

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 6, стр. 425-435
СИНТЕЗ НОВЫХ СОПОЛИМЕРОВ НОРБОРНЕНА И 1,5-БИС-(ГЕКСЕНИЛ)ГЕКСАМЕТИЛТРИСИЛОКСАНА РЕАКЦИЕЙ ОЛЕФИНОВОГО МЕТАТЕЗИСА
А. А. Моронцев a, *, М. Л. Грингольц a, М. П. Филатова a, Ю. И. Денисова a, Я. В. Кудрявцев a, Е. Ш. Финкельштейн a
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
* E-mail: morontsev@ips.ac.ru
Поступила в редакцию 27.04.2020
После доработки 10.06.2020
Принята к публикации 23.07.2020
Аннотация
Получены новые сополимеры норборнена и 1,5-бис-(гексенил-5)-1,1,3,3,5,5-гексаметилтрисилоксана, содержащие в основной цепи гибкие силоксановые и жесткие норборненовые фрагменты. Для их синтеза использованы три разновидности реакции олефинового метатезиса: циклораскрывающая метатезисная полимеризация норборнена, метатезис несопряженного диена 1,5-бис-(гексенил-5)-1,1,3,3,5,5-гексаметилтрисилоксана и межцепная реакция макромолекулярного кросс-метатезиса между полинорборненом и силоксансодержащим полиеном. Последняя реакция изучена впервые. С ее помощью получены и охарактеризованы методами спектроскопии ЯМР 1Н, ЯМР 13С и ИК новые статистические мультиблок-сополимеры норборнена и 1,5-бис-(гексенил-5)-1,1,3,3,5,5-гексаметилтрисилоксана с различной средней длиной блоков. Изучено влияние строения сополимеров на их термические свойства.
ВВЕДЕНИЕ
Широкое использование поликарбосилоксанов обусловлено уникальным сочетанием их полезных свойств, таких как высокая термическая и окислительная стабильность, низкая токсичность, хорошие электрические, оптические, антиадгезионные, газоразделительные и иные характеристики [1–4]. Вместе с тем низкая механическая прочность значительно сужает перспективы их практического применения. Относительно недавно K.B. Wagener с сотрудниками в цикле работ [4–7] продемонстрировали возможности реакции метатезиса несопряженных диенов – ADMET-полимеризации (acyclic diene metathesis) в синтезе широкого ряда поликарбосилоксанов. Для улучшения механических свойств в ADMET-полимеры вводили метоксисилильные [2, 4, 8] и силациклобутановые [6, 9] группы, обеспечивающие дальнейшее сшивание. Другой подход заключался в синтезе сополимеров с жесткими и гибкими блоками по реакции ADMET, в том числе из телехеликов с концевыми диметилсилановыми хлор- или метокси-группами [4, 7, 10–13]. Варьируя соотношение реагентов, получали материалы с широким диапазоном механических характеристик.
В настоящей работе предложен метод синтеза новых сополимеров, содержащих карбосилоксановые и норборненовые блоки, способные сочетать свойства полисилоксанов и полинорборненов. Одним из реагентов выбран метатезисный полинорборнен (ПНБ) – широко известный полимер с температурой стеклования 39°С, обладающий выдающимися виброгасящими, адсорбционными и другими свойствами, выпускаемый под маркой Norsorex [14]. Можно ожидать, что в сополимере эти характеристики и хорошие механические свойства сохранятся за счет присутствия блоков норборнена, а силоксановые блоки придадут сополимеру адгезионные и газоразделительные свойства. Ранее было показано, что введение гибкого силоксанового заместителя в боковую цепь ПНБ обепечивает более эффективное разделение газовых смесей легких углеводородов [15]. Модификация двойных связей основной цепи также заметно влияет на газопроницаемость и селективность газоразделения [16, 17]. Введение гибких силоксановых блоков в относительно жесткую основную цепь ПНБ и его производных может улучшать газопроницаемость пленок из таких сополимеров. Для их синтеза в данной работе использован макромолекулярный кросс-метатезис – новая межцепная реакция, позволяющая получать статистические мультиблок-сополимеры из гомополимеров [18–30]. С помощью макромолекулярного кросс-метатезиса решаются проблемы синтеза сополимеров из мономеров со значительно отличающейся полимеризационной активностью либо полимеризующихся по различным механизмам [18–21, 23]. Синтез сополимеров норборнена (НБ) и карбосилоксановых диенов из мономеров затруднен, так как их гомополимеризация требует существенно различных условий проведения реакции. Так, ROMP (ring-opening metathesis polymerization) напряженной бициклической молекулы НБ – необратимая цепная реакция, которая протекает очень быстро с образованием высокомолекулярного ПНБ [14]. ADMET-полимеризация карбосилоксановых диенов является ступенчатой равновесной реакцией поликонденсации с последовательным образованием ди-, три-, тетрамеров и формированием высокомолекулярных продуктов только на последней стадии. Побочный продукт ADMET – низкомолекулярное соединение (чаще всего этилен), которое необходимо удалять из зоны реакции для смещения равновесия процесса в сторону образования полимера. В связи с этим ADMET осуществляют в глубоком вакууме, при повышенных температурах, иногда с использованием высококипящих растворителей [7, 31].
В научной литературе лишь недавно появились публикации, посвященные макромолекулярному кросс-метатезису. Ранее реакцию метатезиса с участием полимеров рассматривали в основном как нежелательный процесс переноса цепи, приводящий к образованию циклоолигомеров, перегруппировке мономерных звеньев и уширению ММР [14, 24, 32, 33]. В настоящее время показано, что с помощью макромолекулярного кросс-метатезиса можно получать статистические мультиблок-сополимеры из поликарбоната и полиоктенамера (ПЦО), способные к самоорганизации [18], сополимеры из полибутадиена и полиуретана, демонстрирующие улучшенные механические свойства [21], региорегулярные сополимеры из 3-замещенных полиоктенамеров [22]. На примере кросс-метатезиса между ПНБ и ПЦО разработана кинетическая модель реакции [26, 29], продемонстрирована возможность регулирования степени блочности полученных сополимеров за счет выбора условий реакции [24, 25], изучено влияние строения цепи сополимеров на их термические и кристаллические свойства [28, 29]. Кроме того, исследовано влияние гидрокси- [27, 34] и эпокси- [35] заместителей в боковой цепи ПЦО на его кросс-метатезис с ПНБ. Реакцию ПНБ и полибутадиена использовали для оценки каталитической активности рутений-карбеновых комплексов различной природы [36]. Проведена постмодификация полученных мультиблок-сополимеров путем эпоксидирования [37] и гидрирования двойных связей [20]. Все это указывает на широкие возможности и перспективы данного метода. Цель настоящей работы – синтез практически важных кремнийсодержащих сополимеров и расширение круга полимеров, вовлеченных в реакцию макромолекулярного кросс-метатезиса.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты
Все операции с соединениями, чувствительными к воздуху и влаге, проводили на стандартной вакуумной установке и линии Шленка в атмосфере аргона с использованием абсолютированных растворителей. Хлороформ сушили и перегоняли над гидридом кальция, диэтиловый эфир абсолютировали перегонкой над натрием, ТГФ квалификации х.ч. перегоняли над щелочью. Метиловый спирт квалификации х.ч., гидрокарбонат натрия (х.ч.), ингибитор окисления 2,2'-метилен-бис-(6-трет-бутил-4-метилфенол), катализатор Граббса первого (PCy3)2Cl2Ru=CHPh (Г-1,“Sigma–Aldrich”), второго Cl2(PCy3)(Н2IMes)Ru= CHPh (Г-2, “Shaanxi Dideu Medichem”) и третьего поколения (H2IMes)(3-Br-Py)2(Cl)2Ru=CHPh (Г-3, “Sigma–Aldrich”), 6-бромгексен-1 (99.5%, “Hangzhou Yuechen Chemical”), винилэтиловый эфир (99%, “Fluka”), 1,5-дихлор-1,1,3,3,5,5-гексаметил-трисилоксан (“ABCR”), мелкодисперсный порошок магния использовали без дополнительной очистки.
Методы измерений
Спектры ЯМР 1Н и ЯМР 13С{1H} регистрировали на спектрометрах “Bruker Avance 500” с рабочей частотой 500.13 и 125.77 МГц соответственно и “Bruker MSL-300” c рабочей частотой по протонам 300 МГц. Параметры съемки спектров ЯМР 13С{1H}: импульс 30° (3 мкс), время сбора данных 1.3 с, релаксационная задержка 1.4 с, размер фида 64К, размер реального спектра 32К. Химические сдвиги определяли относительно остаточного сигнала растворителя CDCl3 (для спектров ПМР – 7.28 м.д., ЯМР 13C – 77.23 м.д.) и пересчитывали к тетраметилсилану.
Калориметрические измерения выполняли на дифференциальном сканирующем калориметре “Mettler Toledo DSC823e”. Нагревание и охлаждение образцов осуществляли со скоростью 10 град/мин в атмосфере аргона со скоростью потока 70 мл/мин в диапазоне –100…+100°C. Результаты измерений обрабатывали с помощью сервисной программы “STARe”, поставляемой в комплекте с прибором. Точность измерения температуры ±0.3°C, энтальпии ±1 Дж/г.
Молекулярно-массовые характеристики определяли методом ГПХ на жидкостном хроматографе “Waters” с рефрактометрическим детектором и системой последовательно соединенных колонок WAT054460 (“Waters”) и G3000HHR (“TosohBiosep”) со сшитым ПС в качестве наполнителя, элюент ТГФ, скорость 1 мл/мин, температура колонки 25°C, концентрация образца 1 мг/мл, объем пробы 100 мкл, калибровка по ПС-стандартам (“PolymerLabs”).
Комбинированный газохроматографический и масс-спектрометрический (ГХ-МС) анализ проводили с использованием хроматомасс-спектрометра “Thermo Focus DSQ II” с квадрупольным масс-анализатором, энергия электронов 70 эВ, напряжение на электронном умножителе 1244 В, температура источников ионов и интерфейса 280°С, детектирование в режиме регистрации полного ионного тока SIM (selected ion monitoring). Капиллярная колонка со слабо полярной неподвижной жидкой фазой DB-5MS 15 м × 0.25 мм × × 0.25 мкм, газ-носитель гелий, скорость газа-носителя 1 мл/мин, температура инжектора 280°С, объем пробы 0.5 мкл. Режим анализа: начальная температура 40°С, через 5 мин повышение температуры со скоростью 8 град/мин до 280°С, общее время анализа 42 мин.
ИК-спектры регистрировали на ИК-Фурье спектрометре “Thermo Fisher Scientific Nicolet 5700”.
Синтез 1,5-бис-(гексенил-5)-1,1,3,3,5,5-гексаметилтрисилоксана (ГСО)
В трехгорлую круглодонную колбу объемом 500 мл, снабженную магнитной мешалкой, обратным водяным холодильником и капельной воронкой, загружали 3.5 г (0.146 моля) магния. Магний активировали прогреванием в вакууме, охлаждали колбу до комнатной температуры, заполняли аргоном и загружали в нее 150 мл абсолютированного диэтилового эфира. Раствор 6-бромгексена-1 (19.3 мл, 0.144 моля) в 22 мл диэтилового эфира добавляли по каплям в течение 1 ч со скоростью, обеспечивающей равномерное кипение реакционной смеси, после чего кипятили 2 ч и охлаждали до комнатной температуры. К полученному раствору при перемешивании в течение 1 ч прикапывали 16.7 мл (0.077 моля) 1,5-дихлор-1,1,3,3,5,5-гексаметилтрисилоксана, растворенного в 24 мл диэтилового эфира, затем кипятили еще 7 ч. На следующий день отделяли белый осадок. Реакционную массу трижды промывали раствором NaHCO3 (0.53 г в 100 мл дистиллированной воды) и сушили над сульфатом натрия. Смесь перегоняли в вакууме, выделили 18.59 г продукта (выход 83%) с Ткип= 90–93°С при 0.087 мм рт. ст. После повторной перегонки получено 11.6 г целевого продукта с концентрацией 93.8% по данным ГХ-МС. В нем присутствовали 5.6% производных дисилоксана, 0.6% производных тетрасилоксана и незначительное количество силоксанов с алкоксисилильными группами, которым на спектре ЯМР 1Н соответствуют сигналы 3.63 и 3.10 м.д. Появление алкоксисилильных групп может быть результатом взаимодействия хлорсилана с алкоголятом броммагния – продуктом окисления реактива Гриньяра. Перед дальнейшим использованием ГСО выдерживали над гидридом кальция и перегоняли.
ЯМР 1H (300 MГц, CDCl3), δН, м.д.: 5.81 (ш.м., 2H), 5.06–4.89 (ш. м. 4H), 2.05 (м, 4H), 1.38 (ш.м., 8H), 0.52 (м, 4H), 0.07, 0.02 (ш.м., 18H).
ГХ-МС (70 эВ): 15.92 мин m/z = 283.08 (M-CH3)+ 5.6% (1,3-бис-(гексенил)-1,1,3,3-тетраметилдисилоксана); 17.75 мин m/z = 357.09 (M-CH3)+, 93.8% (1,5-бис-(гексенил)-1,1,3,3,5,5-гексаметилтрисилоксана); 19.45 мин m/z = 431.11 (M-CH3)+, 0.6% (1,7-бис-(гексенил)-1,1,3,3,5,5,7,7-октаметилтетрасилоксана).
Синтез поли(1,1,3,3,5,5-гексаметилтрисилоксанил-5-деценилена) (ПГСО)
В отвакуумированный и заполненный аргоном двугорлый реактор объемом 50 мл, снабженный магнитной мешалкой и тефлоновым краном, загружали 0.0035 г (4.25 мкмоля) катализатора Г-1 и 0.43 мл (0.364 г, 0.98 ммоля) ГСО. Смесь трижды дегазировали путем замораживания, вакуумирования и размораживания. Реакцию вели с постепенным повышением температуры до 55°С и углублением вакуума до 0.09 мм рт. ст. После 71 ч нагревания, смесь охлаждали, продукт выделяли высаживанием в метанол, сушили его в вакууме не менее 2 суток. Получили 0.2934 г (выход 87%) маслоподобного продукта.
ЯМР 1H (300 MГц, CDCl3), δН, м.д.: 5.39 (ш.м., 2Н), 1.98 (ш.м., 4Н), 1.36 (ш.м., 8Н), 0.52 (ш.м., 4Н), 0.06, 0.02 (ш.м., 18Н).
ЯМР 13C (126 MГц, CDCl3), δС, м.д.: 130.30 (транс С=С), 129.81 (цис С=С), 33.58, 33.45, 32.40, 29.20, 27.07, 25.45, 23.02, 22.89, 18.28, 18.20, 1.33, 1.23, 0.43, 0.26.
Содержание транс-связей С=С составляет 80–85%, Mw = 9.8 × 103, Đ = 1.6, Tс = –102°С.
Синтез полинорборнена
ПНБ синтезировали метатезисной полимеризацией норборнена на катализаторе Г-1 по методике [37]. Продукт реакции представлял собой волокнистый полимер белого цвета.
Кросс-метатезис ПНБ и ПГСО
В колбе объемом 25 мл с краном и магнитной мешалкой в атмосфере аргона при комнатной температуре готовили раствор 0.0270 г (0.287 ммоля) ПНБ и 0.0977 г (0.284 ммоля) ПГСО в 0.8 мл абсолютированного хлороформа. На следующий день раствор дегазировали 3 раза путем последовательных циклов замораживания–вакуумирования–размораживания смеси. В заполненную аргоном колбу добавляли 0.07 мл отдельно приготовленного 0.007 М раствора Г-2 (0.0027 г, 0.00318 ммоля в 0.47 мл абсолютированного хлороформа). Реакцию вели 24 ч при комнатной температуре, останавливали ее введением 0.1 мл винилэтилового эфира, через 30 мин добавляли 0.05 г ингибитора окисления. Сополимер высаживали в метанол и сушили в вакууме до постоянной массы. Выход 0.1058 г (85%) воскоподобного липкого продукта, Mw = 26.3 × 103, Đ = 1.3, Tс = = – 83°С.
Состав сополимера и доли диад различного типа в нем определяли методом ЯМР 13С. Отнесения сигналов (м.д.) в области углеродов двойных связей выполняли аналогично предыдущим работам [24, 26, 38].
ЯМР 1H (500 МГц, CDCl3), δН, м.д.: 5.39, 5.37, 5.35 (ш. м., 3.54H, СH=), 5.29, 5.28 (ш. м., 0.23H, CH=), 5.22, 5.21 (ш. м., 0.20H, CH=), 3.67, 3.65, 3.64, 3.63, 3.62 (ш. м., 0.15H ГСО–ГСО), 3.52, 3.51, 3.50, 3.49 (ш. м., 0.15H ГСО–ГСО), 2.79 (ш. м., 0.35H; цис-СН, НБ–НБ), 2.45 (ш. м., 1.74H; транс-СН, НБ–НБ), 2.04, 1.98 (ш. м., 4.04H; СН2, ГСО-ГСО), 1.90, 1.88, 1.87, 1.86, 1.84 (ш. м., 1.28H; НБ–НБ), 1.78 (ш. м., 2.20H; НБ–НБ), 1.56 (ш. м., 0.48H НБ–НБ), 1.39, 1.37, 1.36, 1.35, 1.33 (ш. м., 9.64H), 1.10, 1.08, 1.05, 1.03 (ш. м., 1.07H; НБ–НБ), 0.55, 0.53, 0.52 (ш. м., 3.50H; ГСО–ГСО), 0.09, 0.09, 0.08, 0.06, 0.05, 0.03, 0.02 (ш. м., 18H, ГСО–ГСО).
ЯМР 13C (JMOD) (126 МГц, CDCl3), δC, м.д.: 135.60, 135.37, 135.34, 135.21, 135.07, 134.93 (НС3, НБ–ГСО); 134.13, 134.06, 133.91 (НС1,2, цис-НБ–НБ); 133.28, 133.18, 133.03 (НС1,2, транс-НБ–НБ); 130.73, 130.41, 130.29, 130.17(НС5,6, транс-ГСО–ГСО); 129.91 (НС5,6, цис-ГСО–ГСО); 128.67, 128.60, 128.57, 128.44 (НС4 , ГСО–НБ); 66.55, 65.71, 62.33 (СН2, ГСО–ГСО); 43.70, 43.59, 43.46, 43.33 (НС, транс-НБ–НБ); 42.25 (Н2С, НБ–НБ); 42.01, 41.99 (CH), 41.95, 41.93 (H2C); 41.53, 41.5 (Н2С, НБ–НБ); 38.82, 38.57, 38.16 (НС, цис-НБ–НБ); 36.49, 36.40, 36.18, (Н2С, ГСО–ГСО), 35.92 (HC); 33.88 (H2C); 33.70, 33.56 (Н2С, ГСО–ГСО); 33.49 (H2C); 33.06 (Н2С, НБ–НБ); 32.81 (H2C); 32.52 (Н2С, НБ–НБ и ГСО-ГСО); 32.37 (Н2С, НБ–НБ), 29.30 (Н2С, ГСО–ГСО); 27.45 (H2C); 27.18, 25.56, 25.18, 23.61, 22.95, 18.37, 18.29, 18.07 (Н2С, ГСО–ГСО), 1.48, 1.37 (C–(Н3С)2Si–O, ГСО–ГСО); 0.57, 0.39 (O– (Н3С)2Si–O, ГСО–ГСО).
ИК, см–1: 2955, 2922, 2856, 1461, 1447, 1409, 1342, 1257 (инт. с., SiCH3), 1162, 1050 (ш. c. SiOSi), 966 (транс-C=C), 840, 796, 705.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез новых кремнийсодержащих сополимеров норборнена был осуществлен с использованием трех типов реакции олефинового метатезиса: циклораскрывающей метатезисной полимеризации (ROMP), метатезиса несопряженных диенов (ADMET) и межцепной реакции макромолекулярного кросс-метатезиса.
Синтез исходных гомополимеров
Исходный ПНБ синтезировали путем ROMP норборнена в растворе хлороформа под действием катализатора Граббса первого поколения при соотношении [НБ] : [Г-1] = 700 : 1 по описанной ранее методике [37]:
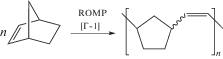
ПНБ содержал преимущественно транс-связи С=С (83%) и имел следующие характеристики: Mw = 3.8 × 105, Đ = 1.8, Tс= 39°С.
Синтез исходного поликарбосилоксана осуществляли из силоксансодержащего диена по схеме метатезисной полимеризации несопряженных диенов [5]. При выборе структуры диена исходили из особенностей протекания ADMET, которая может осложняться побочной реакцией метатезисной циклизации (ring-closing metathesis – RCM), особенно в случаях, если продуктами реакции RCM являются стабильные шести- или семичленные циклы. Низкую активность в ADMET демонстрируют мономеры, содержащие вблизи двойной связи функциональные группы, проявляющие свойства оснований Льюиса, или объемные заместители в аллильном положении [3, 5]. Кроме того, необходимо принимать во внимание ряд ограничений, связанных с реакционной способностью кремнийуглеводородов в метатезисе, а именно неактивность винилсиланов и увеличение реакционной способности двойных связей по мере удаления от атомов кремния [3, 5]. Необходимо также учитывать, что в ходе ADMET для смещения равновесия вакуумированием удаляют легкокипящий продукт реакции – этилен [7]. В связи с этим в качестве мономера мы выбрали высококипящий ГСО, содержащий двойные связи на достаточном удалении от атомов кремния. Ранее ГСО был получен гидросилилированием 1,5-гексадиена гидросиланом – 1,1,3,3,5,5-гексаметилтрисилоксаном в присутствии катализатора Карстеда [8]. Мы синтезировали ГСО из 1,5-дихлор-1,1,3,3,5,5-гексаметилтрисилоксана и реактива Гриньяра, полученного взаимодействием магния с 6-бромгексеном-1:
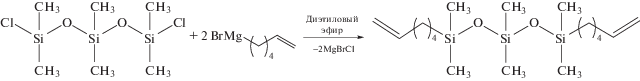
Чистота целевого ГСО, согласно ГХ-МС, составила 93.8%. В нем содержится 5.6% производных дисилоксана и 0.6% производных тетрасилоксана. Наличие силоксановых примесей объясняется их присутствием в исходном 1,5-дихлор-1,1,3,3,5,5-гексаметилтрисилоксане. Согласно ЯМР 1Н, чистота мономера 93%. Перед использованием ГСО выдерживали над гидридом кальция и перегоняли в аргоне.
Полимеризацию ГСО проводили по схеме ADMET на различных Ru-катализаторах Граббса:
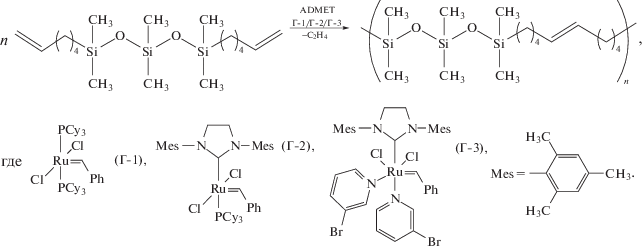
Реакцию вели с постепенным углублением вакуума и последующим повышением температуры от комнатной до 55°С. Варьировали мольное отношение мономер/катализатор и время реакции (табл. 1).
Таблица 1.
Полимеризация ГСО по схеме ADMET на катализаторах Граббса
| Опыт, № | Катализатор | Мольное соотношение [ГСО] : : [катализатор] | Время реакции, ч (при 30–55°C) | Выход, % | Транс-связи C=C, % | Mw × 10–3 | Ð |
|---|---|---|---|---|---|---|---|
| 1 | Г-1 | 230 | 71 | 87 | 82 | 9.8 | 1.6 |
| 2 | Г-1 | 270 | 10 | 91 | 80 | 12.6 | 1.7 |
| 3 | Г-2 | 1000 | 59 | 63 | 74 | 18.0 | 1.4 |
| 4 | Г-2 | 2000 | 32 | 88 | 82 | 5.4 | 1.5 |
| 5 | Г-3 | 300 | 9 | 90 | 80 | 7.8 | 1.5 |
| 6 | Г-3 | 600 | 14 | 79 | 84 | 4.4 | 1.5 |
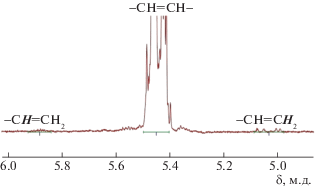
На начальной стадии происходило заметное выделение этилена, который удаляли из зоны реакции вакуумированием. При замедлении выделения этилена постепенно повышали температуру реакции. Ранее ГСО полимеризовали по схеме ADMET только в присутствии катализатора Г-1 при мольном соотношении [мономер] : [Г-1] = 240 в течение 72 ч и при давлении ниже 0.01 мм рт. ст.; получали ПГСО с Mw = 7.6 × 104, Ð = 1.74 [8]. При тех же соотношениях реагентов и времени реакции, но с менее глубоким вакуумом (0.09 мм рт. ст.), нами был синтезирован ПГСО с Mw = 104, Ð = 1.7 (табл. 1, опыт 1). Практически такой же результат был достигнут уже за 10 ч (табл. 1, опыт 2). Вместе с тем молекулярная масса ПГСО, рассчитанная из ПМР-спектра по содержанию концевых групп (рис. 1), составила 1.8 × 105.
Рис. 1.
Область двойных связей С=С в спектре ЯМР 1Н поли(1,1,3,3,5,5-гексаметилтрисилоксанил-5-деценилена) (табл. 1, опыт 2).
Катализатор Г-2 продемонстрировал бòльшую активность: с его участием получен ПГСО с более высокой ММ при значительно меньшей концентрации катализатора (опыт 3). Однако мультиплетность сигналов углеродов С=С в спектре ЯМР 13С поли(1,1,3,3,5,5-гексаметилтрисилоксанил-5-деценилена) (рис. 2, спектр 2) может указывать на протекание изомеризации двойных связей, характерной для процессов, катализируемых Г-2 при длительном нагревании [39]. Для повышения ММ и снижения влияния Г-2 на двойные связи было увеличено соотношение мономер : катализатор и уменьшено время нагревания (опыт 4). В результате мультиплетность спектра действительно уменьшилась (рис. 2, спектр 3), но снижение молекулярной массы ПГСО свидетельствовало о недостаточном количестве катализатора.
Рис. 2.
Спектры ЯМР 13С поли(1,1,3,3,5,5-гексаметилтрисилоксанил-5-деценилена) (область сигналов С=С), синтезированного на катализаторах Граббса Г-1 (1), Г-2 (2, 3 – табл. 1, опыты 3 и 4 соответственно), Г-3 (4).
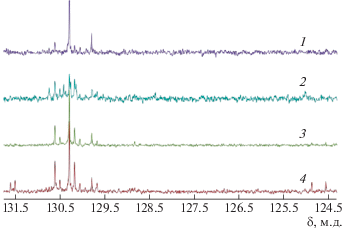
Селективность катализатора Г-3 оказалась меньше, чем у Г-1, но больше, чем у катализатора Г-2 (рис. 2, спектр 4). Низкая молекулярная масса ПГСО, полученного на катализаторе Г-3, подтверждается присутствием сигналов концевых групп даже в спектре ЯМР 13С (сигналы 124.5–125.0 м.д.). Таким образом, для синтеза ПГСО предпочтительно использовать катализатор Г-1.
Синтез силоксансодержащих сополимеров норборнена
Сополимеры НБ–ГСО получали по реакции макромолекулярного кросс-метатезиса между ПНБ и ПГСО в среде хлороформа в присутствии катализаторов Г-1 и Г-2 по методикам, разработанным ранее для кросс-метатезиса между ПНБ и ПЦО или ПНБ и полидодеценамером (ПЦДД) [24, 25, 40]:
Об образовании сополимера судили по появлению сигналов атомов углерода двойных связей гетеродиад в спектре ЯМР 13С (рис. 3). Поскольку двойные связи ПГСО далеко отстоят от атомов кремния, последние практически не оказывают на них воздействия, и сигналы атомов углерода гетеродиад НБ–ГСО оказываются в тех же областях спектра, что и сигналы гетеродиад сополимеров норборнена с циклооктеном, циклопентеном или циклододеценом [26, 33, 41, 42].
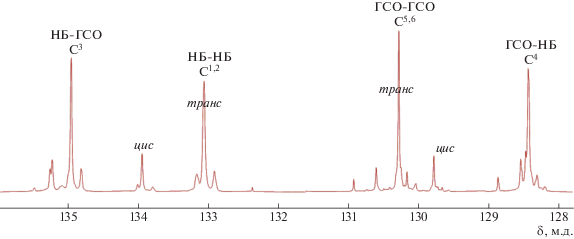
Среднюю длину блоков НБ и ГСО рассчитывали как отношение суммы интегралов атомов углерода в двойных связях гомо- и гетеродиад к интегралу гетеродиад, согласно соотношениям
Ранее нами было показано, что в результате макромолекулярного кросс-метатезиса из гомополимеров образуются статистические мультиблок-сополимеры, при этом степень блочности или среднюю длину блоков можно регулировать, изменяя концентрацию катализатора и полимеров, соотношение исходных полимеров и время реакции. В настоящей работе варьированием указанных параметров были синтезированы сополимеры НБ–ГСО с разной средней длиной блоков (табл. 2, опыты 7–13). Наблюдаемые закономерности макромолекулярного кросс-метатезиса в системе ПНБ–ПГСО сходны с закономерностями, описанными ранее для смесей ПНБ с ПБД, ПЦО и ПЦДД в присутствии катализаторов Граббса. Глубина протекания межцепного обмена возрастает с увеличением количества вводимого катализатора (табл. 2, опыты 7, 8) или с увеличением времени реакции (опыты 9–11), что выражается в уменьшении средней длины блоков L. Изменяя соотношение гомополимеров, можно регулировать соотношение средней длины блоков НБ и ГСО в полученном сополимере (опыт 13). Более активный катализатор Г-2 обеспечивает большую глубину протекания реакции вплоть до образования полностью случайных сополимеров с L = 2 (опыт 12). Согласно предложенному для пары гомополимеров ПНБ–ПЦО механизму реакции [26, 37], на начальных стадиях макромолекулярного кросс-метатезиса при взаимодействии катализатора с гомополимерами происходит “разрезание” макромолекул с образованием полимерных карбенов Ru=ПНБ и Ru=ПЦО, сопровождающееся уменьшением ММ полимеров. В результате при снижении количества вводимого катализатора ММ сополимера увеличивается (опыты 7, 8). Более активный катализатор Г-2 позволяет получить сополимеры с более короткими блоками и большей ММ (опыты 11, 12). Вместе с тем наблюдаются некоторые отличия в поведении ПГСО по сравнению с другими полиалкиленами. Так, в опытах 7–11 длина блока ГСО в 1.5–2 раза меньше длины блока НБ, чего не наблюдали в тех же условиях для ПЦО, но отмечали для систем ПНБ–ПБД–Г-1 [36] и ПНБ–ПЦДД–Г-1 [40].
Таблица 2.
Макромолекулярный кросс-метатезис ПНБ с ПГСО и другими полиалкиленами – ПЦО и ПЦДД
| Опыт, № | Полиалкилен | Мольное соотношение [ПНБ] : : [ПГСО] | Катализатор | Мольное соотношение [полимер]* : : [катализатор] | Время, ч | Выход, % | Mw × × 10–3 | ÐM | LНБ | LГСО |
|---|---|---|---|---|---|---|---|---|---|---|
| 7** | ПГСО | 1 : 1 | Г-1 | 100 | 24 | 48 | 15.7 | 1.5 | 11.5 | 6.2 |
| 8 | 1 : 1 | Г-1 | 300 | 24 | 67 | 32.2 | 2.1 | 26.0 | 19.0 | |
| 9 | 1 : 1 | Г-2 | 600 | 5 | 90 | 15.9 | 1.6 | 16.0 | 12.4 | |
| 10 | 1 : 1 | Г-2 | 600 | 10 | 91 | 14.7 | 1.6 | 7.3 | 5.5 | |
| 11 | 1 : 1 | Г-2 | 600 | 15 | 92 | 15.8 | 1.8 | 4.4 | 3.7 | |
| 12 | 1 : 1 | Г-2 | 1200 | 24 | 85 | 26.3 | 1.3 | 2.1 | 2.1 | |
| 13 | 3.7 : 1 | Г-2 | 1200 | 24 | 88 | 38.8 | 1.3 | 4.7 | 1.4 | |
| 14 | ПЦО [37] | 1 : 1 | Г-1 | 100 | 24 | 92 | 56.0 | 1.5 | 6.8 | 7.3 |
| 15 | ПЦО [24] | 1 : 1 | Г-1 | 300 | 24 | 90 | 37.0 | 2.9 | 14.0 | 15.0 |
| 16 | ПЦО [37] | 2 : 1 | Г-1 | 300 | 24 | 90 | 103.0 | 1.6 | 23.8 | 9.9 |
| 17 | ПЦДД [40] | 1 : 1 | Г-1 | 100 | 24 | 80 | 91.0 | 2.4 | 17.0 | 10.0 |
Эта особенность обусловлена протеканием побочной внутримолекулярной реакции образования циклоолигомеров, которая характерна для линейных макромолекул и практически отсутствует в случае ПНБ [14]. Образующиеся из ПГСО циклоолигомеры отделяются при высаживании сополимеров в спирт, снижая долю гомодиад. В результате состав сополимера, определяемый соотношением средней длины блоков НБ и ГСО в табл. 2, отличается от состава исходной смеси гомополимеров. При этом доля циклоолигомеров выше на начальных стадиях макромолекулярного кросс-метатезиса и снижается по мере протекания межцепного обмена с ростом количества гетеродиад. Соответственно средняя длина блоков НБ и ГСО сближается с увеличением времени реакции (табл. 2, опыты 9–12). Аналогичное поведение отмечали в макромолекулярном кросс-метатезисе между ПНБ и ПЦДД и объясняли меньшей способностью сополимеров к циклообразованию благодаря наличию в них жестких звеньев НБ [40]. В отличие от ПБД, для которого увеличением концентрации полимеров в реакционной смеси до 2 моль/л удалось практически подавить циклообразование, для ПГСО данная реакция заметна даже для концентрированных растворов (табл. 2, опыты 8–10). Особенности поведения ПГСО в макромолекулярном кросс-метатезисе, по-видимому, определяются большей гибкостью молекул ПГСО из-за наличия в мономерном звене силоксанового фрагмента, что способствует протеканию внутримолекулярного метатезиса с образованием циклоолигомеров в отличие от систем, содержащих менее гибкие углеводородные цепи ПБД, ПЦО и ПЦДД.
Таким образом, нами синтезированы новые мультиблок-сополимеры НБ–ГСО, причем различия в строении их цепей обусловлены варьированием условий протекания реакции кросс-метатезиса ПНБ с ПГСО.
Термические свойства сополимеров НБ–ГСО
Термические свойства синтезированных мультиблок-сополимеров НБ–ГСО были изучены методом ДСК (табл. 3, рис. 4). Исходные ПНБ и ПГСО являются полностью аморфными полимерами, что отличает их от частично кристаллических ПБД, ПЦО и ПЦДД. Кристалличность последних определяется наличием в их структуре преимущественно транс-связей С=С. Полученные нами исходные гомополимеры ПНБ и ПГСО содержат более 80% транс-связей, однако не проявляют кристаллических свойств.
Таблица 3.
Термические свойства сополимеров норборнена с ГСО, ЦО и ЦДД
| Опыт, № | Сомономер | Mw × 10–3 | ÐM | LНБ | LГСО | Мольное соотношение [НБ] : [ГСО] | Tс, °C | Tпл, °C | ΔH, Дж/г |
|---|---|---|---|---|---|---|---|---|---|
| 7 | ГСО | 15.7 | 1.5 | 11.5 | 6.2 | 1.9 : 1.0 | –86 | 29 | 5.0 |
| 8 | 32.2 | 2.1 | 26.0 | 19.0 | 1.4 : 1.0 | –8 | 39; 80 | 0.01; 1.0 | |
| 9 | 15.9 | 1.6 | 16.0 | 12.4 | 1.3 : 1.0 | –96 | 33 | 1.5 | |
| 10 | 14.7 | 1.6 | 7.3 | 5.5 | 1.3 : 1.0 | –87; +6 | – | – | |
| 11 | 26.3 | 1.3 | 2.1 | 2.1 | 1.0 : 1.0 | –83 | – | – | |
| 12 | 38.8 | 1.3 | 4.7 | 1.4 | 3.4 : 1.0 | –53 | – | – | |
| 13 | ЦО [37] | 56.0 | 1.5 | 6.8 | 7.3 | 0.9 : 1.0 | –42; +2 | 33; 41 | 3.0; 20.4 |
| 14 | 103.0 | 1.6 | 23.8 | 9.9 | 2.4 : 1.0 | –19 | – | – | |
| 15 | ЦДД [40] | 91.0 | 2.4 | 17.0 | 10.0 | 1.7 : 1.0 | –32 | 74 | 56.0 |
Рис. 4.
а – Кривые ДСК (второе нагревание) исходных гомополимеров ПНБ (1) и ПГСО (2), а также сополимеров НБ-ГСО с различной средней длиной блоков (3–6, табл. 3, опыты 8 (3), 7 (4), 9 (5) и 12 (6)); б – кривые 3 и 4 в увеличенном масштабе.
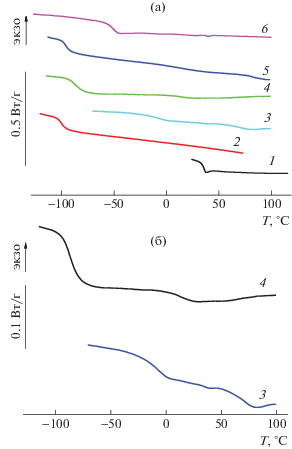
Наличие циклопентанового фрагмента в ПНБ делает его цепь более жесткой по сравнению, например, с полидиенами, вследствие чего Тс оказывается выше комнатной температуры. Большую гибкость цепей ПГСО, приводящую к низкой температуре стеклования, определяет наличие гибкого трисилоксанового фрагмента. Эквимолярная смесь исходных гомополимеров имеет две температуры стеклования 38°С и –102°С, соответствующие значениям Тс ПНБ и ПГСО, что указывает на их термодинамическую несовместимость.
В результате протекания макромолекулярного кросс-метатезиса между ПНБ и ПГСО образуются мультиблок-сополимеры, которые при малой средней длине блоков демонстрируют единственную температуру стеклования и, по-видимому, являются однофазными (опыты 11, 12). Температура стеклования сополимеров НБ–ГСО эквимолярного состава ближе к значению Тс ПГСО из-за значительной разницы молекулярных масс мономеров (344 для ГСО и 94 для НБ), что приводит к существенно большей весовой доле звеньев ГСО в сополимере. Увеличивая долю звеньев НБ, можно повысить Тс сополимеров (опыт 12). На кривых ДСК сополимеров с бòльшей средней длиной блоков ГСО и НБ наблюдаются две температуры стеклования (опыт 10). Аналогичные зависимости Тс от средней длины блоков отмечались ранее для сополимеров НБ–ЦО (табл. 3, опыты 13, 14) [37].
Интересная особенность была обнаружена при дальнейшем увеличении средней длины блоков сополимеров НБ–ГСО (опыты 7–9): на их кривых ДСК в широком интервале температур проявлялись слабые эндотермические эффекты (отметим, что они способны маскировать вторую температуру стеклования). Как видно на рис. 4б, эффект более выражен для сополимера, обогащенного звеньями НБ (опыт 7 в сравнении с опытом 8, кривые 3 и 4). Для сополимера с короткими блоками эндотермические эффекты не проявляются даже в случае значительного избытка звеньев НБ в его составе (опыт 12). Заметим, что для мультиблок-сополимеров НБ–ЦО и НБ–ЦДД также характерно наличие эндотерм на термограммах ДСК (опыты 13–15), обусловленных кристаллическими свойствами блоков ЦО и ЦДД с транс-связями С=С [28, 37, 40]. Более того, степень кристалличности в сополимерах НБ–ЦО в расчете на звено ЦО может быть выше, чем в гомополимере ПЦО, что объясняли увеличением в ходе макромолекулярного кросс-метатезиса содержания звеньев с транс-связями С=С, ответственных за кристалличность в ЦО-блоках [28, 37]. При этом на термограммах видна только одна температура стеклования, поскольку вторая попадает в область плавления. Интересно, что в сополимерах НБ–ЦО увеличение доли звеньев НБ приводит к исчезновению их кристалличности (опыты 13, 14). В отличие от частично кристаллического ПЦО ПГСО является аморфным полимером и в результате макромолекулярного кросс-метатезиса между ПНБ и ПГСО содержание транс-связей С=С практически не изменяется, оставаясь равным 78–83% для гомодиад ГСО и 82–85% для гомодиад НБ. Таким образом, у нас нет веских оснований связывать наблюдаемые эндотермические эффекты с кристалличностью сополимеров НБ–ГСО. Природа данного явления заслуживает отдельного исследования с привлечением структурных методов, что представляется непростой задачей из-за малости теплового эффекта.
ЗАКЛЮЧЕНИЕ
В настоящей работе впервые синтезированы статистические мультиблок-сополимеры, содержащие в основной цепи жесткие блоки норборнена и гибкие блоки 1,5-бис-(гексенил-5)-1,1,3,3,5,5-гексаметилтрисилоксана. Эти сополимеры невозможно получить из соответствующих мономеров вследствие различий в механизме их полимеризации. Нами предложен простой и эффективный путь синтеза таких сополимеров путем макромолекулярного кросс-метатезиса гомополимеров в присутствии катализаторов Граббса первого–второго поколений. Исходный полинорборнен синтезирован по схеме ROMP, в то время как поли(1,1,3,3,5,5-гексаметилтрисилоксанил-5-деценилен) – по схеме ADMET. Изучено влияние условий протекания кросс-метатезиса на строение и характеристики полученных продуктов. Показано, что при изменении средней длины блоков меняются термические свойства сополимеров. По данным ДСК, при нагревании сополимеров со средней длиной блоков в диапазоне 5–11 звеньев наблюдается слабый эндотермический эффект, что указывает на наличие в них организованных структур, отсутствующих в исходных гомополимерах и их смеси.
Авторы благодарят сотрудников ИНХС РАН Р.С. Борисова за проведение анализов методом ГХ-МС, Г.А. Шандрюка за калориметрические измерения, С.А. Легкова за съемку ИК-спектров, а также А.С. Перегудова (ИНЭОС РАН) за съемку спектров ЯМР 13С.
Строение полученных соединений изучено с использованием оборудования Центра коллективного пользования ИНХС РАН и Центра исследования строения молекул ИНЭОС РАН.
Синтез исходных гомополимеров и силоксансодержащих сополимеров норборнена выполнен при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-33-00961-мол-а), остальные исследования – в рамках Государственного задания ИНХС РАН.
Список литературы
Yilgör I., McGrath J.E. // Polysiloxane Copolymers/Anionic Polymerization. Berlin, Heidelberg: Springer, 1988. P. 1.
Matloka P.P., Sworen J.C., Zuluaga F., Wagener K.B. // Macromol. Chem. Phys. 2005. V. 206. № 2. P. 218.
Mukherjee N., Peetz R.M. // Macromolecules. 2008. V. 41. № 18. P. 6677.
Brzezinska K.R., Schitter R., Wagener K.B. // J. Polym. Sci., Polym. Chem. 2000. V. 38. № 9. P. 1544.
Smith D.W., Wagener K.B. // Macromolecules. 1993. V. 26. № 7. P. 1633.
Delgado P.A., Matloka P., Zuluaga F., Wagener K.B. // J. Polym. Sci., Polym. Chem. 2012. V. 50. № 3. P. 431.
Li H., Caire da Silva L., Schulz M.D., Rojas G., Wagener K.B. // Polym. Int. 2017. V. 66. № 1. P. 7.
Matloka P.P., Kean Z., Wagener K.B. // J. Polym. Sci., Polym. Chem. 2010. V. 48. № 9. P. 1866.
Delgado P.A., Zuluaga F., Matloka P., Wagener K.B. // J. Polym. Sci., Polym. Chem. 2009. Vol. 47. № 19. P. 5180.
Małecka E., Marciniec B., Pietraszuk C., Cameron Church A., Wagener K.B. // J. Mol. Catal. A Chem. 2002. V. 190. № 1–2. P. 27.
Brzezinska K.R., Wagener K.B., Burns G.T. // J. Polym. Sci., Polym. Chem. 1999. V. 37. № 6. P. 849.
Smith D.W., Wagener K.B. // Macromolecules. 1993. V. 26. № 14. P. 3533.
Marciniec B., Majchrzak M. // J. Organomet. Chem. 2003. V. 686. № 1–2. P. 228.
Ivin K.J., Mol J.C. Olefin Metathesis and Metathesis Polymerization . London: Acad. Press, 1997.
Finkelshtein E., Gringolts M., Bermeshev M., Chapala P., Rogan Y. // Membrane Materials for Gas and Vapor Separation / Ed.by E. Finkelshtein, Y. Yampolskii/ Chichester: Wiley, 2017. P. 143.
Belov N.A., Gringolts M.L., Morontsev A.A., Starannikova L.E., Yampolskii Y.P., Finkelstein E.S. // Polymer Science B. 2017. V. 59. № 5. P. 560.
Morontsev A.A., Zhigarev V.A., Nikiforov R.Y., Belov N.A., Gringolts M.L., Finkelshtein E.S., Yampolskii Y.P. // Eur. Polym. J. 2018. V. 99. P. 340.
Wagner N.L., Timmers F.J., Arriola D.J., Jueptner G., Landes B.G. // Macromol. Rapid Commun. 2008. V. 29. № 17. P. 1438.
Otsuka H., Muta T., Sakada M., Maeda T., Takahara A. // Chem. Commun. 2009. № 9. P. 1073.
Maeda T., Kamimura S., Ohishi T., Takahara A., Otsuka H. // Polymer (Guildf). 2014. V. 55. № 24. P. 6245.
Ohishi T., Suyama K., Kamimura S., Sakada M., Imato K., Kawahara S., Takahara A., Otsuka H. // Polymer (Guildf). 2015. V. 78. P. 145.
Radlauer M.R., Matta M.E., Hillmyer M.A. // Polym. Chem. 2016. V. 7. № 40. P. 6269.
Daniele S., Mariconda A., Guerra G., Longo P., Giannini L. // Polymer (Guildf). 2017. V. 130. P. 143.
Gringolts M.L., Denisova Y.I., Shandryuk G.A., Krentsel L.B., Litmanovich A.D., Finkelshtein E.S., Kudryavtsev Y.V. // RSC Adv. 2015. V. 5. № 1. P. 316.
Denisova Y.I., Gringolts M.L., Krentsel L.B., Shandryuk G.A., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Ya.V. // Polymer Science B. 2016. V. 58. № 3. P. 292.
Denisova Y.I., Gringolts M.L., Peregudov A.S., Krentsel L.B., Litmanovich E.A., Litmanovich A.D., Finkelshtein E.S., Kudryavtsev Y.V. // Beilstein J. Org. Chem. 2015. V. 11. P. 1796.
Denisova Y.I., Gringolts M.L., Roenko A.V., Shandryuk G.A., Finkelshtein E.S., Kudryavtsev Y.V. // Mendeleev Commun. 2017. V. 27. № 4. P. 416.
Shandryuk G.A., Denisova Y.I., Gringolts M.L., Krentsel L.B., Litmanovich A.D., Finkelshtein E.S., Kudryavtsev Y.V. // Eur. Polym. J. 2017. V. 86. P. 143.
Denisova Yu.I., Gringolts M.L., Krentsel L.B., Shandryuk G.A., Peregudov A.S., Finkelshtein E.Sh., Kudryavtsev Y.V. // Polymer Science B. 2017. V. 59. № 4. P. 412.
Gringolts M.L., Denisova Y.I., Finkelshtein E.S., Kudryavtsev Y. V. // Beilstein J. Org. Chem. 2019. V. 15. P. 218.
Caire da Silva L., Rojas G., Schulz M.D., Wagener K.B. // Prog. Polym. Sci. 2017. V. 69. P. 79.
Bertrand A., Hillmyer M.A. // J. Am. Chem. Soc. 2013. V. 135. № 30. P. 10918.
Monfette S., Fogg D.E. // Chem. Rev. 2009. V. 109. № 8. P. 3783.
Denisova Yu.I., Roenko A.V., Gringolts M.L., Krentsel L.B., Peregudov A.S., Shandryuk G.A., Finkelshtein E.Sh., Kudryavtsev Y.V. // Polymer Science B. 2018. V. 60. № 6. P. 735.
Roenko A.V., Denisova Yu.I., Gringolts M.L., Peregudov A.S., Shandryuk G.A., Finkelshtein E.Sh., Kudryavtsev Y.V. // Polymer Science C. 2019. V. 61. № 1. P. 134.
Morontsev A.A., Gringolts M.L., Filatova M.P., Peregudov A.S., Akmalov T.R., Masoud S.M., Osipov S.N., Denisova Yu.I., Kudryavtsev Y.V. // Polymer Science C. 2019. V. 61. № 1. P. 65.
Morontsev A.A., Denisova Yu.I., Gringolts M.L., Filatova M.P., Shandryuk G.A., Finkelshtein E.Sh., Kudryavtsev Y.V. // Polymer Science B. 2018. V. 60. № 5. P. 688.
Ivin K.J., Lapienis G., Rooney J.J. // Makromol. Chem. 1982. B. 183. № 1. S. 9.
Courchay F.C., Sworen J.C., Wagener K.B. // Macromolecules. 2003. V. 36. № 22. P. 8231.
Denisova Yu.I., Zhigarev V.A., Gringolts M.L., Shandryuk G.A., Peregudov A.S., Finkelshtein E.S., Kudryavtsev Y.V. // Polymer Science C. 2019. V. 61. № 1. P. 120.
Amir-Ebrahimi V., Carvill A.G., Hamilton J.G., Rooney J.J., Tuffy C. // J. Mol. Catal. A. 1997. V. 115. № 1. P. 85.
Ivin K., O’Donnell J., Rooney J., Stewart C. // Makromol. Chem. 1979. B. 180. № 8. S. 1975.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)