

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 6, стр. 447-457
ВЛИЯНИЕ МЕТОДА СИНТЕЗА НА СВОЙСТВА ПРЕКУРСОРОВ УГЛЕРОДНЫХ ВОЛОКОН НА ОСНОВЕ СОПОЛИМЕРОВ АКРИЛОНИТРИЛА И АКРИЛОВОЙ КИСЛОТЫ
Р. В. Томс a, М. С. Балашов a, А. Ю. Гервальд a, Н. И. Прокопов a, А. В. Плуталова b, А. К. Беркович b, Е. В. Черникова b, c, *
a МИРЭА–Российский технологический университет,
Институт тонких химических технологий им. М.В. Ломоносова
119571 Москва, пр. Вернадского, 86, Россия
b Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
c Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский просп., 29, Россия
* E-mail: chernikova_elena@mail.ru
Поступила в редакцию 09.06.2020
После доработки 23.06.2020
Принята к публикации 10.07.2020
Аннотация
Проведено сравнительное исследование термического поведения сополимеров акрилонитрила и акриловой кислоты, синтезированных классической радикальной полимеризацией и полимеризацией с обратимой передачей цепи до глубоких конверсий с разным режимом введения акриловой кислоты в реакцию. Показано, что при близких средних составах “классических” сополимеров различие в их композиционной неоднородности приводит к непрогнозируемому изменению энергии активации циклизации, степени стабилизации и термостойкости сополимеров. Напротив, для сополимеров того же среднего состава, полученных в условиях обратимой передачей цепи, изменение микроструктуры цепи, т.е. распределения звеньев акриловой кислоты по цепи, позволяет управлять скоростью циклизации при сохранении высокой термостойкости сополимеров.
ВВЕДЕНИЕ
Проблема получения качественного прекурсора для производства высокопрочных и высокомодульных углеродных волокон является одной из актуальных задач полимерного материаловедения. Многочисленные попытки ее решения основаны или на использовании разнообразных синтетических подходов [1–5], или на модификации известных либо разработке новых способов формования прекурсоров [4–13]. Наиболее часто для этой цели применяют сополимеры акрилонитрила, которые обладают рядом преимуществ по сравнению с другими известными прекурсорами [14–20].
Анализ литературы показывает, что с точки зрения синтеза задача сводится к созданию сополимера, не содержащего в своей структуре дефектов и обладающего заданным распределением сомономеров вдоль цепи, которое обеспечивает равномерное тепловыделение при термоокислительной стабилизации [21–28]. При этом до сих пор вопрос о природе сомономера и способе его распределения остается открытым.
Использование классической радикальной полимеризации малоперспективно, поскольку даже при поддержании постоянной концентрации мономеров в ходе сополимеризации нельзя избежать образования дефектов: неоднородности сополимеров по ММ и формирования разветвленного полимера за счет передачи цепи на полимер. Кроме того, состав сополимера, а следовательно, и распределение сомономеров, задается в этом случае исключительно составом мономерной смеси и активностью мономеров в сополимеризации.
Решением может быть применение радикальной полимеризации с обратимой деактивацией цепи, основные достижения которой в контролируемом синтезе сополимеров АН проанализированы в работах [4, 5, 29–36]. Наиболее перспективным ее вариантом на текущий момент оказалась радикальная полимеризация с обратимой передачей цепи (ОПЦ); с ее помощью удается синтезировать сополимеры АН с Mn > 105 и относительно узким ММР [31, 35]. Кроме контроля ММР и композиционной однородности сополимеров подобные процессы позволяют осуществлять синтез градиентных сополимеров из мономеров разной активности [37, 38]. Для того, чтобы регулировать распределение сомономеров вдоль цепи можно воспользоваться известным приемом – вводить мономер(-ы) в сополимеризацию с заданной скоростью. Тогда в зависимости от состава мономерной смеси, активности мономеров и скорости их подачи в синтез можно получать сополимеры с необходимым распределением сомономеров. В работе [39] моделированием сополимеризации акрилонитрила и метилметакрилата в условиях обратимого переноса атома с единовременной загрузкой мономеров и при их непрерывной подаче в реакцию с постоянной скоростью было предсказано образование сополимеров разной микроструктуры. На практике подобные эксперименты были проведены в ОПЦ-процессе для смеси акрилонитрила и N-изопропилакриламида [28]. Авторы показали, что при медленном добавлении N-изопропилакриламида в синтез поведение сополимера при термоокислительной стабилизации изменяется: наблюдается понижение тепловыделения при циклизации и расширяется температурный интервал, в котором она происходит. Близкий подход мы реализовали в работе [40] на примере сополимера АН и акриловой кислоты (АК). Было показано, что термическим поведением сополимеров АН можно управлять не только изменяя состав мономерной смеси, но и используя регулируемое с разной скоростью введение АК в ОПЦ-полимеризацию АН.
Цель настоящей работы – подробное сравнительное исследование свойств сополимеров АН и АК, синтезированных разными способами введения АК в реакцию в условиях ОПЦ-процесса и классической радикальной полимеризации. Нам представлялось полезным сравнить “классические” и ОПЦ-сополимеры, полученные при единовременной загрузке мономеров, при добавлении АК в сополимеризацию с постоянной скоростью в течение синтеза и при ее введении через заданное время после начала гомополимеризации АН. В “классических” образцах это должно было привести к образованию сополимеров с разной степенью композиционной неоднородности, а в ОПЦ-сополимерах – к образованию макромолекул с различным распределением звеньев в цепи. Таким образом, анализируя сополимеры близкого среднего состава, но разной молекулярной структуры, можно проследить влияние последней на термическое поведение сополимеров.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Акрилонитрил (99%) и акриловую кислоту (>99%) фирмы “Acros” очищали перегонкой по стандартной методике. ДАК перекристаллизовывали из этанола, хранили в темноте при –3°С. Безводный персульфат калия (ПСК, >98%) и ОПЦ-агент – 2-(додецилтиокарбонилтиоилтио)-2-метилпропионовую кислоту (C12H25–SC(=S)S–C(CH3)2COOH, ЦТК) фирмы “Aldrich” дополнительной очистке не подвергали. ДМСО (99%), ДМФА (HPLC) и метанол фирмы “Fluka” перед использованием перегоняли.
Полимеризацию проводили в трехгорлой колбе объемом 100 мл, снабженной верхнеприводным перемешивающим устройством с мешалкой якорного типа. В колбу помещали раствор, содержащий рассчитанное количество ПСК (10–3 моль/л) и ЦТК (10–3 моль/л) в ДМСО для ОПЦ-полимеризации и ДАК (5.5 × 10–3 моль/л) в ДМСО для классической полимеризации. Затем добавляли АН; АК вводили следующим образом: в начале синтеза; непрерывно со скоростью 0.32 мл/ч и полунепрерывно, т.е. через определенное время от начала синтеза, со скоростью 0.32 или 0.63 мл/ч. Для дозировки АК использовали шприцевой насос BYZ-810; скорость подачи АК составляла 0.32 и 0.63 мл/ч на протяжении 6 и 3 ч полимеризации, соответственно. Колбу продували аргоном (99.99%) в течение 15 мин, закрывали и погружали в баню, разогретую до определенной температуры. В заданное время отбирали пробы для определения конверсии, молекулярно-массовых характеристик и состава сополимеров. По окончании полимеризации реакционную смесь при необходимости разбавляли ДМСО, высаживали в избыток воды, сополимеры фильтровали, промывали водой и сушили при 60°С до постоянной массы. Конверсию мономеров определяли гравиметрически.
Во всех синтезах массовое соотношение АН и АК составляет 90 : 10 (мольное соотношение 92 : 8). ОПЦ-сополимеризацию проводили при 55°С и суммарной концентрации мономеров в растворе 40 мас. %. Классическую радикальную полимеризацию осуществляли при 65°С и суммарном массовом содержании мономеров в растворе 22%.
Молекулярно-массовые характеристики сополимеров АН изучали методом гель-проникающей хроматографии на хроматографе GPC-120 фирмы “PolymerLabs”. Анализ проводили при 50°С в ДМФА, содержащем 0.1 мас. % LiBr, со скоростью потока 1 мл/мин. Для разделения использовали две колонки PLgel 5 µm MIXED С (М = (5 × 102)–(1 × 107)). Для анализа готовили раствор полимера в элюенте с концентрацией полимера, не превышающей 1 мг/мл и не менее 0.7 мг/мл. ММ рассчитывали по стандартам ПММА; для полиакрилонитрила ММ пересчитывали, используя известные из литературы коэффициенты Марка–Куна–Хаувинка (KПАН = 39.4 × 10–4, α = = 0.75, KПMMA = 17.7 × 10–4, α = 0.62 [41]):
Для исследования сополимеров готовили 4%-ный раствор полимеров в ДМСО, наливали его на стеклянную горизонтальную подложку и испаряли растворитель при 80°С до постоянной массы. Готовые пленки снимали с подложки и нарезали квадратные образцы размером 40 × 40 мм; толщина пленки составляла от 10 до 15 мкм.
Тепловые эффекты, наблюдаемые при динамическом нагревании пленок сополимеров, исследовали на дифференциальном сканирующем калориметре “Netzsch DSC 204” фирмы “Netzsch” (Германия) в атмосфере осушенного газа (воздух, аргон) при скорости потока 100 мл/мин в интервале 30–450°С со скоростью нагревания от 5 до 20 град/мин в воздушной атмосфере и 10 град/мин в инертной атмосфере. Для проведения измерений брали приготовленную пленку массой 4–6 мг, помещали в стандартный алюминиевый тигель без крышки. Результаты обрабатывали с помощью программы Netzsch Proteus.
Энергию активации циклизации Ea определяли по методу Киссинджера [42]:
(1)
$ - \frac{{{{E}_{a}}}}{R} = \frac{{d\left[ {\ln \left( {\frac{\varphi }{{T_{p}^{2}}}} \right)} \right]}}{{d\left( {\frac{1}{{{{T}_{p}}}}} \right)}},$Термогравиметрический анализ проводили на термическом анализаторе “STA 449 F3 Jupiter” (“Netzsch”). Потерю массы 15–20 мг образцов сополимеров АН, помещенных в корундовый тигель, анализировали при линейном нагревании со скоростью 10 град/мин от 25 до 450°С в атмосфере аргона со скоростью потока 50 мл/мин.
Состав пленок сополимеров ПАН изучали методом ИК-спектроскопии с помощью ИК-фурье спектрометра “Spectrum Two FT–IR Spectrometer” фирмы “Perkin Elmer” в области 4000–400 см–1. Для количественного определения состава сополимеров в ИК-спектрах использовали калибровочную зависимость, полученную по стандартным смесям гомополимеров АН и АК. Для этого готовили смеси ПАН и ПАК, содержащие 1–15 мас. % ПАК, растворяли в ДМСО, тщательно перемешивали и готовили пленки по методике, описанной выше. Затем регистрировали ИК-спектры стандартных образцов и строили калибровочную зависимость отношения интенсивности оптических полос A1735/A2244 от мольного содержания ПАК, где A1735 отвечает интенсивности характеристических полос поглощения валентных колебаний связей C=O (νC=O = 1735 см–1) для АК и CN (νCN = 2244 см–1) для АН.
Изменения, происходящие в структуре макромолекул при термоокислительной стабилизации, изучали методом ИК-НПВО спектроскопии. Для этого пленку образца помещали в печь на воздухе и отжигали при фиксированной температуре (200, 225, 250°С; точность нагревания контролировали в диапазоне ±1°С) в течение заданного времени, затем регистрировали ИК-спектры при комнатной температуре в режиме НПВО (кристалл алмаз) в области от 4000 до 600 см–1 на ИК-микроскопе “Spectrum Two FT–IR Spectrometer” фирмы “Perkin Elmer” с разрешением 0.5 см–1. Обработку ИК-спектров проводили с использованием программного обеспечения PerkinElmer.
Степень стабилизации Es [43] определяли по уравнению
(2)
${{E}_{S}} = \frac{{{{A}_{{1590\;{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}}}{{{{А}_{{2240\;{\text{с}}{{{\text{м}}}^{{ - 1}}}}}}}},$РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез и характеристика сополимеров акрилонитрила и акриловой кислоты
Классическая радикальная сополимеризация. Синтез сополимеров классической радикальной сополимеризацией осуществляли при суммарной концентрации мономеров 22 мас. %. На рис. 1 приведены зависимости конверсии от времени для сополимеризации АН и АК, инициированной ДАК при 65°С, при единовременной загрузке мономеров, полунепрерывном и непрерывном способе введения АК. Видно, что способ введения АК в синтез влияет на скорость сополимеризации.
Рис. 1.
Зависимость конверсии от времени сополимеризации АН и АК в присутствии ДАК (5.5 × × 10–3 моль/л) при 65°С. 1 – единовременное введение мономеров, 2 – полунепрерывное введении АК через 3 ч после начала полимеризации со скоростью 0.32 мл/ч, 3 – непрерывные введение АК со скоростью 0.32 мл/ч.
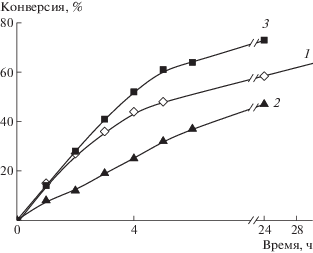
При единовременном введении мономеров в сополимеризацию мольное содержание АК последовательно возрастает с конверсией мономера (рис. 2, кривая 1). При полунепрерывном введении АК вначале, до конверсии примерно 20%, образуется гомополимер АН, и только при более высоких конверсиях формируются макромолекулы, содержащие звенья АК, доля которых увеличивается с повышением конверсии мономера (кривая 2). При непрерывном введении АК в синтез ее доля в сополимере вначале плавно возрастает в ходе процесса (кривая 3), но после конверсии 60% скорость вхождения АК в сополимер увеличивается и средний состав сополимера близок к составу сополимера, образующегося при полунепрерывном способе синтеза. Во всех случаях наблюдается заметное конверсионное изменение состава сополимера, которое наиболее ярко проявляется в случае полунепрерывного способа введения АК. Следовательно, можно предполагать, что продукты, образующиеся на глубоких конверсиях (выше 60%), характеризуются достаточно высокой композиционной неоднородностью.
Рис. 2.
Зависимость мольной доли АК в сополимере от конверсии мономеров для сополимеров АН, полученных радикальной сополимеризацией в ДМСО, инициированной ДАК (5.5 × 10–3 моль/л) при 65°С и содержании мономеров 22 мас. % при единовременном введении мономеров (1), полунепрерывном (2) и непрерывном введении АК (3).
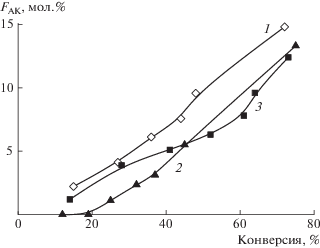
С точки зрения термического поведения сополимеров АН важными характеристиками являются средняя длина последовательности звеньев АН в сополимере (для начальных стадий полимеризации в случае разноактивных мономеров) и конверсионная композиционная неоднородность сополимеров. Их теоретическая оценка возможна на основании значений констант сополимеризации, однако эти данные для исследуемой мономерной пары противоречивые [19]. Нами был проведен синтез сополимеров на начальных конверсиях из мономерной смеси с содержанием АК от 10 до 40 мол. % и анализ состава сополимеров методом ИК-спектроскопии. Константы сополимеризации, определенные методом МНК [45], составляют rАК = 0.82 ± 0.26 и rАН = 0.69 ± 0.05. Однако, если на основании этих констант рассчитать изменение среднего состава сополимера для исследуемого состава мономерной смеси при единовременной загрузке мономеров, то выясняется, что теоретическое значение среднего состава, т.е. мольной доли АК в сополимере, убывает от 0.9 до 0.7 при увеличении конверсии от 2 до 70%. Это противоречит данным эксперимента, приведенного выше (рис. 2, кривая 1) и требует более детального исследования. Таким образом, модель концевого звена оказалась не применима к исследуемой системе для описания изменений состава сополимера. Данный результат может быть вызван следующими обстоятельствами: наличием явного эффекта предконцевого звена, как это наблюдается в сополимеризации АН и стирола [46, 47], или влиянием растворителя, что характерно для полимеризации полярных мономеров [48]. Теоретическое описание изменения среднего и мгновенного состава сополимера в ходе процесса требует дополнительных исследований и выходит за рамки настоящей работы.
С ростом конверсии, т.е. с уменьшением концентрации мономеров, среднечисленная ММ сополимеров уменьшается от ~160 × 103 до ~80 × 103. Одновременно с этим наблюдается увеличение дисперсности Ð сополимеров по ММ от 1.6 до 2.5. Следовательно, образующиеся сополимеры характеризуются неоднородностью не только по составу макромолекул, но и по ММ.
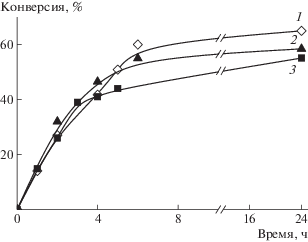
ОПЦ-сополимеризация. Введение ОПЦ-агента позволяет подавить неоднородность макромолекул по составу и ММ. В данном случае благодаря ступенчатому росту макромолекул за счет равновесия между активными и “спящими” цепями ММ полимеров увеличивается в ходе полимеризации. Это позволяет повысить суммарную концентрацию мономеров в растворе, поскольку реакционная система сохраняется гомогенной в более широком интервале концентраций полимера. ОПЦ-сополимеризацию проводили при массовой концентрации мономеров 40%. На рис. 3 приведены кинетические кривые ОПЦ-сополимеризации АН и АК в ДМСО. Видно, что способ введения АК в сополимеризацию (сразу в начале синтеза, полунепрерывно, т.е. через 3 ч после ее начала, или непрерывно в течение полимеризации) практически не влияет на начальную скорость полимеризации. После конверсии ~40% реакция начинает замедляться, темп замедления несколько различается для разных режимов введения АК. Таким образом, в ОПЦ-процессе кинетика сополимеризации оказалась менее чувствительной к способу введения мономеров в реакцию.
Рис. 3.
Зависимость конверсии от времени сополимеризации АН и АК (7 мол. %) в присутствии [ПСК]0 = = [ЦТК]0 =10–3 моль/л при 55°С. 1 – единовременное введение мономеров, 2 – полунепрерывное введение АК через 3 ч после начала полимеризации со скоростью 0.63 мл/ч, 3 – непрерывное введение АК со скоростью 0.32 мл/ч.
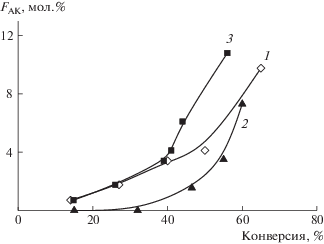
Изменение среднего состава сополимеров с конверсией зависит от режима введения АК в сополимеризацию (рис. 4). Качественно эти изменения близки к описанным выше: мольная доля АК в сополимере увеличивается с ростом конверсии мономера. В отличие от “классических” сополимеров (рис. 2) в данном случае вначале скорость вхождения АК в сополимер ниже, а после конверсии мономеров ~40% она возрастает. Анализ литературы показывает, что в ряде случаев ОПЦ-механизм, ММ сополимера и другие факторы могут влиять на состав сополимера [48].
Рис. 4.
Зависимость мольной доли АК в сополимере FАК от конверсии для сополимеров АН, полученных сополимеризацией в ДМСО при единовременном введении мономеров (1), полунепрерывном (2) и непрерывном введении АК (3). Содержание мономеров 40 мас. %; [ПСК]0 = [ЦТК]0 = 1 × 10–3 моль/л; Т = 55°С.
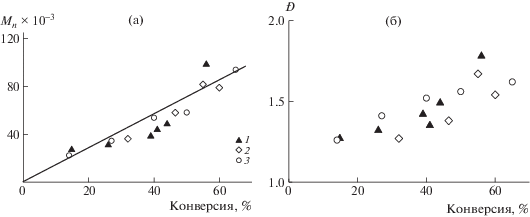
Как следует из данных на рис. 5а, полимеризация в исследуемых системах протекает по ОПЦ-механизму и сопровождается линейным ростом Mn от конверсии мономеров. Сополимеры, образующиеся на начальных и средних конверсиях, характеризуются низкой дисперсностью по ММ (Ð < 1.5) (рис. 5б). С увеличением продолжительности реакции растет доля “мертвых” цепей, образующихся за счет распада ПСК, что приводит к незначительному повышению значения Ð по сравнению с дисперсностью сополимеров, образующихся в классической радикальной полимеризации.
Рис. 5.
Зависимости Mn (а) и дисперсии Ð (б) сополимеров АН и АК от конверсии мономеров при сополимеризации в присутствии [ПСК]0 = [ЦТК]0 = 10–3 моль/л при единовременном введении мономеров (1), непрерывном (2) и полунепрерывном введении АК в полимеризацию (3). Т = 55°С.
Преимуществом использования ОПЦ-процесса является подавление композиционной неоднородности сополимеров за счет постоянного “оживления” макромолекул и их участия в реакции роста цепи. В этом случае в полунепрерывной ОПЦ-сополимеризации происходит образование блок-статистического сополимера ПАН–блок–П(АН–со–АК), а в непрерывной ОПЦ-сополимеризации и при единовременном введении мономеров в синтез – статистического сополимера П(АН–со–АК) с разным распределением звеньев АК в сополимере вследствие разных мгновенных составов мономерных смесей.
Отталкиваясь от полученных результатов, методами классической радикальной сополимеризации и ОПЦ в условиях разных способов введения АК в сополимеризацию были синтезированы шесть сополимеров, которые использовали в дальнейшем для изучения их термического поведения. Характеристики сополимеров приведены в табл. 1.
Таблица 1.
Характеристики бинарных сополимеров АН и АК для исследования свойств
| Образец, № | ОПЦ-агент | Введение АК | Mn × 10–3 | Ð | FАК, мол. % | |
|---|---|---|---|---|---|---|
| способ | скорость, мл/ч | |||||
| 1 | – | Единовременно | – | 68 | 2.14 | 14.8 |
| 2 | – | Полунепрерывно | 0.32 | 73 | 2.40 | 13.3 |
| 3 | – | Непрерывно | 0.32 | 76 | 2.02 | 12.4 |
| 4 | ЦТК | Единовременно | – | 94 | 1.62 | 9.8 |
| 5 | ЦТК | Полунепрерывно | 0.63 | 78 | 1.54 | 7.8 |
| 6 | ЦТК | Непрерывно | 0.32 | 98 | 1.80 | 10.8 |
Термические свойства пленок синтезированных сополимеров
Можно было ожидать, что различие в неоднородности сополимеров по составу и ММ, а также в микроструктуре цепи будет влиять на процессы термических превращений в сополимерах АН.
На рис. 6 приведены термограммы “классических” и ОПЦ-сополимеров, зарегистрированные в инертной атмосфере. В обоих случаях способ введения АК в синтез влияет на свойства сополимеров, однако термическое поведение сополимеров зависит также и от механизма полимеризации.
Рис. 6.
Термограммы сополимеров АН и АК, зарегистрированные в инертной атмосфере, при скорости нагревания 10 град/мин. Здесь и на рис. 7 и 8 номера кривых соответствуют номерам образцов в табл. 1.
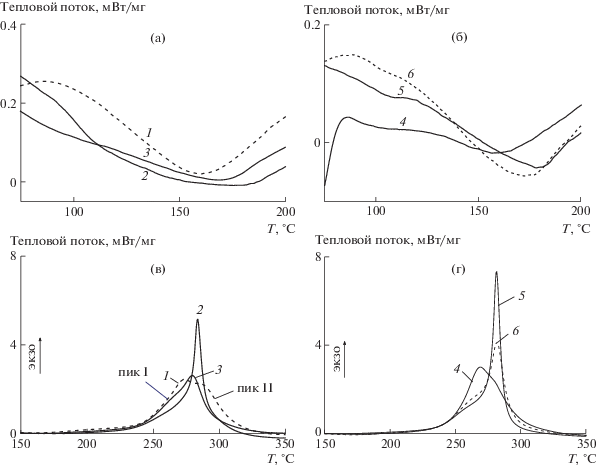
Дрейф базовой линии в области температур 75–150°С обусловлен расстекловыванием. Видно, что для ОПЦ-сополимеров этот процесс происходит в более узком интервале температур и выражен ярче вследствие более высокой композиционной однородности сополимеров.
На всех термограммах наблюдается два пика, соотношение интенсивностей тепловыделения которых определяется условиями синтеза сополимеров. В инертной атмосфере тепловыделение связано с протеканием реакции циклизации, которое может протекать по радикальному механизму (пик II) в случае ПАН и его сополимеров с инертными мономерами, например алкилакрилатами [49, 50]:
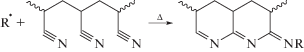
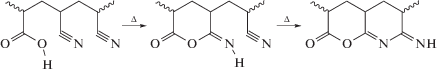
При наличии в сополимере АН звеньев кислотных мономеров при более низких температурах может реализовываться ионный механизм циклизации (пик I) [51, 52]:
На термограммах “классических” сополимеров, полученных при единовременном (кривая 1) и непрерывном (кривая 3) введении АК в сополимеризацию интенсивности пиков I и II сопоставимы, т.е. вклад обоих механизмов циклизации близкий. При полунепрерывном введении АК в синтез (кривая 2) образуется, по-видимому, наиболее композиционно-неоднородный сополимер, в котором заметную долю составляют макромолекулы ПАН. В результате преобладает механизм радикальной циклизации. Тепловой эффект реакции (–ΔH) и величина (–ΔH/ΔТ), показывающая количество тепла, которое выделяется за температурный диапазон прохождения реакции циклизации, симбатно уменьшаются в ряду образцов 1–2–3 (табл. 2).
Таблица 2.
Анализ термограмм сополимеров АН и АК с разным распределением звеньев, зарегистрированных в инертной атмосфере
| Образец, № | Tпик, °С | –ΔH*, Дж/г | –ΔH/ΔT | Тепловой поток, Вт/г | Ea**, кДж/моль | |
|---|---|---|---|---|---|---|
| пик I | пик II | |||||
| 1 | 274, 284 | 770 | 4.3 | 2.44, 2.26 | 110 | 290 |
| 2 | 264, 284 | 680 | 4.0 | 1.11, 5.15 | 170 | 150 |
| 3 | 267, 280 | 585 | 3.6 | 1.78, 2.61 | 120 | 200 |
| 4 | 269, 280 | 700 | 4.7 | 3.00, 2.38 | 125 | 230 |
| 5 | 260, 282 | 710 | 4.8 | 1.22, 7.33 | 140 | 160 |
| 6 | 264, 282 | 680 | 4.8 | 1.63, 4.13 | 100 | 170 |
** Энергия активации рассчитана по уравнению (1).
Отличительной чертой ОПЦ-сополимеров является более интенсивное тепловыделение при циклизации (рис. 6б). Можно предположить, что это обусловлено меньшей дефектностью макромолекул, т.е. большей их однородностью по составу и ММ. При полунепрерывном синтезе (кривая 6) вследствие блочного строения сополимера (ПАН–блок–П(АН–со–АК)) преобладает радикальный механизм циклизации. При единовременном введении АК (кривая 4) больший вклад в экзо-эффект вносит механизм ионной циклизации, а при непрерывном – радикальный. Тепловой эффект реакции уменьшается в ряду образцов 4–5–6, однако значение (–ΔH/ΔТ) практически не изменяется.
Более подробный анализ термограмм образцов 1–6, зарегистрированных в инертной атмосфере при разной скорости нагревания образцов, по уравнению 1 позволил оценить энергии активации низкотемпературной (пик I) и высокотемпературной (пик II) циклизации (табл. 2).
В случае “классических” сополимеров закономерность в соотношении энергии активации ионной и радикальной циклизации при близких средних составах сополимеров, но разных способах введения АК в сополимеризацию, не прослеживается. Для ОПЦ-сополимеров характерно более низкое значение энергии активации циклизации по ионному, чем по радикальному механизму. Последнее хорошо согласуется с нашими предыдущими результатами [40] и литературными данными для сополимеров АН и итаконовой кислоты [53]. Можно предположить, что обнаруженное различие связано с композиционной неоднородностью “классических” сополимеров.
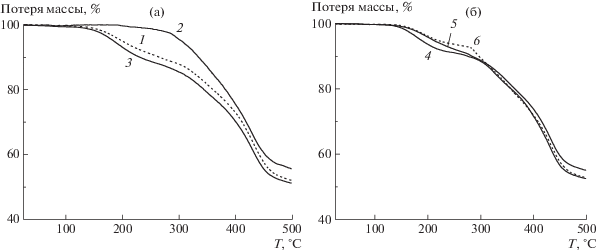
В инертной атмосфере, если в структуре макромолекул нет дефектов, должны протекать исключительно реакции циклизации. Согласно данным ТГА, оказалось, что сополимеры АН и АК, полученные методом ОПЦ, обладают более высокой термостойкостью (рис. 7). Исключение составляет образец 2, характеризующийся наибольшей композиционной неоднородностью.
Рис. 7.
Зависимости потери массы образцов сополимеров АК и АН, полученных классической (а) и ОПЦ сополимеризацией (б) от температуры нагревания. Атмосфера азота, скорость нагревания 10 град/мин.
Аналогичные эксперименты с использованием метода ДСК были проведены и в атмосфере воздуха (рис. 8). Здесь механизм процесса более сложный и он включает наряду с реакциями циклизации, реакции дегидрирования и окисления. И в этом случае поведение ОПЦ-сополимеров зависит от метода синтеза и способа введения АК в сополимеризацию.
Рис. 8.
Термограммы сополимеров АН и АК, зарегистрированные в атмосфере воздуха, при скорости нагревания 10 град/мин.
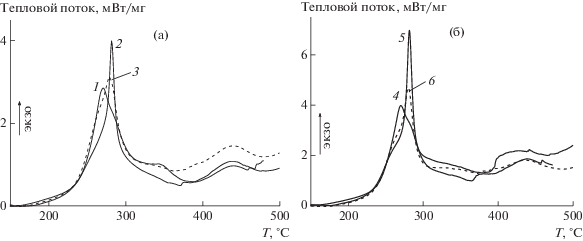
Видно, что процессы термоокислительной стабилизации в “классических” сополимерах начинаются при тех же температурах (рис. 8а), что и в ОПЦ-сополимерах (рис. 8б), но и интенсивность тепловыделения в первом случае ниже.
Количественную оценку изменения химической структуры в процессе термоокислительной стабилизации проводили методом ИК-спектроскопии, рассчитывая долю непрореагировавших нитрильных групп φCN и степень стабилизации Es по уравнениям (2) и (3). Пленки сополимеров вначале были выдержаны на воздухе при фиксированной температуре в течение заданного времени, а затем при комнатной температуре были зарегистрированы их ИК-спектры. Типичная трансформация ИК-спектров сополимеров при нагревании во времени приведена на рис. 9.
Рис. 9.
ИК-спектры пленок сополимера 5 после стабилизации на воздухе при 250°С в течение 0 (1), 30 (2), 60 (3), 150 (4), 240 (5) и 480 мин (6).
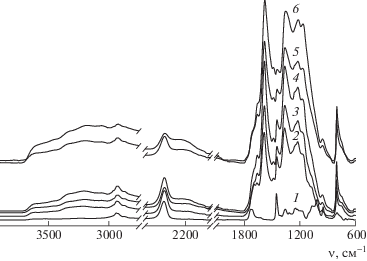
В исходном спектре присутствуют полосы, характеризующие функциональные группы сомономеров. Валентные колебания СН проявляются в области 2900–2860 см–1, нитрильной группы νCN – при 2243 см–1, карбоксильной группы νС=О – при 1727 см–1, деформационные δСНН – при ~1450 и 1360 см–1, смешанные δСНН и маятниковые ${{\gamma }_{{{\text{C}}{{{\text{H}}}_{2}}}}}$ при ~1073 см–1, слабые полосы – при 1250 и 762 см–1 [4, 52]. Наличие карбоксильной группы в структуре макромолекулы может проявляться по-разному в ИК-спектре в зависимости от окружения и ионизации этой группы.
После нагревания пленок в спектрах появляются новые полосы поглощения при 3350 (νNH), 1581–1650 (ν–C=N–, νC=C, νN–H, νC=О), 1481 (ν–C=N–), 1363 (δC–H, CH), 1243 и 1172 (νC–C, νC–N, νC–O), 800 см–1 (δ–C=C–H), которые могут быть отнесены к образующимся в системе сопряжения связям. Со временем происходит рост интенсивности указанных полос и понижение интенсивности полосы поглощения валентных колебаний нитрильной группы (νC≡N), которая уширяется и расщепляется на две полосы с максимумами при 2243 см–1 (нитрильная группа в исходном терполимере) и при 2200 см–1 (нитрильная группа, участвующая в сопряжении). Одновременно уменьшается интенсивность полосы поглощения деформационных колебаний СH2-групп основной цепи δСНН при 1450 см–1, а также валентных колебаний СН при 2940 (νsC–H) и 2870 см–1 (νasC–H). Все эти изменения указывают на формирование достаточно хорошо развитой системы полисопряженных связей за счет реакций циклизации нитрильных групп, дегидрирования метиленовых и метиновых групп основной цепи. Обращает на себя внимание поведение полосы поглощения, отвечающей карбоксильной группе νС=О. При нагревании она смещается в длинноволновую область и регистрируется в виде плеча при ~1722 см–1. Одновременно появляется новая полоса при 1650 см–1 в виде плеча полосы валентных колебаний –C=N– (1581 см–1). Согласно литературным данным, эти полосы относят к колебаниям карбонильной группы нафтиридинового и акридонового колец. Их появление связано с инициированием реакции циклизации карбоксильными группами акриловой кислоты и образованием соответствующих циклических структур.
На основании результатов ИК-спектроскопии по уравнению (2) была рассчитана степень стабилизации Es как соотношение интенсивности полос поглощения иминных –C=N– и нитрильных групп –C≡N соответственно (рис. 10). Видно, что скорость циклизации ОПЦ-сополимеров несколько ниже, чем образцов, синтезированных классической радикальной сополимеризацией (а); это различие усиливается при повышении температуры. Начальная скорость изменения степени стабилизации не зависит от способа введения мономеров в синтез в случае “классических” сополимеров. Однако для образца 2, для которого характерна наибольшая композиционная неоднородность, при увеличении температуры наблюдается резкое возрастание степени стабилизации.
Рис. 10.
Зависимости степень стабилизации Es от времени при нагревании пленок сополимеров на воздухе при постоянной температуре: 200 (1–6), 225 (1'–6') и 250°С (1''–6''). а: образец 1 (1, 1', 1''), 2 (2, 2', 2'') и 3 (3, 3', 3''); б: образец 4 (4, 4', 4''), 5 (5, 5', 5'') и 6 (6, 6', 6'').
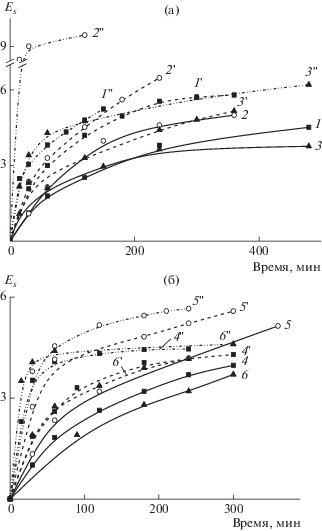
В случае ОПЦ-сополимеров степень стабилизации зависит от микроструктуры цепи, т.е. от способа введения АК в синтез. Для блочного распределения звеньев она выше и затем уменьшается при переходе от единовременного способа введения АК к непрерывному, т.е. по мере увеличения равномерности распределения АК в сополимере. С ростом температуры это различие нивелируется. В целом однородность макромолекул по составу способствует более равномерному процессу циклизации в сополимерах АН.
ЗАКЛЮЧЕНИЕ
В настоящей работе мы провели подробное исследование влияния метода синтеза на термическое поведение сополимеров АН и акриловой кислоты. Использование различного способа введения АК в синтез в условиях “классической” и ОПЦ-полимеризации позволило сравнить, как влияет композиционная неоднородность и распределение звеньев в цепи на процессы циклизации. Аналогично нашим предыдущим исследованиям, мы показали, что в ОПЦ-сополимерах циклизация характеризуется более интенсивным тепловыделением. Однако величина (–ΔH/ΔТ), характеризующаяся количеством тепла, которое выделяется за температурный диапазон прохождения реакции циклизации, практически не зависит от микроструктуры цепи (ОПЦ-сополимеры), но чувствительна к неоднородности сополимеров по составу (“классические” сополимеры). Для ОПЦ-сополимеров степень стабилизации ниже, чем для “классических” сополимеров. При этом в первых микроструктура оказывает влияние на скорость циклизации, а во вторых – начальная скорость изменения степени стабилизации одинакова независимо от способа введения АК в синтез.
Таким образом, очевидно, что метод ОПЦ с варьируемым способом введения мономеров в синтез действительно привлекателен с точки зрения управления свойствами сополимеров АН.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-29-17004-мк).
Список литературы
Zhang S., Yin L., Wang J., Zhang W., Zhang L., Zhu X. // Polymers. 2017. V. 9. № 12. P. 26.
Jiang J., Lu X., Lu Y. // J. Appl. Polym. Sci. 2010. V. 116. P. 2610.
Krishnan G.S., Burkanudeen A., Murali N., Phadnis H. // Green Chemistry. 2012. V. 14. № 6. P. 1778.
Chernikova E.V., Toms R.V., Prokopov N.I., Duflot V.R., Plutalova A.V., Legkov S.A., Gomzyak V.I. // Polymer Science B. 2017. V. 59. № 1. P. 28.
Grishin D.F., Grishin I.D. // Fibre Chem. 2019. V. 50. № 6. P. 514.
Estrin Y.I., Tarasov A.E., Grishchuk A.A., Chernyak A.V., Badamshina E.R. // RSC Adv. 2016. V. 6. № 108. P. 106064.
Kaur J., Millington K., Smith S. // J. Appl. Polym. Sci. 2016. V. 133. № 38. P. 43963.
Gao Q., Jing M., Wang C., Zhao S., Chen M., Qin J. // J. Macromol. Sci. B. 2017. V. 1. P. 128.
Kirsten M., Meinl J., Schönfeld K., Michaelis A., Cherif C. // J. Appl. Polym. Sci. 2016. V. 133. № 29. P. 43698.
Tian Y., Han K., Zhang W. // Mater. Lett. 2013. V. 92. P. 119.
Batchelor B.L., Mahmood S.F., Jung M. // Carbon. 2016. V. 98. P. 681.
Sayar S., Moskowitz J., Fox B., Wiggins J., Wallace G. // J. Appl. Polym. Sci. 2019. P. 47932.
Morris E.A., Weisenberger M.C., Bradley S.B., Abdallah M.G., Mecham S.J., Pisipati P., McGrath J.E. // Polymer. 2014. V. 55. № 25. P. 6471.
Carbon Fibers and Their Composites / Ed. by P. Morgan. New York: Taylor and Francis, 2005.
Chand S. // J. Mater. Sci. 2000. V. 5. P. 1303.
Carbon Fiber Composites / Ed. by D.L.Chung. Newton: Butterworth–Heinemann, 1994.
Huang X. // Materials. 2009. V. 2. P. 2369.
Edie D.D. // Carbon. 1998. V. 36. P. 345.
Bajaj P., Paliwal D.K., Gupta A.K. // J. Appl. Polym. Sci. 1993. V. 49. P. 823.
Bansal R.C., Donnet J.B. // Compr. Polym. Sci. 1990. V. 6. P. 501.
Liu J., He L., Ma S., Liang J., Zhao Y., Fong H. // Polymer. 2015. V. 61. P. 20.
Dalton S., Heatley F., Budd P.M. // Polymer. 1999. V. 40. P. 5531.
Gupta A., Harrison I.R. // Carbon. 1997. V. 35. P. 809.
Beltz L.A., Gustafson R.R. // Carbon. 1996. V. 34. P. 561.
Wangxi Z., Jie L., Gang W. // Carbon. 2003. V. 41. P. 2805.
Bansal R.C., Donnet J.B. // Compr. Polym. Sci. 1990. V. 6. P. 501.
Saha B., Schatz G.C. // J. Phys. Chem. B. 2012. V. 116. P. 4684.
Moskowitz J.D., Wiggins J.S. // Polymer. 2016. V. 84. P. 311.
Гришин Д.Ф., Гришин И.Д. // Успехи химии. 2015. Т. 84. № 7. С. 712.
Chernikova E.V., Poteryaeva Z.A., Belyaev S.S., Nifant’ev I.E., Shlyakhtin A.V., Kostina Yu.V., Cherevan’ A.S., Efimov M.N., Bondarenko G.N., Sivtsov E.V. // Polymer Science B. 2011. V. 53. № 7–8. P. 391.
Chernikova E.V., Toms R.V., Gervald A.Yu., Proko-pov N.I. // Polymer Science C. 2020. V. 62. № 1. P. 17.
Spörl J.M., Ota A., Beyer R., Lehr T., Müller A., Hermanutz F., Buchmeiser M.R. // J. Polym. Sci., Polym. Chem. 2014. V. 52. P. 1322.
Spörl J., Hermanutz F., Buchmeiser M. // Int. Fiber J. 2014. V. 28. № 6. P. 24.
Chernikova E.V., Kishilov S.M., Plutalova A.V., Kostina Yu.V., Bondarenko G.N., Baskakov A.A., Il’in S.O., Nikolaev A.Yu. // Polymer Science B. 2014. V. 56. № 5. P. 553.
Moskowitz J.D., Abel B.A., McCormick C.L., Wiggins J.S. // J. Polym. Sci., Polym. Chem. 2016. V. 54. P. 553.
Cai J., McDonnell J.Y., Brackley C., O’Brien L. // Mater. Today Commun. 2016. V. 9. P. 22.
Chernikova E.V., Sivtsov E.V. // Polymer Science B. 2017. V. 59. № 2. P. 117.
Rizzardo E., Chiefari J., Mayadunne R.T.A., Moad G., Thang S.H. // ACS Symp. Ser. 2000. V. 768 P. 278.
Al-Harthi M., Khan M.J., Abbasi S.H., Soares J.B.P. // Macromol. React. Eng. 2009. V. 3. P. 148.
Toms R.V., Balashov M.S., Shaova A.A., Gerval’d A.Y., Prokopov N.I., Plutalova A.V., Chernikova E.V. // Polymer Science B. 2020. V. 62. № 2. P. 102.
Polymer Handbook / Ed. by J. Brandrup E. H. Immergut, E.A. Grulke. NewYork: Wiley, 1999.
Kissinger H.E. // Anal. Chem. 1957. V. 29 P. 1702.
Ouyang Q., Cheng L., Wang H., Li K. // Polym. Degrad. Stab. 2008. V. 93. P. 1415.
Collins G.L., Thomas N.W., Williams G.E. // Carbon. 1988. V. 26. P. 671
Езриелев А.И., Брохина Э.О., Роскин Е.С. // Высокомолек. соед. А. 1969. Т. 11. № 8. С. 1670.
Cieplak P., Megiel E., Kaim A. // J. Polym. Sci., Polym. Chem. 2002. V. 40. P. 3592.
Alam M.M., Peng H., Jack K.S., Hill D.J.T., Whittaker A.K. // J. Polym. Sci., Polym. Chem. 2017. V. 55. I. 5. P. 919.
Madruga E.L. // Prog. Polym. Sci. 2002. V. 27. P. 1879.
Hao J., Liu Y., Lu C. // Polym. Degrad. Stab. 2018. V. 147. P. 89.
Bahrami S.H., Bajaj P., Sen K. // J. Appl. Polym. Sci. 2003. V. 88. № 3. P. 685.
Devasia R., Nair C.P.R., Sivadasan P., Katherine B.K., Ninan K.N. // J. Appl. Polym. Sci. 2003. V. 88. № 4. P. 915.
Sivy G.T., Coleman M.M. // Carbon. 1981. V. 19. № 2. P. 127.
Loginova E.V., Mikheev I.V., Volkov D.S., Proskurnin M.A. // Analyt. Methods. 2016. V. 8. I. 2. P. 371.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)