

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 5, стр. 372-381
СИНТЕЗ БЛОК-СОПОЛИМЕРОВ АКРИЛОВОЙ КИСЛОТЫ И н-БУТИЛАКРИЛАТА В УСЛОВИЯХ ОБРАТИМОЙ ПЕРЕДАЧИ ЦЕПИ В ВОДНО-СПИРТОВОЙ СРЕДЕ
Н. С. Серхачева a, *, Е. В. Черникова b, Н. И. Прокопов a, В. В. Огай a, К. О. Минеева b, Д. С. Целоусов a, И. О. Плюснина a
a МИРЭА – Российский технологический университет
Институт тонких химических технологий имени М.В. Ломоносова
119571 Москва, пр. Вернадского, 86, Россия
b Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: natalia-serkhacheva@yandex.ru
Поступила в редакцию 18.03.2020
После доработки 18.04.2020
Принята к публикации 13.05.2020
Аннотация
Исследованы закономерности дисперсионной полимеризации н-бутилакрилата в водно-спиртовой среде в присутствии гидрофильного полимерного агента обратимой передачи цепи на основе полиакриловой кислоты с тритиокарбонатной группой внутри цепи. Показано, что порционное введение мономера в синтез позволяет достичь более высоких предельных конверсий. При этом образующиеся блок-сополимеры характеризуются относительно узким молекулярно-массовым распределением, а их среднечисленная молекулярная масса линейно растет с увеличением концентрации мономера. Полученные дисперсии триблок-сополимера полиакриловая кислота–блок–поли(н-бутилакрилат)–блок–полиакриловая кислота имеют унимодальное распределение частиц по размерам, которое сохраняется после диализа против воды. Показана возможность применения синтезированных дисперсий в качестве полимерной матрицы для иммобилизации наночастиц оксида цинка и в качестве полимерного прекурсора в процессе затравочной полимеризации стирола.
ВВЕДЕНИЕ
На протяжении последних лет синтез самоорганизующихся амфифильных наноразмерных объектов на основе блок-сополимеров вызывает повышенный интерес со стороны многих исследователей. Такие объекты в зависимости от природы блоков могут найти применение в различных областях техники, технологии и биомедицины [1].
Самоорганизованные структуры на основе амфифильных блок-сополимеров можно получить двумя способами [2]: мицеллобразованием ранее синтезированных амфифильных блок-сополимеров в селективном растворителе по отношению к одному из блоков [3] и в процессе гетерофазной полимеризации, вызывающей самоорганизацию [4]. В первом случае амфифильный блок-сополимер сначала растворяют в общем растворителе, затем добавляют селективный растворитель, который индуцирует мицеллообразование. Таким способом получены самоорганизованные структуры на основе диблок-сополимеров стирола и акриловой кислоты (АК), стирола и этиленгликоля, триблок-сополимеров на основе стирола, АК и метакриловой кислоты [5, 6] и другие. Например, в работе [7] диблок-сополимер АК и стирола сначала растворяют в ДМФА, затем добавляют по каплям воду, изменяя термодинамическое качество растворителя, что вызывает мицеллообразование. С ростом длины гидрофобного блока полистирола средний диаметр ассоциатов увеличивается, однако сферическая морфология частиц остается неизменной (хотя для амфифильных блок-сополимеров возможны и морфологические переходы). Этот метод имеет ряд недостатков. Одним из наиболее значимых является большая длительность процесса и использование достаточно разбавленных суспензий (обычно менее 1 мас. %) [8–10].
В основе второго способа лежит образование самоорганизованных частиц в результате проведения полимеризации с обратимой деактивацией цепи, например с обратимой передачей цепи (ОПЦ) в гетерогенных системах (дисперсионная и эмульсионная полимеризация). При этом в качестве стабилизатора образующихся полимерно-мономерных частиц применяют заранее синтезированный, как правило, гидрофильный полимерный ОПЦ-агент. Полимеризацию проводят в растворителе, в котором растворим полиОПЦ-агент, а гомополимер, образующийся из второго мономера, в нем нерастворим. В качестве таких растворителей зачастую используют водные среды, алканы [11], низшие спирты [12], ионные жидкости [13], сверхкритический CO2 [14] и другие. Когда в процессе полимеризации длина цепи растущего блока достигает критического значения, начинается самоорганизация частиц с образованием наноассоциатов, размер которых увеличивается с конверсией мономера.
Характерные для частиц амфифильных блок-сополимеров переходы от сферической морфологии, например ядро–оболочка, до морфологии более высоких порядков зависят не только от длины обоих блоков в сополимере, что было показано во многих работах [15–18], но и от природы и концентрации инициатора, температуры, природы растворителя, концевой группы полиОПЦ-агента, скорости перемешивания и т.д. [19]. Свойства блока, формирующего ядро, определяются типом мономера и могут быть скорректированы путем сополимеризации с мономерами разной полярности/строения. Архитектура такого блока – еще один ключевой параметр, влияющий на морфологию частиц, образующихся в процессе полимеризации. В работах [20, 21] наблюдали изменение морфологии при синтезе полистирольных червеобразных структур и везикул эмульсионной ОПЦ-полимеризацией стирола в присутствии сополимеров МАК с полиэтиленгликольметилакрилатом (ПЭГМА). Влияние состава исходного полимерного прекурсора было отмечено в работе [22]. Авторы изучали влияние топологии полиОПЦ-агента за счет введения звеньев ПЭГМА в разные положения вдоль цепи полиОПЦ-агента и обнаружили, что морфология частиц изменяется от сфер к везикулам, когда гидрофильные боковые цепи ПЭГМА были близки к гидрофобному блоку ПС.
В большинстве экспериментов в гетерофазной ОПЦ-полимеризации с участием полимерных ОПЦ-агентов получают гидрофобные блоки линейного строения, но в последнее время стали появляться публикации, в которых описаны макромолекулы более сложной структуры. Так, в работе [23] был синтезирован звездообразный амфифильный блок-сополимер ПЭГ и поли(диацетон акриламида) дисперсионной полимеризацией диацетон акриламида в воде с использованием бифункционального полиОПЦ-агента на основе ПЭГ. Звездообразные блок-сополимеры способны формировать морфологию более высокого порядка (червеобразную и везикулы) при значительно более низкой длине гидрофобного блока по сравнению с линейными блок-сополимерами.
Относительно высокая концентрация полимера, универсальность, легкость аппаратурного оформления, потенциальная масштабируемость делают гетерофазную ОПЦ-полимеризацию перспективной для промышленного производства различных функциональных материалов. Кроме того, данному методу присущи достоинства, характерные для традиционных гетерофазных процессов, например низкая вязкость и легкий теплоотвод.
Наибольшее распространение получила дисперсионная ОПЦ-полимеризация. Этот процесс сначала происходит по гомогенному механизму, как полимеризация в растворе, а затем по гетерогенному [26, 27]. На начальной (гомогенной) стадии полимеризация протекает гораздо медленнее, чем на более поздней (гетерогенной) стадии, и обычно характеризуется длительным индукционным периодом, особенно при полимеризации малоактивных мономеров, таких как стирол. Как правило, продолжительность индукционного периода зависит от строения и длины цепи полимерного ОПЦ-агента, природы растворителя и концентрации мономера [28–30].
Ранее нами уже были изучены закономерности гетерофазной полимеризации (эмульсионной, миниэмульсионной и дисперсионной) акриловых мономеров в условиях обратимой передачи цепи, где роль ОПЦ-агента выполняла полиакриловая кислота с тритиокарбонатной группой внутри цепи (ПАКТК) [31–35]. Для выявления общих закономерностей процесса на примере полимеризации н-бутилакрилата (БА) было изучено влияние таких параметров, как концентрация мономера, полимерного ОПЦ-агента, длина гидрофобного блока, природа дисперсионной среды и природа инициатора. Положение тритиокарбонатной группы за счет роста цепи в оба конца обеспечивает образование триблок-сополимера, ПАК–блок–ПБА–блок–ПАК. Было показано, что полимеризация состоит из двух стадий, на первой стадии формируются узкодисперсные амфифильные блок-сополимеры с коротким гидрофобным блоком и их самоорганизация. На второй стадии происходила полимеризация внутри ПМЧ, в процессе которой эти блок-сополимеры выполняют функции не только ОПЦ-агента, но и в большей степени стабилизатора ПМЧ.
В результате эмульсионной и миниэмульсионной полимеризации полученные блок-сополимеры образуют устойчивые дисперсии с практически унимодальным РЧР, но имеют бимодальное молекулярно-массовое распределение, что дает нам основание полагать, что оба процесса протекают по смешанному механизму. Иными словами, ПМЧ могут формироваться не только из частиц, сформированных вследствие самоорганизации амфифильных блок-сополимеров, но и из частиц, возникающих из микрокапель мономера, на поверхности которых адсорбировалась ПАКТК.
Однако более перспективной с точки зрения контроля ММР оказалась дисперсионная ОПЦ-полимеризация в водно-спиртовой среде. За счет варьирования компонентов рецептуры и условий синтеза были подобраны оптимальные параметры проведения полимеризации. Полимеризацию осуществляли в избытке ПАКТК по отношению к инициатору. Было показано, что наиболее эффективным инициатором является ДАК, а наилучший контроль молекулярно-массовых характеристик достигается при полимеризации БА в водно-спиртовой среде (объемное соотношение метанол : вода = 4 : 1). Вместе с тем оказалось, что для дисперсионной полимеризации характерен продолжительный индукционный период и невысокие предельные конверсии мономера.
В целом, были найдены условия, при которых удается получить триблок-сополимеры на основе АК и БА, самоорганизующиеся в водных и водно-спиртовых средах в устойчивые частицы со структурой ядро–оболочка (ядро – поли(н-бутилакрилат), оболочка – полиакриловая кислота).
Чтобы использовать синтезированные дисперсии амфифильных блок-сополимеров в качестве затравочных частиц для дальнейшей полимеризации с сохранением функции ОПЦ-агента, наиболее подходящими оказались суспензии, полученные дисперсионной полимеризацией, но остался ряд вопросов, требующих дополнительных исследований. Например, необходимо уменьшить продолжительность индукционного периода, что сократит период формирования ПМЧ, и повысить ММ блок-сополимеров и контроль ММР. Возможным решением этой проблемы является порционное введение мономера в полимеризацию, которое мы опробовали ранее в эмульсионной сополимеризации БА с фторалкилакрилатами в условиях ОПЦ [35]. Так, было показано, что самоорганизация амфифильных блок-сополимеров, имеющих короткий гидрофобный блок за счет порционного введения мономера в реакционную среду, позволяет достичь высоких конверсий мономера с сохранением контроля над ММР.
Цель настоящей работы состоит в изучении процесса гетерофазной полимеризации БА в водно-спиртовой среде в присутствии ПАКТК в качестве полимерного ОПЦ-агента в условиях порционного введения мономера в начале процесса. Как мы полагаем, это позволит улучшить эффективность полимерного ОПЦ-агента, сформировать ПМЧ на более ранних стадиях и получить триблок-сополимер с сохранением ОПЦ-механизма.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Мономеры – акриловую кислоту, н-бутил-акрилат, стирол (99%, “Aldrich”) перед использованием перегоняли под вакуумом. Инициатор ДАК перекристаллизовывали из метанола. Растворители очищали по стандартным методикам. ОПЦ-агент – дибензилтритиокарбонат (БТК) синтезировали, как описано ранее, и охарактеризовывали методом ЯМР [36]. Дигидрат ацетата цинка (98%, “Acros”), гидроксид калия (KOH) (“Реахим”) использовали без дополнительной очистки.
Полиакриловую кислоту с тритиокарбонатной группой (ПАКТК) синтезировали следующим образом. Навески БТК (5 × 10–2 моль/л) и ДАК (1 × 10–3 моль/л) растворяли в 1,4-диоксане, к полученному раствору добавляли акриловую кислоту. Объемное отношение акриловая кислота : 1,4-диоксан составляло 1 : 1. Полимеризацию проводили при 80°С до предельной конверсии. Полученный полимер лиофильно высушивали из 1,4-диоксана, растворяли в воде, диализовали против воды с использованием целлюлозных мембран MEMBRA-CEL®, MWCO 3500 и лиофильно сушили. По данным ГПХ, Mn = 9.6 × 103, Ð = 1.31.
Для проведения полимеризации БА в смеси метанола и воды (объемное отношение мета-нол : вода составляло 4 : 1) растворяли заданное количество ПАКТК и ДАК. Полученный раствор переносили в трехгорлую колбу объемом 100 мл, погруженную в масляную баню, снабженную якорной мешалкой с двигателем и стеклянным холодильником Либиха, продували азотом в течение 30 мин, добавляли мономер, воду (объемное отношение БА : водно-метанольная смесь составляло 1.0 : 7.5). Готовую смесь продували азотом при постоянном перемешивании со скоростью 80 об./мин в течение 60 мин и полимеризовали при 65°С в течение 3 ч. Использовали следующие способы введения мономера: сразу, по каплям в течение полимеризации и порционно. В последнем случае сначала добавляли первую порцию мономера (10 или 15%) и через заданное время (5 или 10 мин) – вторую (85 или 90%).
Для проведения затравочной полимеризации полученные частицы очищали диализом против воды от непрореагировавшего мономера и ПАКТК. По данным ГПХ, Mn = 17.1 × 103, Ð = 1.63.
Синтез наночастиц оксида цинка ZnO проводили по стандартной методике [37]. Раствор КОН в метаноле (0.12 моль/л) готовили при интенсивном перемешивании в течение 30 мин при 60°С. К 150 мл раствора небольшими порциями при постоянном перемешивании приливали 50 мл заранее приготовленного раствора дигидрата ацетата цинка в метаноле (0.16 моль/л). Суспензию перемешивали в течение 2 ч при 60°С, затем центрифугировали при скорости 3000 об./мин в течение 10 мин. Полученный осадок многократно промывали метанолом для удаления побочных продуктов реакции и редиспергировали в метаноле.
Синтез композиционных частиц осуществляли по описанной выше методике полимеризации, только для приготовления дисперсионной среды вместо метанола использовали дисперсию ZnO в метаноле с концентрацией 2 мас. %.
Затравочную полимеризацию стирола проводили в трехгорлой колбе объемом 100 мл. В колбу, продутую азотом, помещали дисперсию затравочных частиц (3 мас. %), приливали стирол (массовое соотношение стирол : затравочные частицы = 1.5 : 1), оставляли при постоянном перемешивании в течение 20 мин в атмосфере азота. Затем добавляли заранее приготовленный раствор ДАК (5.5 × 10–4 моль/л) в метаноле и переносили колбу в масляную баню, заранее разогретую до 65°С. Полимеризацию осуществляли в течение 3 ч.
Кинетику полимеризации изучали методом гравиметрии, через заданные промежутки времени отбирали пробы объемом 2 мл, охлаждали на ледяной бане, добавляли гидрохинон для предотвращения дальнейшей полимеризации и высушивали каждую пробу до постоянной массы при 45°С.
Средний диаметр частиц полимерных дисперсий определяли методом динамического рассеяния света с помощью лазерного анализатора частиц “ZetasizerNano-ZS” фирмы “Malvern” (Великобритания), снабженного гелий-неоновым лазером с длиной волны 633 нм и мощностью источника света 5 мВт. Диапазон измерения частиц 0.6–6000 нм. Угол детектирования рассеяния света 173°. Полученные дисперсии предварительно разбавляли водно-метанольной смесью (объемное отношение метанол : вода составляло 4 : 1). Измерения проводили при комнатной температуре по методике, рекомендованной производителем.
Молекулярно-массовые характеристики полимеров оценивали методом ГПХ. Все полученные образцы содержали карбоксильные группы АК, поэтому были предварительно метилированы диазометаном [38]. Измерения проводили на хроматографе GPC-120 фирмы “PolymerLabs”, оборудованном рефрактометрическим и вискозиметрическим детекторами. Образцы блок-сополимеров анализировали при 50°С со скоростью потока 1 мл/мин. В качестве элюента использовали ДМФА, содержащий 0.1 мас. % LiBr. Хроматограф снабжен двумя колонками PLgel 5 µm MIXED B (М = (5 × 102)–(1 × 107)). Для калибровки применяли узкодисперсные стандарты полиметилметакрилата.
УФ-спектры поглощения растворов синтезированных блок-сополимеров в тетрагидрофуране (1 мг/мл) регистрировали при комнатной температуре на УФ-спектрометре “Unico 2804” (США).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
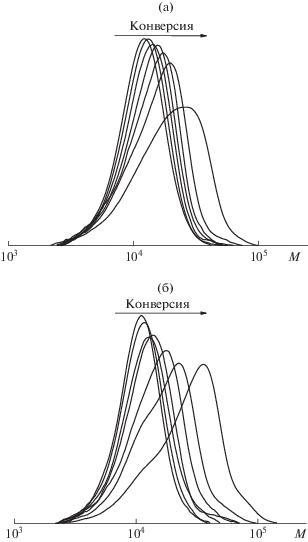
Одним из недостатков дисперсионной ОПЦ-полимеризации является невысокая скорость процесса, поэтому на первом этапе данную задачу решали путем увеличения концентрации инициатора. Как видно на рис. 1, на кривых зависимости конверсии мономера от времени индукционный период не наблюдается. Повышение концентрации ДАК приводит к закономерному росту скорости полимеризации и к несущественному уширению ММР (рис. 2). Для всех систем наблюдается сдвиг кривых ГПХ в сторону больших молекулярных масс в ходе полимеризации.
Рис. 1.
Зависимость конверсии мономера от времени при полимеризации БА в присутствии ПАКТК. [ДАК] = 5.5 × 10–4 (1), 8.3 × 10–4 (2) и 1.1 × 10–3 моль/л (3). Здесь и на рис. 2–7 объемное соотношение метанол : : вода = 4 : 1 и БА : дисперсионная среда = 1.0 : 7.5; [ПАКТК] = 1.6 × 10–3 моль/л; Т = 65°С.
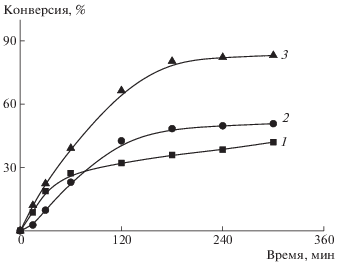
Рис. 2.
Кривые ГПХ метилированных образцов блок-сополимеров, полученных дисперсионной полимеризацией БА в присутствии ПАКТК. [ДАК] = 5.5 × × 10–4 (а), 8.3 × 10–4 (б) и 1.1 × 10–3 моль/л (в).
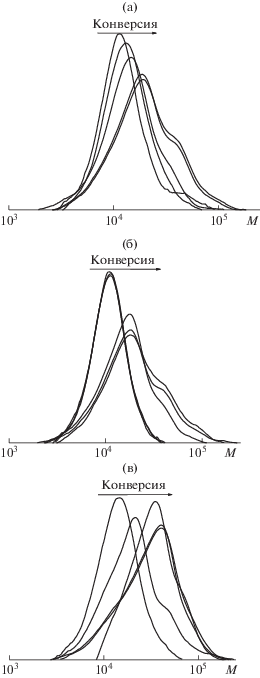
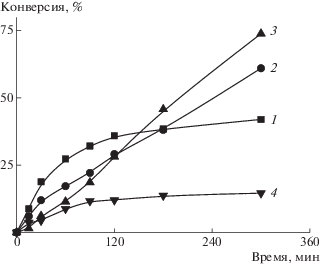
Однако дисперсность синтезированных блок-сополимеров по ММ все же растет с повышением конверсии. Мы предположили, что усилить контроль над ММР можно за счет порционного добавления мономера. В последующих экспериментах использованы различные способы введения мономера: с единовременной загрузкой всего объема, порционное и по каплям (скорость подачи – 10 мл/ч). Порционное и непрерывное (по каплям) добавление мономера может способствовать более эффективному формированию ПМЧ, благодаря образованию более короткого гидрофобного блока на ранних стадиях полимеризации. Как и ожидалось, при порционном и непрерывном введении БА начальная скорость дисперсионной полимеризации меньше, чем при единовременном способе (рис. 3). Однако в первом случае удается достичь более высоких предельных конверсий БА (кривые 2 и 3) по сравнению с единовременной загрузкой (кривая 1).
Рис. 3.
Зависимость конверсии мономера от времени при полимеризации БА в присутствии ПАКТК. [ДАК] = 5.5 × 10–4 моль/л, способ введения мономера: единовременная загрузка (1), порционное введение − 15% БА сразу и 85% БА через 5 (2) или 10 мин (3), а также по каплям (4).
Зависимость конверсии БА от времени при порционном введении мономера и различных начальных объемах БА приведена на рис. 4а. Вначале в реакционную смесь добавляли 10 или 15% мономера и через 5 мин после начала полимеризации оставшиеся 90 или 85% БА. Начальная скорость в обоих экспериментах была одинаковой, но предельная конверсия выше во втором опыте. Стоит отметить, что время введения второй порции мономера практически не влияет на кинетику полимеризации (рис. 4б), но влияет на ММР образующегося триблок-сополимера (рис. 5). При добавлении второй порции мономера через 10 мин ММР заметно уширяется, но в обоих случаях наблюдается рост ММ образующегося полимера. Можно предположить, что более длительный промежуток между введением первой и второй порций мономера приводит к появлению новых цепей или новых ПМЧ, растущих в разных условиях, и как следствие к уширению ММР (рис. 5).
Рис. 4.
Зависимости конверсии мономера от времени при полимеризации БА в присутствии ПАКТК. [ДАК] = 5.5 × 10–4 моль/л, порционное введение мономера: а – 10% БА сразу и 90% БА через 5 мин (1); 15% БА сразу и 85% БА через 5 мин (2); б – 15% БА сразу и 85% БА через 5 (1) или 10 мин (2).
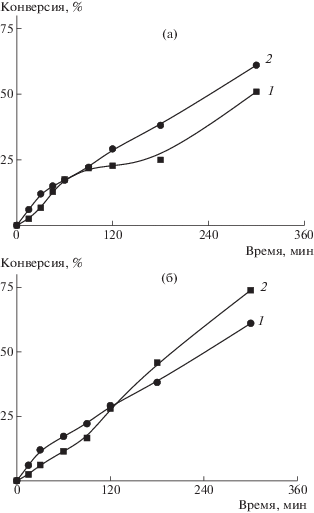
Рис. 5.
Кривые ГПХ метилированных образцов сополимеров, полученных дисперсионной полимеризацией БА в присутствии ПАКТК. [ДАК] = 5.5 × × 10–4 моль/л. Порционное введение мономера: 15% БА сразу и 85% БА через 5 (а) или 10 мин (б).
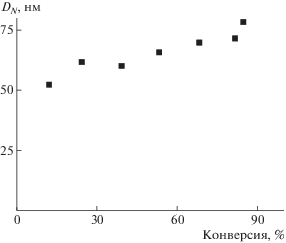
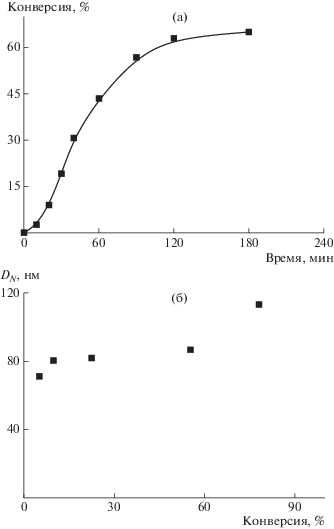
Изменение размера частиц во времени изучали на примере полимеризации БА при концентрации ДАК, равной 5.5 × 10–4 моль/л. Из зависимости среднего диаметра частиц от конверсии мономера (рис. 6) следует, что в процессе полимеризации диаметр ПМЧ повышается от 50 до 80 нм, что соответствует закономерностям традиционной дисперсионной полимеризации, когда рост частиц происходит за счет последовательного поступления мономера в ПМЧ [39]. При достижении конверсии ∼70% наблюдается легкая опалесценция реакционной системы, которая постепенно усиливается, что свидетельствует об увеличении размера частиц.
Рис. 6.
Зависимость среднего диаметра частиц DN от конверсии мономера при дисперсионной полимеризацией БА. [ДАК] = 5.5 × 10–4 моль/л.
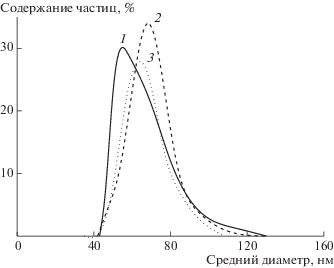
Концентрация ДАК практически не влияет на средний диаметр частиц, он варьируется в диапазоне 55–80 нм, но влияет на ширину РЧР. При увеличении концентрации инициатора РЧР сужается, что указывает на очень короткий период формирования ПМЧ (рис. 7). В процессе полимеризации наибольшей опалесценцией характеризовалась система с более высокой концентрацией ДАК.
Рис. 7.
Среднечисленное распределение частиц по размерам, полученных дисперсионной полимеризацией БА в присутствии ПАКТК. [ДАК] = 5.5 × 10–4 (1), 8.3 × 10–4 (2) и 1.1 × 10–3 моль/л (3).
Все полученные дисперсии частиц триблок-сополимера ПАК–блок–ПБА–блок–ПАК были очищены диализом от остаточного мономера и непрореагировавшей ПАКТК. Стоит отметить, что после диализа средний диаметр частиц практически не изменился. Даже при введении в систему метилового спирта в разном количестве средний диаметр остается постоянным, что подтверждает высокую стабильность образовавшихся частиц.
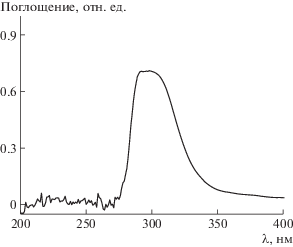
Синтезированный триблок-сополимер, согласно ОПЦ-механизму, должен содержать внутри цепи тритиокарбонатный фрагмент и, следовательно, сохранять функцию полимерного ОПЦ-агента. Для доказательства этого был зарегистрирован спектр поглощения образца триблок-сополимера в ТГФ (1 мг/мл) методом УФ-спектроскопии (рис. 8). Видно, что наблюдается интенсивный максимум при 305–307 нм, соответствующий поглощению тритиокарбонатной группы [40]. Таким образом, полученные суспензии пригодны для осуществления затравочной полимеризации и формирования мультиблок-сополимеров.
Рис. 8.
УФ-спектр триблок-сополимера, полученного дисперсионной полимеризацией БА в присутствии ПАКТК, в ТГФ.
Чтобы дисперсии частиц амфифильных блок-сополимеров могли быть использованы в качестве матрицы для иммобилизации неорганических частиц или затравочных частиц при проведении гетерофазной полимеризации с обратимой передачей цепи, полученный триблок-сополимер должен отвечать следующим требованиям: частицы дисперсии должны иметь узкое распределение по размерам и в триблок-сополимере должно быть достаточное количество “живых” цепей. Синтезированные дисперсии полностью отвечают всем этим требованиям.
В качестве примера приведем ОПЦ-полимеризацию БА в присутствии наночастиц ZnO. Синтез композиционных частиц осуществляли по описанной выше методике, но в качестве дисперсионной среды использовали смесь устойчивой метанольной дисперсии ZnO и воды. Как мы полагаем, введение наночастиц в дисперсионную среду будет способствовать локализации наночастиц на поверхности, а не в объеме полимерной частицы. Исходя из данных рис. 9, на котором представлены кривые зависимости конверсии БА от времени в отсутствие и в присутствии наночастиц, можно отметить появление более длинного индукционного периода и замедления процесса во втором случае. Причиной могут служить диффузионные затруднения, возникающие в процессе формирования ПМЧ со стороны наночастиц, расположенных на поверхности ПМЧ и препятствующих диффузии мономера в место протекания полимеризации. Можно предположить, что гидроксильные группы, находящиеся на поверхности наночастиц, кооперативно взаимодействуют с карбоксильными группами блоков ПАК, стабилизирующих частицы блок-сополимеров, подобно типичным полимер-полимерным комплексам с участием поликислот [41]. Косвенно данное предположение согласуется с тем фактом, что синтезированные композиционные частицы имеют бимодальное РЧР, средний диаметр и содержание первой фракции составили 93 нм и 89%, а второй – 159 нм и 11%. Этот процесс будет нами более подробно описан в следующих публикациях.
Рис. 9.
Зависимость конверсии мономера от времени при полимеризации БА в отсутствие (1) и в присутствии ZnO (2). Объемное соотношение БА : дисперсионная среда = 1.0 : 7.5, [ПАКТК] = 1.6 × 10–3 моль/л, [ДАК] = 5.5 × 10–4 моль/ л.
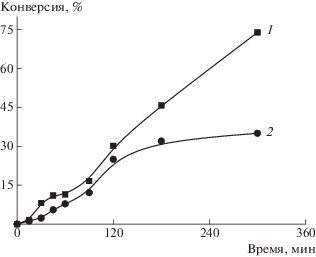
Синтезированные частицы (дисперсию со средним диаметром частиц 57 нм) использовали в качестве затравочных при полимеризации стирола. Полимеризация протекает с высокой скоростью, в начале процесса наблюдается небольшой индукционный период в течение 7 мин и через 3 ч конверсия мономера составляет 65% (рис. 10а). Как видно на рис. 10б, с повышением конверсии стирола средний диаметр частиц увеличивается до 115 нм, а РЧР остается унимодальным. Это позволяет полагать, что новые полистирольные частицы не образуются. По данным просвечивающей электронной микроскопии видно, что частицы имеют сферическую форму (рис. 11).
Рис. 10.
Зависимости конверсии мономера от времени (а) и среднего диаметра частиц от конверсии стирола (б). Здесь и на рис. 11 и 12: объемное соотношение вода : метанол = 3 : 1 и стирол : дисперсионная среда = 1 : 25. Массовое соотношение стирол : затравочные частицы = 2 : 1. [Стирол] = 0.34 моль/л, [ДАК] = 5.5 × 10–4 моль/л. T = 65°С.

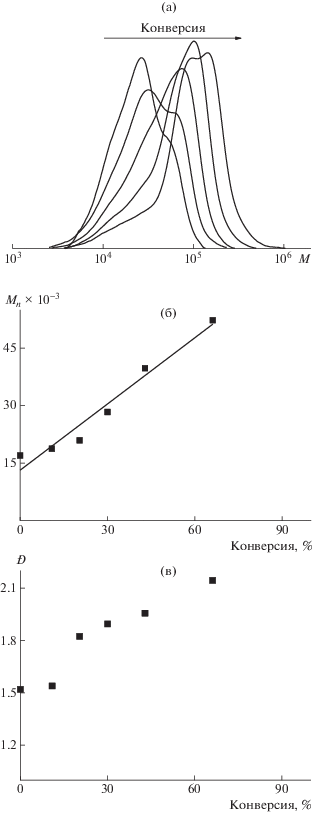
Анализ молекулярно-массовых характеристик полученного блок-сополимера свидетельствует о сохранении ОПЦ-механизма процесса. С повышением конверсии мономера кривые ГПХ (рис. 12а) сдвигаются в область более высоких молекулярных масс, при достижении конверсии более 60% наблюдается небольшая бимодальность (Ð = 2.1) (рис. 12в), зависимость Mn от конверсии имеет линейный характер (рис. 12б). Следовательно, в ходе синтеза образуются пентаблок-сополимеры следующего строения ПАК–блок–ПБА–блок–ПС–блок–ПБА–блок–ПАК.
Рис. 12.
Кривые ГПХ продуктов затравочной полимеризации стирола (а), а также зависимости Mn (б) и Ð (в) блок-сополимеров.
Были определены краевые углы смачивания для синтезированных образцов сополимеров: каплю воды поместили на пленки, полученные из 3%-ных растворов сополимеров в ДМФА на тефлоновой или стеклянной подложке. Величина краевого угла смачивания на тефлоновой подложке для затравочных частиц составила 39.7°, а после полимеризации стирола – 98.3°, а на стеклянной – 25.4° и 63.1° соответственно, что подтверждает включение блока полистирола в полимерную цепь и эффективное протекание затравочной полимеризации.
ЗАКЛЮЧЕНИЕ
Таким образом, в настоящей работе продемонстрировано влияние концентрации маслорастворимого инициатора, порядка введения мономера в синтез на закономерности ОПЦ-полимеризации БА в водно-спиртовой среде в присутствии ПАКТК.
Полученные результаты свидетельствуют о том, что порционное введение мономера позволяет повысить эффективность полимерного ОПЦ-агента и обеспечить формирование ПМЧ на более ранних стадиях процесса. Методом УФ-спектроскопии установлено наличие “живых” цепей в образующемся амфифильном триблок-сополимере.
Полученные дисперсии сополимеров АК и БА имеют унимодальное РЧР, сохраняют устойчивость в течение длительного времени даже при замене водно-метанольной среды на водную или спиртовую и могут быть использованы в качестве как полимерной матрицы для иммобилизации наночастиц ZnO, так и полимерного ОПЦ-агента для затравочной полимеризации стирола. Было показано, что в процессе полимеризации нового мономера частицы сохраняют функцию ОПЦ-агента. При наращивании более гидрофобного блока полистирола сферическая морфология частиц не изменяется.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-33-00386).
Список литературы
Charleux B., Delaittre G., Rieger J., D’Agosto F.// Macromolecules. 2012. V. 45. P. 6753.
Mai Y., Eisenberg A. // Chem. Soc. Rev. 2012. V. 41. P. 5969.
Blanazs A., Armes S.P., Ryan A.J. // Macromol. Rapid Commun. 2009. V. 30. P. 267.
Cockram A.A., Neal T.J., Derry M.J., Mykhaylyk O.O., Williams N.S.J., Murray M.W., Emmett S.N., Armes S.P. // Macromolecules. 2017. V. 50. № 3. P. 796.
Choucair A., Lavigueur C., Eisenberg A. // Langmuir. 2004. V. 20. P. 3894.
Hales K., Chen Z., Wooley K.L., Pochan D.J. // Nano Lett. 2008. V. 8. P. 2023.
Khan H., Cao M., Duan W., Ying T., Zhang W. // Polymer. 2018. V. 150. P. 204.
Delaittre G., Save M., Gaborieau M., Castignolles P., Rieger J., Charleux B. // Polym. Chem. 2012. V. 3. P. 1526.
Kim K.H., Kim J., Jo W.H. // Polymer. 2005. V. 46. P. 2836.
Kitayama Y., Moribe H., Kishida K., Okubo M. // Polym. Chem. 2012. V. 3. P. 1555.
Derry M.J., Fielding L.A., Armes S.P. // Prog. Polym. Sci. 2016. V. 52. P. 1.
Lowe A.B. // Polymer. 2016. V. 106. P. 161.
Zhang Q., Zhu S. // ACS Macro Lett. 2015. V. 4. P. 755.
Zetterlund P.B., Aldabbagh F., Okubo M. // J. Polym. Sci., Polym. Chem. 2009. V. 47. P. 3711.
Tan J., He J., Li X., Xu Q., Huang C., Liu D., Zhang L. // Polym. Chem. 2017. V. 8. P. 6853.
Liu H., Gao C., Ding Z., Zhang W. // Macromol. Chem. Phys. 2016. V. 217. P. 467.
Zhou J., Zhang W., Hong C., Pan C. // Polym. Chem. 2016. V. 7. P. 3259.
He W.D., Sun X.L., Wan W.M., Pan C.Y. // Macromolecules. 2011. V. 44. P. 3358.
Yeow J., Boyer C. // Adv. Sci. 2017. V. 4. P. 1700137.
Zhang W., D’Agosto F., Boyron O., Rieger J., Charleux B. // Macromolecules. 2012. V. 45. P. 4075.
Boisse′ S., Rieger J., Belal K., Di-Cicco A., Beaunier P., Li M.H., Charleux B. // Chem. Commun. 2010. V. 46. P. 1950.
Lesage de la Haye J., Zhang X., Chaduc I., Brunel F., Lansalot M., D’Agosto F. // Angew Chem. Int. Ed. 2016. V. 55. P. 3739.
Wang X., Figg C.A., Lv X., Yang Y., Sumerlin B.S., An Z. // ACS Macro Lett. 2017. V. 6. P. 337.
Sugihara S., Armes S.P., Lewis A.L. // Angew. Chem. Int. Ed. 2010. V. 49. P. 3500.
Wang X., An Z. // Macromol. Rapid Commun. 2018. V. 40. P. 1800325.
Blanazs A., Madsen J., Battaglia G., Ryan A.J., Armes S.P. // J. Am. Chem. Soc. 2011. V. 133. P. 16581.
Sugihara S., Armes S.P., Blanazs A., Lewis A.L. // Soft Matter. 2011. V. 7. P. 10787.
Su Y., Xiao X., Li S., Dan M., Wang X., Zhang W. // Polym. Chem. 2014. V. 5. P. 578.
Dan M., Huo F., Zhang X., Wang X., Zhang W. // J. Polym. Sci., Polym. Chem. 2013. V. 51. P. 1573.
Xiao X., He S., Dan M., Su Y., Huo F., Zhang W. // J. Polym. Sci., Polym. Chem. 2013. V. 51. P. 3177.
Chernikova E.V., Plutalova A.V., Mineeva K.O., Nasimova I.R., Kozhunova E.Yu., Bolshakova A.V., Tolkachev A.V., Serkhacheva N.S., Zaitsev S.D., Prokopov N.I., Zezin A.B. // Polymer Science B. 2015. V. 57. № 6. P. 547.
Chernikova E.V., Serkhacheva N.S., Smirnov O.I., Prokopov N.I., Plutalova A.V., Lysenko E.A., Kozhu-nova E.Yu. // Polymer Science B. 2016. V. 58. № 6. P. 629.
Serkhacheva N.S., Prokopov N.I., Chernikova E.V., Kozhunova E.Y., Lebedeva I.O., Borisov O.V. // Polym. Int. 2019. V. 68. P. 1303.
Serkhacheva N.S., Plutalova A.V., Kozhunova E.Yu., Prokopov N.I., Chernikova E.V. // Polymer Science B. 2018. V. 60. № 2. P. 204.
Serkhacheva N.S., Smirnov O.I., Tolkachev A.V., Prokopov N.I., Plutalova A.V., Chernikova E.V., Kozhunova E.Yu., Khokhlov A.R. // RSC Adv. 2017. V. 7. P. 24522.
Chernikova E.V., Terpugova P.S., Bui C., Charleux B. // Polymer. 2003. V. 44. № 5. P. 4101.
Dazhi S., Minhao W., Luyi S., Yuntao L., Nobuo M., Hung-Jue S. // J. Sol-Gel Technol. 2007. V. 43. P. 237.
Giordanengo R., Viel S., Hidalgo M., Allard-Breton B., Thévand A., Charles L. // J. Am. Soc. Mass Spectrometry. 2010. V. 21. № 6. P. 1075.
Handbook of Radical Polymerization / Ed by K. Matyjaszewski, T.P. Davis. Weinheim: Wiley, 2002.
Chernikova E.V., Plutalova A.V., Garina E.S., Vishnevetsky D.V. // Polym. Chem. 2016. V. 7. P. 3622.
Yu X., Tanaka A., Tanaka K., Tanaka T. // J. Chem. Phys. 1992. V. 97. № 10. P. 7805.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)