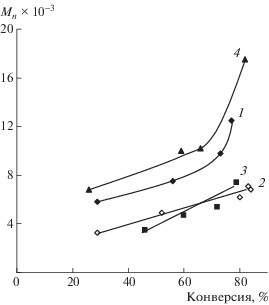
На рис. 2 представлены зависимости ММ синтезированных образцов от конверсии. Как следует из представленных данных, при формировании алкоксиаминов in situ в присутствии 0.5 мол. % МДН или БДН, обладающих более высокой акцептирующей способностью, на первоначальном этапе не наблюдается линейного роста ММ с конверсией (рис. 2а, начальные участки кривых 1 и 2), что может быть связано с активным акцептированием образующихся радикалов роста МДН и БДН. В дальнейшем процесс синтеза полимеров сопровождается линейным возрастанием ММ с конверсией. При увеличении концентрации нитронов до 3.0 мол. % зависимость ММ от конверсии становится линейной для всех использованных динитронов, независимо от их строения (рис. 2б). Причем, чем выше концентрация динитронов, тем ниже ММ синтезированных образцов на первоначальных степенях превращения мономера в полимер.
Рис. 2.
Зависимости от конверсии Mn образцов ПС, полученных в присутствии 1.0 мол. % ДАК при Т = = 90°С, а также 0.5 (а) и 3.0 мол. % динитронов (б): 1 – МДН, 2 – БДН, 3 – ФДН.
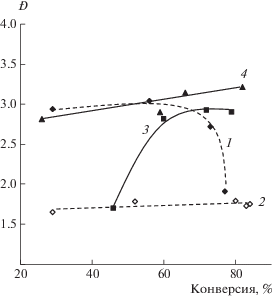
Значения полидисперсности образцов, синтезированных в присутствии динитронов, зависят как от структуры и акцептирующей способности нитрона, так и от их концентрации (рис. 3). В случае использования в качестве источника нитроксильных радикалов МДН в концентрации 0.5 мол. % происходит резкое увеличение коэффициентов полидисперсности с возрастанием степени превращения мономера в полимер. При более высокой концентрации МДН (3.0 мол. %) полидисперсность образцов ПС увеличивается до определенного значения, а затем на более глубоких степенях превращения наблюдается ее незначительное снижение. Высокие значения Ð для синтезированных с участием МДН полимеров и отсутствие линейной зависимости ММ с ростом конверсии (рис. 2а, кривая 1), характерной для полимеризации в режиме “живых” цепей [2–5], могут быть связаны с одновременно протекающими в системе несколькими процессами. Приведенные на рис. 4 кривые ММР полимерных образцов косвенно подтверждают это предположение.
Рис. 3.
Зависимости от конверсии Ð образцов ПС, полученных в присутствии 1.0 мол. % ДАК при Т = = 90°С, а также 0.5 (а) и 3.0 мол. % динитронов (б): 1 – МДН, 2 – БДН, 3 – ФДН.
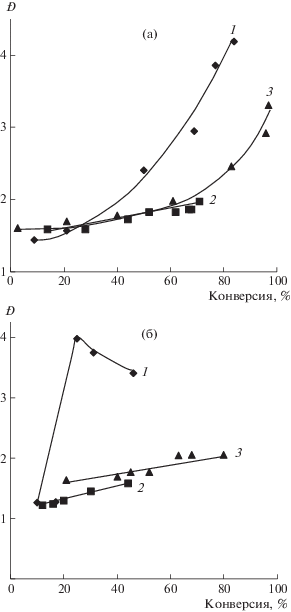
Рис. 4.
Кривые ММР образцов ПС, синтезированных в присутствии 3.0 мол. % МДН и 1.0 мол. % ДАК при Т = 90°С (1–3) при автополимеризации стирола (4). Конверсия 10 (1), 25 (2), 46 (3) и 56% (4).
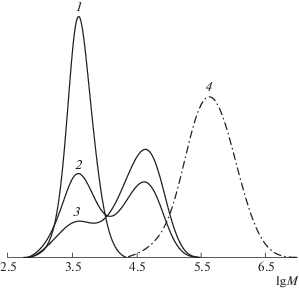
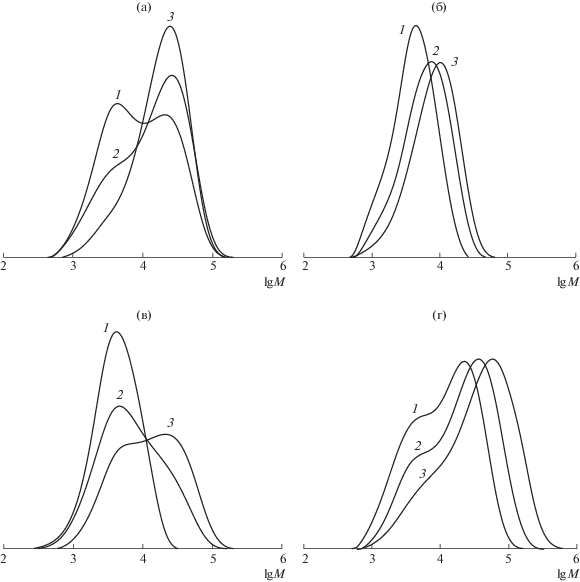
Кривые ММР образцов, синтезированных с использованием МДН, бимодальны (рис. 4). При этом вторая мода появляется при конверсии мономера более 20–30% и возрастает по ходу процесса. Образование двух мод может быть связано с разными механизмами обрыва полимерной цепи на начальных и глубоких значениях конверсии. В начале процесса обрыв растущих цепей, главным образом, происходит за счет их акцептирования динитроном. Затем в системе начинает преобладать бимолекулярный обрыв цепей, что выражается в появлении второй моды на кривых ММР.
В случае синтеза ПС в присутствии БДН независимо от его концентрации, коэффициент полидисперсности ПС не превышает значения 2.0. При концентрации динитронов 3.0 мол. % Ð составляет 1.22–1.58 (рис. 3). Кривые ММР образцов ПС, синтезированных в присутствии различной концентрации БНД, являются унимодальными (рис. 5). При этом с ростом степени превращения мономера максимум моды последовательно смещается в высокомолекулярную область. Однако при использовании 3.0 мол. % БДН смещения низкомолекулярного плеча практически не наблюдается, что приводит к некоторому возрастанию полидисперсности.
Рис. 5.
Кривые ММР образцов ПС, синтезированных в присутствии 0.5 (а) и 3.0 мол. % БДН (б) при Т = = 90°С в присутствии 1.0 мол. % ДАК. а – Конверсия 14 (1), 44 (2) и 68% (3); б – конверсия 16 (1), 30 (2) и 44 (3).

При синтезе ПС в присутствии ФДН при повышении его количества абсолютные значения полидисперсности уменьшаются и при концентрации 3.0 мол. % не превышают 2.0 (см. рис. 3б). Кривые ММР образцов ПС, полученных в данных условиях, представлены на рис. 6. Как следует из приведенных зависимостей, в отличие от динитронов с алкильными заместителями в своем составе, ММР образцов ПС, полученных в присутствии ФДН, унимодальное, и с ростом степени превращения мономера наблюдается смещение кривых ММР в область более высоких значений ММ, что в целом характерно для процессов, протекающих в режиме обратимого ингибирования [16–18].
Рис. 6.
Кривые ММР образцов ПС, синтезированных в присутствии 0.5 (а) и 3.0 мол. % ФДН (б) при Т = = 90°С в присутствии 1.0 мол. % ДАК. а – Конверсия 3 (1), 40 (2) и 96% (3); б – конверсия 21 (1), 52 (2) и 68% (3).
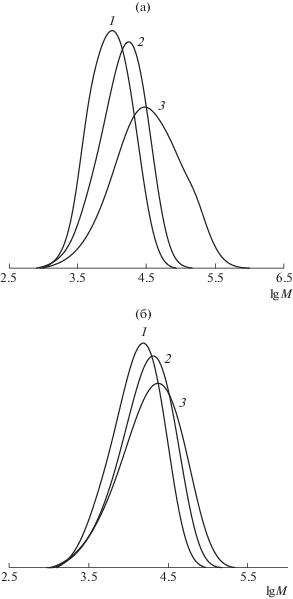
Таким образом, проведение полимеризации стирола в присутствии динитронов на основе глиоксаля, выступающих в качестве акцепторов свободных радикалов и источников нитроксилов in situ при температуре 90°С, позволяет регулировать молекулярно-массовые характеристики ПС. Отметим, что структура динитронов превалирует в акцептировании активных радикалов роста и возможности осуществления полимеризации в условиях обратимого ингибирования. Так, МДН и БДН достаточно эффективно улавливают свободные радикалы. Однако, как свидетельствуют полученные экспериментальные результаты, в данном температурном режиме указанные динитроны выступают в качестве ингибиторов полимеризации, обратимое взаимодействие образующихся нитроксилов с радикалами роста не наблюдается. В отличие от МДН и БДН, меньшей акцептирующей способностью обладает ФДН. Вместе с тем, образующиеся in situ на его основе нитроксильные радикалы способны к обратимому взаимодействию с радикалами роста при температуре 90°С, о чем свидетельствует четкое смещение всей кривой ММР с конверсией в область более высоких значений ММ.
Синтез ПС в присутствии динитронов в высокотемпературном режиме
Известно [3, 5, 16], что эффективность нитроксильных радикалов и алкоксиаминов как регуляторов радикальной полимеризации определяется не только их строением, но и температурными условиями протекания процесса. В этой связи и с учетом рассмотренных выше особенностей полимеризации стирола при 90°С были исследованы закономерности синтеза ПС с участием динитронов различного строения при температуре 130°С.
Анализ кинетических кривых, приведенных на рис. 7, показывает, что независимо от структуры использованных динитронов процесс полимеризации стирола на начальных значениях конверсии протекает практически с одинаковой скоростью. При этом сравнительный анализ полимеризации стирола в присутствии МДН в массе и в растворе ДМСО указывает на то, что в среде растворителя процесс полимеризации протекает до более глубокой конверсии при практически сопоставимых значениях скорости на начальной стадии.
Рис. 7.
Полулогарифмическая кинетическая зависимость полимеризации стирола при Т = 130°С в присутствии 1.0 мол. % ДАК и 1.5 мол. % динитролов: 1 – МДН в массе мономера, 2 – МДН в ДМСО (1 : 1), 3 – БДН в массе мономера, 4 – ФДН в массе мономера.
Молекулярная масса ПС, синтезированного в присутствии МДН в массе мономера, до глубокой конверсии растет линейно (рис. 8). При этом абсолютные значения ММ образцов ПС, синтезированных в присутствии динитронов, в несколько раз ниже чем у образцов, формирующихся при термическом инициировании полимеризации (М = 150 × 103). Однако в случае синтеза полимера в массе после 70% конверсии наблюдается выраженный рост ММ. Аналогичная закономерность прослеживается и для ПС, полученного с участием ФДН в высокотемпературном режиме (рис. 8).
Рис. 8.
Зависимость от конверсии Mn образцов ПС, полученных при Т = 130°С в присутствии 1.0 мол. % ДАК и 1.5 мол. % динитронов: 1 – МДН в массе мономера, 2 – МДН в ДМСО (1 : 1), 3 – БДН в массе мономера, 4 – ФДН в массе мономера.
В случае проведения полимеризации стирола в присутствии БДН и МДН в среде ДМСО молекулярные массы образцов имеют более низкие значения, чем при синтезе полимера без растворителя. При этом линейная зависимость роста ММ с конверсией соблюдается вплоть до 90%-ной степени превращения мономера в полимер.
Несмотря на идентичные кинетические закономерности и особенности роста ММ с конверсией в присутствии исследуемых систем в высокотемпературном режиме, зависимость коэффициентов полидисперсности от конверсии имеет индивидуальный характер, определяемый структурой нитрона (рис. 9). Так, коэффициенты полидисперсности образцов ПС, полученных в присутствии МДН и ФДН в массе мономера, имеют на начальных степенях превращения более высокие значения (Ð = 2.8–3.0), как и их ММ, в отличие от полимеризации в присутствии БДН и МДН в ДМСО как растворителе (Ð = 1.65–1.70). Данный параметр для систем на основе ФДН остается высоким вплоть до глубоких степеней превращения мономера в полимер. В случае БДН, несмотря на линейный рост ММ с конверсией, индекс полидисперсности значительно возрастает и на предельных степенях превращения имеет значение ~3.0. Такой существенный рост полидисперсности в присутствии БДН, вероятно, обусловлен реализацией ряда процессов, в том числе осуществлением полимеризации в присутствии нитроксилов различной природы. В частности, в работе [34] выявлена возможность распада нитроксильного радикала, образующегося в присутствии БДН при высоких значениях температуры, на 2-метил-2-нитрозопропан и трет-бутилвинилнитроксил. Первый также является эффективной спиновой ловушкой, способной взаимодействовать с активными радикальными центрами с образованием нитроксильных радикалов in situ.
Рис. 9.
Зависимость от конверсии Ð образцов ПС, полученных при Т = 130°С в присутствии 1.0 мол. % ДАК и 1.5 мол. % динитронов: 1 – МДН в массе мономера, 2 – МДН в ДМСО (1 : 1), 3 – БДН в массе мономера, 4 – ФДН в массе мономера.
В отличие от ФДН и БДН их менее пространственно замещенный аналог МДН позволяет синтезировать ПС при температуре 130°С с относительно низкими значениями коэффициентов полидисперсности (рис. 9). Что же касается синтеза ПС, то в массе мономера полидисперсность понижается к концу процесса, а при осуществлении полимеризации в среде ДМСО – остается невысокой (Ð < 1.75) во всем интервале конверсии.
Поскольку коэффициенты полидисперсности ПС, полученного в присутствии БДН и ФДН, имеют высокие значения, то кривые ММР для данных систем достаточно широкие (рис. 10). При этом в случае БДН, который нестабилен при высоких значениях температуры, наблюдается уширение ММР с конверсией, и на кривых появляется бимодальность (рис. 10в). Кривые ММР полимеров, полученных в массе мономера с участием термически более стабильных МДН и ФДН, имеют бимодальный вид на начальных этапах процесса. С течением полимеризации моды кривых смещаются в область более высоких значений ММ. Вместе с тем уменьшается доля низкомолекулярных продуктов, что в случае МДН влечет образование узкодисперсного образца, характеризующегося унимодальным распределением (рис. 10а). В случае полимеризации стирола в среде ДМСО в присутствии МДН кривые ММР образцов ПС унимодальны и четко смещаются в высокомолекулярную область с конверсией (рис. 10б).
Рис. 10.
Кривые ММР образцов ПС, синтезированных при Т = 130°С в присутствии 1.5 мол. % динитронов и 1.0 мол. % ДАК: а – МДН в массе мономера, конверсия 29 (1), 56 (2) и 77% (3); б – МДН в среде ДМСО (1 : 1), конверсия 29 (1), 52 (2) и 89% (3); в – БДН в массе мономера, конверсия 46 (1), 60 (2) и 79% (3); г – ФДН в массе мономера, конверсия 26 (1), 66 (2) и 82% (3).
Таким образом, закономерности полимеризации стирола в присутствии динитронов в высокотемпературном режиме существенно отличаются от проведения процесса при 90°С. В присутствии БДН и ФДН контроль молекулярно-массовых характеристик ПС ухудшается, что связано с акцептирующей способностью, устойчивостью указанных динитронов при температуре 130°С и стабильностью образующихся продуктов в процессе акцептирования радикалов роста динитронами. МДН, содержащий в своем составе менее пространственно-затрудненные группы, в высокотемпературном режиме проявляет бόльшую эффективность в качестве контролирующего агента молекулярно-массовых характеристик ПС, о чем свидетельствуют достаточно низкие значения коэффициентов полидисперсности образцов ПС, синтезированных в среде ДМСО.
Постполимеризация с участием алкоксиаминов, синтезированных в присутствии динитронов
Как было изложено выше, высокомолекулярные алкоксиамины, полученные in situ на основе динитронов, в зависимости от их исходной структуры и акцептирующей способности позволяют регулировать полимеризацию стирола по механизму обратимого ингибирования в различных температурных режимах. С целью более детального доказательства реализации полимеризации стирола в условиях обратимого ингибирования в настоящей работе была проведена постполимеризация стирола на высокомолекулярных алкоксиаминах, полученных in situ полимеризаций стирола в присутствии динитронов. Основные результаты постполимеризации обобщены в табл. 2.
Таблица 2.
Постполимеризация стирола в присутствии высокомолекулярных алкоксиаминов на основе динитронов
| Образец, № | Алко- ксиамин | Условия | Т, °С | Время, ч | Конве-рсия, % | Mn × × 10–3 | Mw × × 10–3 | Ð |
|---|---|---|---|---|---|---|---|---|
| 1* | 1МДН | Стирол + 1.0 мол. % ДАК + 1.5 мол. % МДН | 130 | 15 | 89 | 6.4 | 11.1 | 1.73 |
| 2** | − | Стирол + 0.1 мол. % алкоксиамин 1МДН | 70 | 100 | 25 | 32.0 | 419.0 | 13.1 |
| 3** | − | Стирол + 0.1 мол. % алкоксиамин 1МДН | 130 | 17 | 82 | 111.0 | 174.0 | 1.57 |
| 4* | 2МДН | Стирол + 1.0 мол. % ДАК + 3.0 мол. % МДН | 90 | 5 | 24 | 3.8 | 9.3 | 2.45 |
| 5** | − | Стирол + 0.1 мол. % алкоксиамин 2МДН | 130 | 17 | 91 | 53.0 | 93.0 | 1.75 |
| 6* | 1БДН | Стирол + 1.0 мол. % ДАК + 1.5 мол. % БДН | 90 | 60 | 47 | 6.2 | 10.5 | 1.69 |
| 7** | − | Стирол + 0.1 мол. % алкоксиамин 1БДН | 70 | 100 | 42 | 55.0 | 332.0 | 6.05 |
| 8** | − | Стирол + 0.1 мол. % алкоксиамин 1БДН | 130 | 17 | 83 | 50.0 | 120.0 | 2.40 |
| 9* | 2БДН | Стирол + 1.0 мол. % ДАК + 3.0 мол. % БДН | 90 | 60 | 30 | 4.8 | 7.0 | 1.46 |
| 10** | − | Стирол + 0.1 мол. % алкоксиамин 2БДН | 70 | 100 | 41 | 39.0 | 173.0 | 4.43 |
| 11** | − | Стирол + 0.1 мол. % алкоксиамин 2БДН | 110 | 57 | 92 | 89.0 | 135.0 | 1.52 |
| 12** | − | Стирол + 0.1 мол. % алкоксиамин 2БДН | 130 | 17 | 91 | 60.0 | 102.0 | 1.70 |
| 13* | 1ДФН | Стирол + 1.0 мол. % ДАК + 3.0 мол. % ФДН | 90 | 0.8 | 45 | 10.7 | 19.0 | 1.78 |
| 14** | − | Стирол + 0.1 мол. % алкоксиамин 1ФДН | 70 | 100 | 52 | 51.0 | 217.5 | 4.26 |
| 15** | − | Стирол + 0.1 мол. % алкоксиамин 1ФДН | 90 | 100 | 88 | 73.0 | 282.5 | 3.87 |
| 16** | − | Стирол + 0.1 мол. % алкоксиамин 1ФДН | 130 | 17 | 91 | 36.4 | 99.6 | 2.74 |
Результаты постполимеризации подтверждают особенности гомополимеризации стирола с участием динитронов. Так, в случае проведения процесса в низкотемпературном интервале формируются образцы с достаточно высокой полидисперсностью (табл. 2, образцы 2, 7, 10 и 14). При этом ММР продукта полимеризации имеет бимодальный вид для алкоксиаминов на основе МДН и БДН, а для алкоксиаминов на базе ФДН наблюдается достаточно интенсивное низкомолекулярное плечо. На рис. 11а представлено ММР постполимера, синтезированного в низкотемпературном режиме на примере алкоксиамин–1МДН.
Рис. 11.
ММР постполимеров, синтезированных в присутствии алкоксиаминов на основе динитронов: 1 – исходный алкоксиамин, 2 – постполимер, 3 – полимер, образующийся в результате термического инициирования. Молекулярно-массовые характеристики алкоксиаминов представлены в табл. 2. Условия синтеза постполимеров: а – 1МДН, Т = 70°С; б – 1МДН, Т = 130°С; в – 2БДН, Т = 110°С; г – 1ФДН, Т = 90°С.
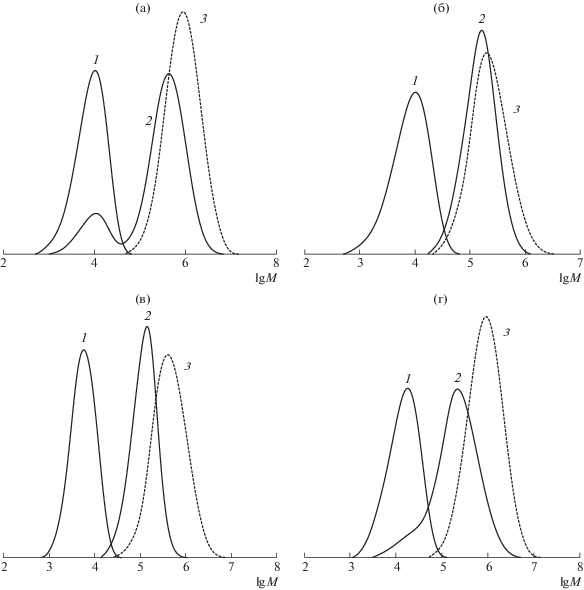
Повышение температуры постполимеризации до 110–130°С, как и в случае гомополимеризации стирола, позволяет осуществлять процесс в контролируемом режиме. Так, для всех исследуемых систем можно видеть увеличение ММ постполимеров по сравнению с исходными ММ используемых алкоксиаминов, при этом в присутствии МДН и БДН возможно образование полимеров с достаточно низкими значениями индекса полидисперсности (табл. 2). ММР таких продуктов унимодально (рис. 11б, 11в). В случае синтеза постполимеров в присутствии алкоксиаминов на основе ФДН температурный режим процесса не играет столь существенной роли как для МДН-, так и БДН-содержащих алкоксиамины. Независимо от температурного режима полимеризации образуется продукт с более высокими значениями ММ по сравнению с исходным алкоксиамином, но полидисперсность образцов достаточно высокая (табл. 2, образцы 13–15). При этом ММР постполимера всегда имеет низкомолекулярное плечо, независимо от температурного режима (рис. 11г).
Таким образом, полученные данные по постполимеризации стирола однозначно свидетельствуют о возможности протекания полимеризации с участием динитронов и алкоксиаминов на их основе в условиях обратимого ингибирования. Достаточно высокие значения коэффициентов полидисперсности указывают на протекание побочных процессов, приводящих к гибели нитроксильных радикалов и высокомолекулярных алкоксиаминов с образованием “мертвого” полимера.
Гидродинамические характеристики ПС, синтезированного в присутствии динитронов
Для ряда синтезированных в присутствии динитронов полимеров были исследованы молекулярно-массовые и гидродинамические характеристики макромолекул методами статического и динамического рассеяния света в среде толуола. Средневесовую молекулярную массу Mw, второй вириальный коэффициент А2 и радиус инерции Rg определяли методом статического рассеяния света, а гидродинамический радиус Rh макромолекул оценивали исходя из найденных значений коэффициентов диффузии D0 динамическим методом рассеяния света по уравнению Стокса–Эйнштейна:
где D0– коэффициент диффузии, k – константа Больцмана, T – абсолютная температура, η0 – вязкость растворителя.Молекулярно-массовые характеристики представлены в табл. 3. Как видно, для растворов ПС в толуоле коэффициент А2 имеет достаточно большие положительные значения, что свидетельствует о хорошем термодинамическом качестве растворителя. Следует отметить, что значения ММ, полученные методом статического светорассеяния, выше значений, оцениваемых исходя из ГПХ-анализа. На основании показателей Rg и Rh определено значение форм-фактора (ρ = Rg/Rh), характеризующее степень “асимметричности” макромолекулы в растворе. Так, для ряда конформаций макромолекул вычислены значения ρ. Принято считать [35], что для твердой сферы ρ = 0.778, для гауссова клубка полидисперсного линейного полимера в θ-растворителе данный параметр принимает значение 1.73, а в хорошем растворителе ρ = 2.05. Значения ρ > 2.05 характерны для эллипсоида и жесткостержневых полимеров. В случае полидисперсных звездообразных полимеров с числом лучей, равным четырем, в θ-растворителе ρ = 1.534. С увеличением числа лучей или разветвлений в макромолекуле данный параметр уменьшается до 1.225. Из данных табл. 3 видно, что в случае синтеза ПС, синтезированного с участием 0.5 мол. % БДН и 3.0 мол. % ФДН, значения форм-фактора близки к значениям для гауссова клубка линейного полидисперсного полимера. В присутствии 0.5 мол. % МДН и ФДН данный параметр близок к показателю твердой сферы или сверхразветвленных полимеров. Увеличение концентрации МДН и БДН по анализируемому параметру может повлечь формирование разветвленных и звездообразных полимеров (схема 2).
Таблица 3.
Молекулярно-массовые и гидродинамические характеристики ПС, синтезированных в присутствии динитронов при Т = 90°С
| Динитрон | Концен-трация, мол. % | Mn* × 10–3 | Mw* × 10–3 | Ð | Mw** × × 10–3 | A2 × 104, см3 моль/г2 | Rg, нм | Rh, нм | Rg/Rh |
|---|---|---|---|---|---|---|---|---|---|
| − | – | 12.4 | 32.2 | 2.60 | 45.0 | 6.03 | 10.30 | 4.81 | 2.14 |
| МДН | 0.5 | 11.6 | 64.4 | 5.55 | 72.7 | 6.30 | 6.64 | 8.54 | 0.78 |
| 3.0 | 9.8 | 26.6 | 2.71 | 38.8 | 3.50 | 8.44 | 6.11 | 1.38 | |
| БДН | 0.5 | 12.3 | 23.1 | 1.88 | 27.3 | 3.01 | 10.10 | 5.41 | 1.87 |
| 1.5 | 8.0 | 14.0 | 1.75 | 26.0 | 4.49 | 8.81 | 5.35 | 1.65 | |
| ФДН | 0.5 | 16.0 | 39.4 | 2.46 | 49.6 | 5.51 | 9.01 | 9.02 | 0.96 |
| 3.0 | 14.9 | 30.6 | 2.10 | 33.1 | 5.20 | 12.8 | 6.48 | 1.98 |
Такие отличающиеся результаты создания различной конформационной структуры макромолекул в случае синтеза ПС в присутствии соответствующих динитронов при различных значениях концентрации обусловлены акцептирующей способностью нитронов, а также стабильностью их и образующихся in situ алкоксиаминов. Действительно, формирования звездообразных структур можно было бы ожидать при низких значениях концентрации нитронов, но в случае незначительной акцептирующей способности динитронов или малой стабильности алкоксиаминов будет в основном образовываться линейный полимер с конформацией гауссова клубка. В таких случаях повышение концентрации может приводить к формированию разветвленных (трех- или четырехлучевых звездообразных) макромолекул, что наблюдается в случае БДН. Отметим, при проведении полимеризации in situ будут образовываться асимметричные звездообразные структуры с неравноценными лучами. Напротив, повышение концентрации динитронов может привести к превалированию реакции акцептирования нитроном радикалов роста с формированием только мононитроксилов, и, как следствие, к образованию линейных структур в форме статистического клубка; эта закономерность четко наблюдается при повышении концентрации ФДН. Также в случае МДН и ФДН наблюдается увеличение радиуса инерции с ростом концентрацией динитронов.
ЗАКЛЮЧЕНИЕ
В результате проведенных исследований установлено, что динитроны способны выступать в качестве регуляторов радикальной полимеризации стирола. При этом концентрационные и температурные параметры процесса полимеризации наряду со строением динитронов играют важную роль в возможности реализации механизма обратимого ингибирования. Так, оптимальным условием для протекания синтеза ПС по схеме обратимого ингибирования в присутствии N,N–диметилглиоксальдинитрона (наименее пространственно-затрудненного из используемых динитронов) является высокотемпературный режим (130°С). В случае использования N,N–дифенилглиоксальдинитрона, имеющего в своем составе объемные фенильные радикалы, процесс полимеризации стирола в условиях обратимого ингибирования может протекать при 90°С. Оценкой некоторых гидродинамических характеристик синтезированного ПС установлено, что варьированием концентрации используемых динитронов как акцепторов активных радикалов возможно формирование звездообразных макромолекул.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 20-03-00150) с использованием оборудования Центра коллективного пользования “Новые материалы и ресурсосберегающие технологии” (ННГУ им. Н.И. Лобачевского).
Список литературы
Moad G. The Chemistry of Radical Polymerization. Amsterdam: Elsevier Science, 2018.
Reversible Deactivation Radical Polymerization: Mechanisms and Synthetic Methodologies / Eds by K. Matyjaszewski, H. Gao, B.S. Sumerlin, N.V. Tsarevsky. Washington: American Chemical Society, 2018.
Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science / Ed. by D. Gigmes. UK: The Royal Society of Chemistry, 2016
Zaremski M.Yu. // Polymer Science C. 2015. V. 57. № 1. P. 65.
Nicolas J., Guillaneuf Y., Lefay C., Bertin D., Gigmes D., Charleux B. // Prog. Polym. Sci. 2013. V. 38. № 1. P. 63.
Edeleva M., Audran G., Marque S., Bagryanskaya E. // Materials. 2019. V. 12. № 5. P. 688.
Bao Y., Guégain E., Nicolas V., Nicolas J. // Chem. Commun. 2017. V. 53. P. 4489.
Wang H.C., Li Y., Yang M.J. // Sensors Act. B. 2007. V. 124. № 2. P. 360.
Lindner S.M., Kaufmann N., Thelakkat M. // Org. Electronics. 2007. V. 8. P. 69.
Destarac M. // Polym. Chem. 2018. V. 9. № 40. P. 4947.
Milan M. // Curr. Org. Chem. 2018. V. 22. № 13. P. 1264.
Edeleva M.V., Parkhomenko D.A., Morozov D.A., Dobrynin S.A., Trofimov D.G., Kanagatov B., Kirilyuk I.A., Bagryanskaya E.G. // J. Polym. Sci., Polym. Chem. 2014. V. 52. P. 929.
Ballard N., Aguirre M., Simula A., Agirre A., Leiza J.R., Asua J.M., S. van Es // Acs Macro Lett. 2016. V. 5. № 9. P. 1019.
Simula A., Ballard N., Aguirre M., Leiza J.R., S. van Es, Asua J.M. // Eur. Polym. J. 2019. V. 110. P. 319.
Sciannamea V., Jérôme R., Detrembleur C. // Chem. Rev. 2008. V. 108. № 3. P. 1104.
Kolyakina E.V., Grishin D.F. // Russ. Chem. Rev. 2009. V. 78. № 6. P. 535.
Grishin D.F., Kolyakina E.V., Polyanskova V.V., Grishin I.D. // Russ. J. Appl. Chem. 2007. V. 80. № 1. P. 122.
Zaremski M.Yu., Orlova A.P., Garina E.S., Olenin A.V., Lachinov M.B., Golubev V.B. // Polymer Science A. 2003. V. 45. № 6. P. 502.
Grishin D.F., Semenycheva L.L., Bulgakova S.A., Mazanova L.M., Kolyakina E.V. // Polymer Science A. 2003. V. 45. № 4. P. 305.
Grishin D.F., Semenycheva L.L., Sokolov K.V., Kolyakina E.V. // Polymer Science B. 2000. V. 42. № 7–8. P. 189.
Zaremski M.Yu., Reznichenko A.L., Grinevich Yu.V., Garina E.S., Lachinov M.B., Golubev V.B. // Polymer Science A. 2009. V. 47. № 6. P. 5365.
Grishin D.F., Kolyakina E.V., Pavlovskaya M.V., Lazarev M.A., Shchepalov A.A. // Controlled/Living Radical Polymerization: Progress in RAFT, DT, NMP & OMRP. ACS Symp. Ser. Am. Chem. Soc. / Ed. by K. Matyjiaszewski. Washington, 2009. V. 1024. Ch. 7. P. 95.
Sciannamea V., Guerrero-Sanchez C., Schubert U.S., Catala J.-M., Jerome R., Detrembleur C. // Polymer. 2005. V. 46. № 23. P. 9632.
Sciannamea V., Catala J.M., Jerome R., Detrembleur C. // J. Polym. Sci., Polym. Chem. 2007. V. 45. № 7. P. 1219.
Kolyakina E.V., Alyeva A.B., Grishin D.F. // Appl. Solid State Chem. 2018. V. 2. P. 29.
Kolyakina E.V., Alyeva A.B., Sazonova E.V., Shchepalov A.A., Grishin D.F // Russ. Chem. Bull. 2019. V. 68. № 8. P. 1585.
Энциклопедия полимеров / Под ред. В.А. Кабанова. М.: Советская энциклопедия, 1972. Т. 1.
Синтезы органических препаратов / Под ред. Б.А. Казанского. М.: Изд-во иностр. лит., 1953. Т. 4.
Вайсберг А., Проскауэр Э., Риддик Дж., Тупс Э. Органические растворители. М: Изд-во иностр. лит. 1958.
Raspertova I., Osetska O., Gubina K., Lampeka R. // Polyhedron. 2011. V. 30. P. 2320.
Беленький Б.Г., Виленчик Л.З. Хроматография полимеров. М.: Химия, 1978.
Эскин В.Е. Рассеяние света растворами полимеров. Л.: Наука, 1986.
Yi Xu, Yan Gu, Steven Y. Qian // Int. J. Mol. Sci. 2012. V. 13. № 11. P. 14648.
Detlef Rehorek, Edward G. Janzen // J. für Praktische Chemie. 1985. V. 327. № 6. P. 968.
Burchard W. // Adv. Polym. Sci. 1999. V. 143. P. 113.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)