

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 5, стр. 356-363
СИНТЕЗ И СВОЙСТВА СЕТЧАТЫХ ПАРНЫХ ПОЛИМЕРОВ НА ОСНОВЕ ПОЛИ-N-ВИНИЛПИРРОЛИДОНА
В. Н. Кижняев a, *, Ф. А. Покатилов a, Е. В. Акамова a
a Иркутский государственный университет
664003 Иркутск, ул. К. Маркса, 1, Россия
* E-mail: kizhnyaev@chem.isu.ru
Поступила в редакцию 13.01.2020
После доработки 10.03.2020
Принята к публикации 30.03.2020
Аннотация
Осуществлено реакционное смешение поли-N-винилпирролидона, модифицированного введением “якорных” оксирановых фрагментов, с полимерами различной природы, содержащими в структуре N–Н незамещенные тетразольные циклы. Получены амфифильные и полиэлектролитные сетчатые парные полимеры типа “conetworks”, способные к ограниченному набуханию в водных средах с образованием гидрогелей, проявляющих чувствительность к изменению рН, ионной силы и температуры окружающей среды.
ВВЕДЕНИЕ
Гидрогели на основе сетчатых полимеров одновременно сочетают в себе свойства растворов (незначительное содержание полимерной компоненты на фоне большого содержания иммобилизованного растворителя, высокие значения коэффициентов диффузии и т.д.) и твердых тел (сохранение формы, относительная механическая прочность и т.д.). Это предопределило широкое использование гидрогелевых материалов в различных областях науки, производства, сельского хозяйства и медицины в качестве суперабсорбентов воды, сорбентов ценных и экологически опасных веществ, носителей лекарственных субстанций, мягких и биосовместимых материалов, металлонаполненных наноматериалов и т.д. [1–6]. В последнее время особый интерес исследователей возник к так называемым “умным” полимерным гелям, которые могут направленно и обратимо изменять свои свойства под воздействием внешних факторов, например при варьировании состава, ионной силы, рН и температуры окружающей среды [4, 7–14]. К числу гелевых материалов, проявляющих подобные способности, относятся гидрогели на основе сетчатых полимеров амфифильной и полиэлектролитной природы; среди них особое место занимают макромолекулярные образования, получившие название “amphiphilic conetworks” (амфифильные блок-сополимерные сетки) [15]. Они представляют собой сшитые блок-сополимеры, которые в зависимости от природы составляющих их цепочечных фрагментов обладают различными типами чувствительности к изменению параметров внешней среды, в том числе и рН- и термочувствительностью [15–17]. К типу “conetworks” можно отнести и сетчатые структуры, представляющие собой парные полимеры, получаемые посредством реакционного смешения высокомолекулярных соединений различной природы [4, 18–20]. Формирование сетчатых парных полимеров происходит в результате взаимодействия обладающих химическим сродством “якорных” функциональных групп (например, N–Н незамещенных тетразольных и оксирановых гетероциклов), расположенных случайным образом в структуре разнородных макромолекул, что приводит к их ковалентному связыванию [21, 22]. Получаемая подобным способом трехмерная сетка может объединять цепочечные фрагменты самых разных по природе полимеров – ионизующихся и неиногенных, гидрофильных и гидрофобных, чувствительных к различным изменениям в окружающей среде.
Одним из перспективных полимерных компонентов для создания парных полимеров является поли-N-винилпирролидон (ПВП), проявляющий широкий спектр ценных в практическом отношении свойств: гидрофильность, отсутствие токсичности, биосовместимость, комплексообразующую способность по отношению к ионам металлов и различным органическим веществам, включая лекарственные субстанции [23]. Варьируя природу второго участника реакционного смешения, например используя ионизующийся полимер, можно получать сетчатые полимерные продукты, обладающие еще более высокими по сравнению с ПВП сорбционными характеристиками по отношению к воде и другим веществам, чувствительными на изменения рН, ионной силы и температуры среды. При сочетании с гидрофобным полимерным реагентом, например с ПС, возможно образование макромолекулярных систем “amphiphilic conetworks”, с проявлением свойств, присущих данному типу сетчатых материалов.
В настоящей работе представлены результаты исследования процесса реакционного смешения ПВП, модифицированного посредством введения в его макромолекулы небольших количеств звеньев 2-(винилоксиэтокси)метилоксирана (коммерческое название “Винилокс”) (ВО)

с тетразолсодержащими полимерами различной природы. В качестве ионогенного полимерного реагента был выбран сополимер 5-винилтетразола (ВТ) с акрилонитрилом эквимольного состава (ПВТ(АН)), для которого продукты реакционного смешения проявляют полиэлектролитные свойства. Гидрофобным полимерным реагентом для получения амфифильных парных полимеров служил полистирол, модифицированный посредством введения в его макромолекулы небольших количеств звеньев ВТ (ПС(ВТ)) для обеспечения межмакромолекулярного ковалентного связывания с ПВП(ВО):
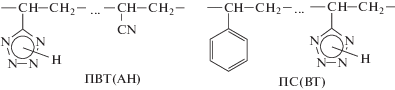
Рассмотрены некоторые свойства получаемых сетчатых парных полимеров и гидрогелей на их основе.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали коммерческий ПВТ(АН) (производства Открытого акционерного общества “Сибреактив”, Россия), синтезированный азидированием полиакрилонитрила. Состав сополимера рассчитывали на основании данных по содержанию кислотных N–Н незамещенных тетразольных циклов, определяемых методом потенциометрического титрования растворов полимера 0.1 М раствором NaOH в водной среде. Образцы ПВП(ВО) с содержанием мономерных звеньев винилового эфира 0.06, 0.085, 0.10 и 0.17 мол. долей получали сополимеризацией N-винилпирролидона с ВО в среде этанола под действием ДАК (0.5 мас. % от суммы момномеров) при температуре 60°С и продолжительности 48 ч. Полученные полимерные продукты высаживали и промывали диэтиловым эфиром, сушили под вакуумом до постоянной массы. Введение в макромолекулярную цепь полистирола тетразолсодержащих фрагментов в количестве 0.06, 0.11 и 0.16 мол. долей осуществляли посредством совместной полимеризации стирола с ВТ в среде ДМФА под действием ДАК (0.5 мас. % от суммы мономеров) при температуре 60°С и продолжительности 24 ч. Продукты сополимеризации высаживали и промывали этанолом, сушили под вакуумом до постоянной массы. Соотношение мономерных звеньев в полимерных продуктах определяли из данных элементного анализа, который выполняли с использованием CHN-анализатора FLASH ВА 1112 Series, химического анализа на содержание эпоксидных групп [24] (модифицированный ПВП) и кислотных N–Н незамещенных тетразольных циклов (модифицированный ПС). Все аналитические методы продемонстрировали удовлетворительное соответствие результатов оценки состава сополимеров.
Взаимодействие ПВП(ВО) с ПВТ(АН) и ПС(ВТ) осуществляли в среде ДМФА при различных суммарных концентрациях и соотношениях полимеров, температуре и продолжительности. Для проведения процесса при комнатной температуре смешивали предварительно приготовленные растворы в ДМФА оксиран- и тетразолсодержащих полимеров. Затем реакционную смесь загружали в ампулы, продували аргоном, запаивали и выдерживали в термостате при указанной температуре и продолжительности. Реакция между полимерами сопровождалась гелеобразованием; время потери текучести системой τ фиксировали визуально. Полученную в результате реакции гелеобразную массу выдерживали в течение недели под периодически заменяемым ДМФА с целью удаления непрореагировавших исходных полимерных реагентов и достижения парным полимером равновесного набухания. Далее в зависимости от целей исследования из геля удаляли растворитель с целью определения выхода гель-фракции (другими словами, выхода парного полимера) и степени его набухания в ДМФА, либо гели выдерживали в течение трех недель под периодически заменяемой дистиллированной водой для удаления ДМФА. Таким образом, получали гидрогели на основе парных полимеров, содержащих неионизованные тетразольные фрагменты. С целью перевода кислотных N–Н незамещенных тетразольных циклов в солевую форму

образцы гидрогелей парного полимера ПВП(ВО)–ПВТ(АН) погружали в 0.1 М водный раствор NaOH до равновесного набухания, после чего ионизованный гидрогель выдерживали в течение недели под периодически заменяемой дистиллированной водой до нейтральной рН реакции промывных вод.
Степень набухания полимерных образцов в различных растворителях определяли гравиметрическим методом и вычисляли по формуле
где mг и mп – масса набухшего гидрогеля и сухого полимера соответственно. Задаваемую температуру при исследовании процессов набухания поддерживали с помощью воздушного термостата “Memmert” (точность термостатирования ±0.5°С). Варьирование рН среды осуществляли добавлением в систему соляной кислоты или гидроксида натрия; контроль за рН среды проводили с использованием иономера ЭВ-74.Пленки смесей полимеров отливали из реакционной массы до точки гелеобразования на стеклянную подложку с последующим удалением растворителя. Микрофотографии пленок в отраженном УФ-свете получали с использованием флуоресцентного микроскопа “Биоптик С-400” при увеличении 80. ИК-спектры пленок ПВП(ВО) и его смесей с ПВТ(АН) регистрировали на ИК-фурье-спектрометре “Инфралюм ФТ-801”. Для определения поверхностных свойств парных полимеров пленки готовили поливом на стеклянную подложку с последующим выдерживанием при температуре 80°С в течение двух недель. Затем пленки отмывали от непрореагировавших полимеров выдерживанием под ДМФА и сушили под вакуумом. Краевой угол смачивания полимерных пленок водой определяли методом сидячей капли. Вискозиметрическое титрование раствора ПВП(ВО) раствором ПВТ(АН) в ДМФА (концентрация полимеров 1 г/дл) проводили с помощью капиллярного вискозиметра с висячим уровнем при 25°С. Расчет аддитивных значений приведенной вязкости растворов смесей полимеров при их различном соотношении выполняли в соответствии с методикой [25].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
ПВП является неионогенным водорастворимым полимером. Введение в его макромолекулы небольшого количества звеньев ВО не сказывается на совместимости полимера с водой. С целью получения на основе ПВП(ВО) парных полимеров амфифильной и полиэлектролитной природы в качестве второго участника реакционного смешения были выбраны гидрофобный ПС, содержащий в структуре небольшое количество “якорных” тетразольных фрагментов, и сополимер ВТ с АН эквимольного состава. Во втором случае тетразольные циклы выполняют роль не только “якорных” групп, обеспечивающих в результате реакции с оксирановыми циклами ковалентное связывание разнородных макромолекул, но и роль функциональных фрагментов, благодаря которым парные полимеры приобретают свойства полиэлектролитов кислотной природы. Реакционное смешение ПВП(ВО) с ПС(ВТ) и ПВТ(АН) осуществляли в среде общего по отношению ко всем исходным полимерам растворителя ДМФА. Смеси реагирующих полимеров в ДМФА оставались гомогенными в течение всего процесса формирования парных полимеров.
Особенностью системы ПВП(ВО)–ПВТ(АН) является то, что реакции между “якорными” тетразольными и оксирановыми циклами, протекающей с заметной скоростью при повышенных температурах, предшествует образование уже при смешении растворов полимеров интерполимерного комплекса (ИПК), стабилизированного водородными связями между тетразольным и пирролидоновым циклами, принадлежащих разнородным макромолекулам:
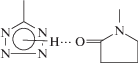
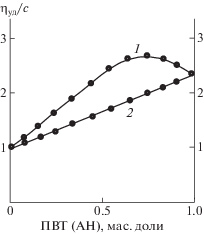
На участие карбонильной группы лактамного цикла в образовании водородных связей указывает смещение в ИК-спектрах смеси ПВП(ВО) с ПВТ(АН) полосы поглощения валентных колебаний связи С=О на 20 см–1 в более низкочастотную область (1674 → 1654 см–1) по сравнению с аналогичной полосой в спектре ПВП(ВО). Другим доказательством интерполимерного взаимодействия между ПВП(ВО) и ПВТ(АН) служат результаты вискозиметрии. Экспериментально определяемые значения приведенной вязкости растворов смесей полимеров в ДМФА превосходят расчетные аддитивные значения вязкости при любых соотношениях компонентов (рис. 1), что, согласно работе [25], свидетельствует о существовании ИПК.
Рис. 1.
Приведенная вязкость растворов смесей ПВП(ВО) с ПВТ(АН) в ДМФА при 25°С в зависимости от массовой доли тетразолсодержащего полимера. 1 – эксперимент; 2 – соответствующая расчетная аддитивная зависимость.
Отмеченное интерполимерное взаимодействие определило и выбор ионогенного тетразолсодержащего участника реакционного смешения с ПВП(ВО). Содержание звеньев ВТ в ПВТ(АН) 0.5 мол. долей является максимальным, при котором смеси тетразол- и пирролидонсодержащих полимеров в ДМФА остаются гомогенными во всем диапазоне изученных концентраций. При большем содержании звеньев ВТ в сополимере ПВТ(АН) (как и при использовании гомополимера ВТ) при любых концентрациях полимеров образующийся между ПВП(ВО) и ПВТ(АН) интерполимерный комплекс выделяется из раствора в ДМФА в отдельную фазу. При реакционном смешении ПВП(ВО) с модифицированным ПС(ВТ), содержащим незначительные количества звеньев ВТ, проявления интерполимерного взаимодействия между участниками реакции не отмечено.
При повышенных температурах в смесях ПВП(ВО) с ПС(ВТ) и ПВТ(АН) протекает процесс реакционного смешения полимеров посредством ковалентного связывания разнородных макромолекул вследствие реакции “якорных” тетразольных и оксирановых функциональных фрагментов.
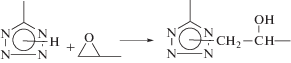
Об этом свидетельствует, например, возрастание однородности пленок, отлитых из реакционной смеси ПВП(ВО)–ПС(ВТ), с увеличением продолжительности реакции между полимерами (рис. 2). ПВП(ВО) и ПС(ВТ) – несмешивающиеся полимеры, поэтому, несмотря на гомогенность их смесей в среде ДМФА, после удаления растворителя каждый из полимеров образует свою конденсированную фазу. Пленки, отлитые перед самым моментом гелеобразования в реакционной системе, представляют собой визуально прозрачные образования, поскольку смешение полимеров происходит на молекулярном уровне.
Рис. 2.
Фотографии (1–3) и микрофотографии (×80) в отраженном УФ-свете (4–6) пленок, отлитых из смеси ПВП(ВО)–ПС(ВТ) (1, 4) и реакционных смесей ПВП(ВО)–ПС(ВТ), полученных за 1 (2, 5) и 3 суток (3, 6). ПВП(ВО) : ПС(ВТ) = 1 : 1, Σспл = 10 г/дл, Т = 80°С.
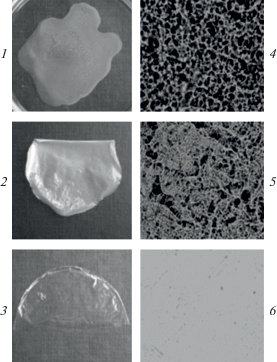
Реакционное смешение полимеров проводили при суммарных концентрациях полимерных реагентов (3–10 г/дл), превышающих критическую концентрацию образования в системе флуктуационной физической сетки зацеплений между макромолекулярными клубками (область кроссовера) [26]. Как следствие, при реализации ковалентного связывания макромолекул в системе формировалась единая пространственная химическая сетка. Таким образом, реакция между полимерами сопровождалась гелеобразованием реакционной смеси, а продуктами реакции были сетчатые парные полимеры. Процесс формирования пространственной макромолекулярной сетки можно охарактеризовать двумя параметрами: временем потери текучести системой τ и содержанием гель-фракции (т.е. выходом сетчатого парного полимера), которые в свою очередь зависят от природы смешиваемых компонентов и условий осуществления реакционного смешивания полимеров. Следует отметить, что выход гель-фракции во всех опытах оценивали за одинаковый промежуток времени (через 7 суток) после точки гелеобразования реакционной смеси. К этому моменту на основании постоянства указанного параметра и коэффициента набухания Кнаб продуктов реакции во времени через пятые сутки после потери текучести реакционной системой (рис. 3) формирование сетчатой структуры парного полимера следует считать полностью завершенным.
Рис. 3.
Выход гель-фракции при реакционном смешении (1, 2) и степень набухания (3, 4) парных полимеров ПВП(ВО)–ПВТ(АН) (1, 3) и ПВП(ВО)–ПС(ВТ) (2, 4) в ДМФА при 25°С в зависимости от продолжительности синтеза. ПВП(ВО) : ПВТ(АН)(ПС(ВТ)) = 1 : 1; Σспл = = 10 г/дл; Т = 80°С; мольная доля звеньев ВО в ПВПД – 0.06, звеньев ВТ в ПС – 0.11.

В системах ПВП(ВО)–ПС(ВТ) и ПВП(ВО)–ПВТ(АН) ускорению процесса гелеобразования и возрастанию выхода гель-фракции способствует увеличение содержания “якорных” оксирановых фрагментов в модифицированном ПВП(ВО) и повышение суммарной концентрации полимерных реагентов (рис. 4). Однако если количественные значения выхода парного полимера от условий реакции для обеих систем практически совпадают (кривые 3, 4, 9 и 10), то временные характеристики гелеобразования в реакционных системах сильно разнятся (кривые 1, 2, 7 и 8). Вероятно, отмеченное выше интерполимерное взаимодействие между ПВП(ВО) и ПВТ(АН) в какой-то степени сохраняется и при повышенных температурах, способствуя лучшей совместимости компонентов, поэтому времена потери текучести при реакции данной пары полимеров измеряются минутами или часами. В случае системы ПВП(ВО)–ПС(ВТ) временные характеристики гелеобразования измеряются сутками. Кроме того, образование сетчатого парного полимера при реакции ПВП(ВО) с ПВТ(АН) отмечено при температурах ниже 80°С (вплоть до комнатной) (рис. 4, кривые 5, 6). Общие закономерности проявляются и во влиянии соотношения полимерных реагентов на рассматриваемые параметры процесса реакционного смешения. Для обеих систем ПВП(ВО)–ПС(ВТ) и ПВП(ВО)–ПВТ(АН) наименьшие величины времен потери текучести реакционными смесями (отличия только в абсолютных значениях) и максимальный выход гель-фракции (92–97%) отмечены при массовой доле ПВП(ВО) в исходной смеси 0.5–0.7 (рис. 5). По-видимому, это оптимальные соотношения компонентов, при которых с наибольшей скоростью и конверсией формируется сетчатая структура парного полимера. Значительное преобладание одного из полимерных реагентов в смеси влечет за собой замедление процесса сшивания разнородных макромолекул и существенное снижение выхода гель-фракции.
Рис. 4.
Время потери текучести τ (1, 2, 5, 7, 8) и выход гель-фракции (3, 4, 6, 9, 10) в реакционных системах ПВП(ВО)–ПВТ(АН) (1–6) и ПВП(ВО)–ПС(ВТ) (7–10) в ДМФА в зависимости от мольного содержания звеньев ВО (1, 3, 7, 9), температуры реакции (5, 6) и суммарной концентрации компонентов (2, 4, 8, 10). ПВП(ВО) : ПВТ(АН)(ПС(ВТ)) = 1 : 1; Σспл = 10 г/дл (1, 3, 5, 6, 7, 9); Т = 80°С (1–4, 7–10); мольная доля звеньев ВО в ПВП – 0.06 (2, 4, 5, 6, 8, 10), звеньев ВТ в ПС – 0.11 (7–10).

Рис. 5.
Время потери текучести τ (1, 3) и выход гель-фракции (2, 4) в реакционных системах ПВП(ВО)–ПВТ(АН) (1, 2) и ПВП(ВО)–ПС(ВТ) (3, 4) в ДМФА в зависимости от массовой доли ПВП(ВО) в исходной смеси полимеров. Σспл= 10 г/дл; Т = 80°С; мольная доля звеньев ВО в ПВПД – 0.06, звеньев ВТ в ПС – 0.11.

Как отмечено выше, при реакции ПВП(ВО) с ПС(ВТ) и ПВТ(АН) процесс формирования пространственной сетки не заканчивается в момент потери текучести реакционной системой, а завершается приблизительно на пятые сутки после гелеобразования. В результате образуются соответственно амфифильные и полиэлектролитные сетчатые парные полимеры, склонные к ограниченному набуханию в ДМФА и в воде. Сорбционная способность (Кнаб) по отношению к указанным жидкостям определяется как структурой пространственной сетки, так и природой образующих ее полимеров. Для обоих типов парных полимеров снижению степени набухания (вследствие формирования более жесткой сетки) способствует увеличение содержания “якорных” оксирановых фрагментов в исходном ПВП(ВО), повышение суммарной концентрации полимерных реагентов при синтезе (рис. 6) и, в случае парного полимера ПВП(ВО)–ПВТ(АН), рост в его структуре доли макромолекул ПВП(ВО) (рис. 7, кривые 1–3). При этом сорбционная способность парных полимеров ПВП(ВО)–ПВТ(АН) изученного диапазона составов даже в неионизованном состоянии по отношению к воде превосходит таковую по отношению к ДМФА. Ионизация макромолекул ПВТ(АН) способствует еще более существенному набуханию парных полимеров в водной среде с максимальным значением Кнаб образующихся гидрогелей при рН, равном 8.5 (рис. 7, кривая 4). И, наоборот, подавление диссоциации кислотной компоненты (уменьшение значения pH среды) сопровождается резким коллапсом гидрогелей. Аналогичное резкое снижение сорбционных свойств вызывает возрастание ионной силы водной среды, вызванное добавлением небольших количеств низкомолекулярной соли (рис. 7, кривая 5). Таким образом, гидрогели на основе парных полимеров ПВП(ВО)–ПВТ(АН) проявляют классические полиэлектролитные свойства, характеризующиеся высокой амплитудой изменения сорбционных свойств в зависимости от рН и ионной силы водной среды.
Рис. 6.
Степень набухания парных полимеров ПВП(ВО)–ПВТ(АН) (1, 2) и ПВПД(ВО)–ПС(ВТ) (3, 4) в ДМФА при 25°С в зависимости от мольного содержания звеньев ВО (1, 3) и суммарной концентрации компонентов (2, 4). ПАК : ПВП(ВО) : : ПВТ(АН)(ПС(ВТ)) = 1 : 1; Σспл = 10 г/дл (1, 3); Т = = 80°С; мольная доля звеньев ВО в ПВПД – 0.06 (2, 4), звеньев ВТ в ПС – 0.11.
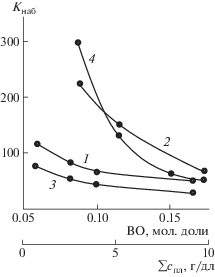
Рис. 7.
Степень набухания парных полимеров ПВП(ВО)–ПВТ(АН) (1–5) и ПВП(ВО)–ПС(ВТ) (6–8) в ДМФА (1, 6) при 25°С, в воде при 25 (2, 4, 5, 7) и 60°С (3, 8) в зависимости от массовой доли ПВП(ВО) в парном полимере (1–3, 6–8), рН среды (4), концентрации Na2SO4 в водном растворе (5). ПВП(ВО) : ПВТ(АН)(ПС(ВТ)) = 1 : 1 (4, 5); Σспл = 10 г/дл; Т = 80°С; мольная доля звеньев ВО в ПВПД – 0.06, звеньев ВТ в ПС – 0.11.

В случае амфифильных парных полимеров ПВП(ВО)–ПС(ВТ) при избытке ПС преобладает сорбционная способность по отношению к органическому растворителю, и лишь при избыточном содержании макромолекул ПВП(ВО) значения Кнаб парных полимеров в воде становятся выше аналогичных значений в ДМФА (рис. 7, кривые 6, 7). Кроме того, следует отметить экстремальный вид кривых с минимумом зависимости степени набухания от соотношения компонентов в полимерном продукте, даже в водной среде. Можно было ожидать, что с увеличением доли макромолекул гидрофильного ПВП(ВО) в парном полимере его степень набухания в воде будет монотонно возрастать. Однако, как отмечено выше, парные полимеры, синтезируемые при соотношениях полимерных реагентов близких к эквивалентному, характеризуются наиболее жесткой пространственной сеткой. Вероятно, минимальные значения Кнаб при указанном соотношении компонентов объясняются превалированием эффекта ужесточения сетчатой структуры парного полимера над увеличением гидрофильности по мере повышения содержания в его структуре макромолекул ПВП(ВО). Амфифильный характер парных полимеров ПВП(ВО)–ПС(ВТ) проявляется и в снижении сорбционной способности по отношению к воде с ростом температуры. Причина подобного эффекта заключается в проявлении макромолекулярными цепями ПС(ВТ) гидрофобных взаимодействий между структурными фрагментами сетки в водных средах. С повышением температуры гидрофобные взаимодействия усиливаются, увеличивая жесткость полимерной сетки, что и вызывает снижение степени набухания гидрогелей.
Соотношение компонентов сказывается и на поверхностных свойствах пленочного материала на основе парных полимеров ПВП(ВО)–ПС(ВТ). Как и ожидалось, увеличение доли гидрофильной компоненты в парных полимерах способствует уменьшению значений углов смачивания пленок водой (рис. 8), т.е. повышению сродства к воде пленочного материала.
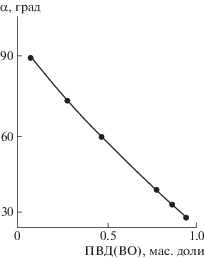
ЗАКЛЮЧЕНИЕ
Представленный вариант реакционного смешения гидрофильного ПВПД с ионогенным и гидрофобным полимерами позволил получить водосовместимые сетчатые парные полимеры полиэлектролитной и амфифильной (типа “conetworks”) природы. Особенности строения продуктов реакционного смешения, а именно, сочетание разнородных цепочечных фрагментов, связанных ковалентными связями только в местах взаимопроникновения макромолекулярных клубков, обусловливают и особенности поведения в водных средах. Так, парные полимеры ПВП(ВО)–ПВТ(АН) разного состава, содержащие даже неионизованные тетразольные фрагменты, обладают сродством к воде, образуя гидрогели с высокой степенью набухания. В то же время интерполимерные комплексы ПВП с гомополимером ВТ, стабилизированные водородными связями, или статистические сополимеры N-винилпирролидона с ВТ в неионизованном состоянии в воде не растворяются и не набухают. Гидрогели парных полимеров ПВПД(ВО)–ПВТ(АН) проявляют присущую полиэлектролитным системам чувствительность к изменению рН окружающей среды и присутствию низкомолекулярных солей. Гидрогели на основе парных полимеров ПВП(ВО)–ПС(ВТ), подобно другим амфифильным полимерным системам, характеризуются термочувствительностью, выражающейся в уменьшении коэффициентов водопоглощения с повышением температуры окружающей среды. В целом вариантов построения сетчатых полимерных систем на основе ПВП посредством предложенного подхода реакционного смешения может быть достаточно много. При этом, варьируя подбором второго участника смешения гидрофильно-гидрофобный баланс получаемых парных полимеров, в перспективе можно добиться проявления гидрогелями так называемого “умного” поведения, заключающегося в обратимом переходе из набухшего в сколлапсированное состояние в узком интервале изменений параметров окружающей среды.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований и Правительства Иркутской области (код проекта 17-43-380005).
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов, требующего раскрытия в данной статье.
Список литературы
Chaterji S., Kwon I.K., Park K. // Prog. Polym. Sci. 2007. T. 32. C. 1083.
Biorelated Polymers and Gels / Ed.by N. Okano. San Diego: Acad Press, 1998.
Kopecek J., Yang J. // Polym. Int. 2007. V. 56. № 9. P. 1078.
Pavlyuchenko V.N., Ivanchev S.S. // Polymer Science A. 2009. V. 51. № 7. P. 743.
Ozay H., Kubilay S., Aktas N., Sahiner N. // Int. J. Polym. Mater. Polym. Biomater. 2011. V. 60. № 2. P. 163.
Ozay O., Aktas N., Inger E., Sahiner N. // Int. J. Hydrogen Energy. 2011. V. 36. № 3. P. 1998.
Philippova O.E. // Polymer Science C. 2000. V. 42. № 2. P. 208.
Horkay F., Tasaki I., Basser P. // Biomacromolecules. 2000. V. 1. № 1. P. 84.
Aseyev V.O., Tenhu H., Winnik F.M. // Adv. Polym. Sci. 2006. V. 196. P. 1.
Kramarenko E.Yu., Philippova O.E., Khokhlov A.R. // Polymer Science C. 2006. V. 48. № 1. P. 1.
Chaterji S., Kwon I.K., Park K. // Prog. Polym. Sci. 2007. V. 32. P. 1083.
Galaev I., Mattiasson B. Smart Polymers: Applications in Biotechnology and Biomedicine. New York: CRC Press, 2007.
Aguilar M.R., Elvira C., Gallardo A., Vazquez B., Roman J.S. // Topics in Tissue Engineering. 2007. V. 3. P. 1.
Starodubtsev S.G., Vasilevskaya V.V., Khokhlov A.R. // Smart Polymers: Applications in Biotechnology and Biomedicine, Second Edition / Ed. by Galaev I., Mattiasson BO. London; New York: CPC Press, 2008. P. 81.
Erdodi G., Kennedy J.P. //Progr. Polym. Sci. 2006. V. 31. № 1. P. 1.
Tobis J., Thomann Y., Tiller J.C. // Polymer. 2010. V. 51. № 1. P. 35.
Rikkou M.D., Loizou E., Patrickios C.S., Porcar L. // Eur. Polym. J. 2010. V. 46. № 3. P. 441.
Baranov A.O., Kotova A.V., Zelenetskii A.N., Prut E.V. // Russ. Chem. Rev. 1997. V. 66. № 10. P. 877.
Askadskii A.A. // Russ. Chem. Rev. 1999. V. 68. № 4. P. 317.
Plate N.A., Litmanovich A.D., Kudryavtsev Ya.V. Macromolecular Reactions in Polymer Melts and Blends. Moscow: Nauka, 2008.
Kizhnyaev V.N., Pokatilov F.A., Zhitov R.G., Proidakov A.G., Krakhotkina E.A. // Polymer Science B. 2015. V. 57. № 5. P. 504.
Kizhnyaev V.N., Pokatilov F.A., Shabalin A.I., Zhitov R.G. // e-Polymer. 2019. V. 19. P. 421.
Kirsh Y.E. Water Soluble Poly-N-vinylamides. Weinheim: Wiley-VCH, 1998.
Торопцева А.М., Белогородская К.В., Бондаренко В.М. Лабораторный практикум по химии и технологии высокомолекулярных соединений. Л.: Химия, 1972.
Bel'nikevich N.G., Budtova T.V., Nikolaeva O.V., Vesnebolotskaya S.A.// Polymer Science B. 2002. V. 44. № 1–2. P. 27.
Бартенев Г.М., Френкель С.Я. Физика полимеров. Л.: Химия, 1990.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)