

Высокомолекулярные соединения (серия Б), 2021, T. 63, № 1, стр. 55-65
СИНТЕЗ ЗВЕЗДООБРАЗНЫХ ПОЛИ(2-АЛКИЛ-2-ОКСАЗОЛИН)ОВ НОВОГО ТИПА НА ОСНОВЕ СУЛЬФОХЛОРИРОВАННОГО КАЛИКС[8]АРЕНА
А. Н. Блохин a, *, А. Б. Разина a, А. Э. Бурсиан a, А. В. Теньковцев a
a Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр., 31, Россия
* E-mail: 44stuff44@gmail.com
Поступила в редакцию 26.06.2020
После доработки 01.09.2020
Принята к публикации 15.09.2020
Аннотация
Предложен новый подход к синтезу сульфонилхлоридных инициаторов катионной полимеризации 2-оксазолинов с каликсареновым ядром. Изучена кинетика полимеризации 2-этил-2-оксазолина в сульфолане при 100°С на синтезированном октафункциональном инициаторе. Установлено, что в системе наблюдается замедленное иницирование полимеризации, константа скорости роста цепи составляет kp = 1.20 × 10–3 л/моль с. Методом катионной полимеризации 2-алкил-2-оксазолинов с раскрытием цикла получены звездообразные термочувствительные полимеры с использованием каликс[8]арена, функционализированного сульфонилхлоридными группами в нижнем ободе, и изучены молекулярно-массовые характеристики полимеров. Определена критическая концентрация мицеллообразования звездообразного поли(2-изопропил-2-оксазолин)а (СККМ = 6.25 × 10–5 г/мл) и, на примере гидрофобного красителя куркумина, исследована солюбилизирующая способность полученных полимеров.
ВВЕДЕНИЕ
Катионная полимеризация 2-алкил-2-оксазолинов позволяет легко получать блок-сополимеры и полимеры с разветвленной структурой, такие как молекулярные щетки и звездообразные полимеры. Образующиеся амфифильные N-ацилполиэтиленимины (полиоксазолины) являются биосовместимыми полимерами [1, 2], проявляющими термочувствительность в водных средах [3]. Так называемые “умные” материалы на основе поли(2-алкил-2-оксазолин)ов могут быть использованы в различных биомедицинских приложениях.
В качестве инициаторов полимеризации 2-оксазолинов применялись соединения различных классов, полифункциональные инициаторы и макроинициаторы [4]. Представляет интерес получение звездообразных полимеров с ядром, являющимся не только центром ветвления, но и способным к комплексообразованию. Полимеры такой структуры могут быть задействованы в системах целевой доставки лекарственных препаратов, радионуклидов и флуоресцентных меток.
Одним из используемых в этом случае подходов можно назвать синтез мультифункциональных инициаторов на основе макроциклов. Здесь выделяются каликс[n]арены – циклические олигомеры, служащие продуктами конденсации фенола с формальдегидом. Известно, что каликсарены образуют соединения включения типа “гость−хозяин” с низкомолекулярными органическими соединениями, ионами металлов [5] и могут быть задействованы в системах молекулярного распознавания [6]. Низкая токсичность каликсаренов позволяет применять их в составе материалов в живых средах [7].
Звездообразные поли(2-оксазолин)ы с каликс[n]ареновым ядром, функционализированным по нижнему ободу, были синтезированы на основе макроинициаторов алкилбромидного типа и достаточно подробно изучены [8–11]. Также получены региоизомерные структуры макроинициаторов с каликсареновым фрагментом, функционализированным по верхнему ободу инициирующими сульфонилхлоридными группами [12].
Настоящая работа ставит своей задачей разработку новой схемы синтеза каликс[8]аренового инициатора с введением в его нижний обод сульфонилхлоридных заместителей с целью сравнения эффективности использования сульфонилхлоридных групп в двух разных типах инициаторов и выявления различий в применении ароматических и алифатических сульфонилхлоридов для проведения полимеризации. Кроме того, в работе уделяется внимание процессам ассоциации в водных растворах звездообразных поли(2-оксазолин)ов нового типа. В частности, поставлена задача установления критической концентрации мицеллообразования в растворах синтезированных полимеров, которая не определялась ранее для звездообразных поли(2-оксазолин)ов с каликсареновым ядром. Также в работе проведена оценка солюбилизирующей способности полимеров в водных растворах на примере гидрофобного красителя куркумина.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и реагенты
Растворителями служили хлорбензол фирмы “J&K”, бензол и петролейный эфир компании “Вектон”, которые использовали без специальной очистки. Дихлорметан (“Вектон”) перегоняли над пятиокисью фосфора, сульфолан (“Aldrich”) − в вакууме (Tкип = 166°С, p = 1 мм рт. ст.). Пирролидин очищали простой перегонкой. Этилцеллозольв (“Вектон”) и тетрагидрофуран очищали от пероксидов кипячением и перегонкой над хлоридом меди(I), после чего повторно перегоняли над гидридом кальция. Мономеры 2-метил-, 2-этил-, 2-изопропил-2-оксазолин (“Aldrich”) перегоняли над гидридом кальция. Пара-трет-бутилкаликс[8]арен синтезировали по методике [13].
Синтез 11-сульфоундеканоата калия
В колбу, снабженную обратным холодильником, помещали 30 г (0.11 моля) 11-бромундекановой кислоты, 21.4 г (0.17 моля) сульфита натрия, 9.6 г (0.11 моля) гидрокарбоната натрия, 300 мл дистиллированной воды. Смесь перемешивали и нагревали на водяной бане в течение 20 ч. После охлаждения до комнатной температуры в раствор добавляли 8.4 г (0.11 моля) хлорида калия, затем 10 мл концентрированной соляной кислоты при интенсивном перемешивании. Осажденный продукт фильтровали, промывали поочередно охлажденным водным раствором хлорида калия 5%, этиловым спиртом, диэтиловым эфиром, сушили. Продукт перекристаллизовывали из водного раствора хлорида калия 10%, подкисленого 2−3 мл концентрированной соляной кислоты. Выход продукта составил 20.1 г (67%). Tпл = = 144°С. Спектр ЯМР 1H (400 MГц, D2O, 20°С): δ (м.д.) 2.82 (t, 2H), 2.30 (t, 2H), 1.64 (quint, 2H), 1.52 (quint, 2H), 1.38‒1.17 (m, 12H).
Синтез хлорангидрида 11-хлорсульфонилундекановой кислоты
В колбу, снабженную обратным холодильником, помещали 15 г (0.053 моля) 11-сульфоундеканоата калия, 50 мл (0.69 моля) хлористого тионила. В смесь добавляли несколько капель диметилформамида в качестве катализатора, затем кипятили при перемешивании в течение 3 ч. После охлаждения до комнатной температуры раствор фильтровали, избыток хлористого тионила отгоняли в вакууме (100°C, 20 мм рт. ст.). Продукт перегоняли в вакууме (200°C, 0.1 мм рт. ст.). Выход продукта составил 5.1 г (32%). Tкип = 200°C (0.1 мм рт. ст.). Спектр ЯМР 1H (400 MГц, СDCl3, 20°С): δ (м.д.) 3.54 (t, 2H), 2.90 (t, 2H), 1.82‒1.67 (m, 4H), 1.55‒1.25 (m, 12H).
Синтез инициатора 5,11,17,23,29,35,41,47-окта(трет-бутил)каликс [8]арил-49,50,51,52,53,54,55,56-окта(11-хлорсульфонилундеканоат)а
В колбу, снабженную обратным холодильником, помещали 2 г (1.54 × 10–3 моль) 4-трет-бутилкаликс[8]арена и 10 мл хлорбензола. Смесь насыщали сухим аргоном в течение 15 мин, затем добавляли 7.5 г (0.0246 моля) хлорангидрида 11-хлорсульфонилундекановой кислоты и нагревали при 120°C в течение 48 ч в атмосфере аргона. Растворитель отгоняли на роторном испарителе, после чего остаток в колбе взбалтывали с 20 мл горячей воды и эквивалентным объемом бензола. Органическую фазу отделяли от водной, бензол отгоняли. Остаток в колбе очищали от побочного продукта (11-хлорсульфонилундекановая кислота) двукратной экстракцией в 20 мл горячего этилового спирта. Для удаления следов влаги продукт растворяли в безводном дихлорметане и упаривали досуха. В качестве альтернативного способа очистки может быть использована колоночная хроматография: сорбент – силикагель, элюент – петролейный эфир (дихлорметан) в соотношении 9 : 1. Выход продукта составил 4.2 г (80%). Спектр ЯМР 1H (400 MГц, СDCl3, 20°С): δ (м.д.) 6.92 (s, 16H), 3.63 (s, 16H), 3.54 (t, 16H), 2.41 (t, 16H), 1.77 (quint, 16H), 1.66 (quint, 16H), 1.54‒1.21 (m, 96H), 1.06 (s, 72H).
Тестовая реакция фенола с хлорангидридом 11-хлорсульфонилундекановой кислоты
В колбу, снабженную обратным холодильником, помещали 1 г (0.0106 моля) фенола, 6.5 г (0.0212 моля) хлорангидрида 11-хлорсульфонил-ундекановой кислоты и 5 мл тетрахлорэтана. Смесь насыщали сухим аргоном, затем нагревали до температуры кипения и перемешивали в течение 4 ч. После охлаждения отбирали пробу для анализа методом спектроскопии ЯМР 1H.
Исследование кинетики полимеризации 2-этил-2-оксазолина
В пробирку помещали 0.5 г (5.04 × 10–3 моль) 2-этил-2-оксазолина, 0.5 г (4.16 × 10–3 моль) сульфолана и 0.0721 г (2.10 × 10–5 моль) инициатора окта-(трет-бутил)каликс[8]арил-окта-(11-хлорсульфонилундеканоат)а. Смесь нагревали при температуре 100°С, пробы реакционной смеси отбирали в ампулы для ЯМР и охлаждали до 0°С.
Синтез звездообразных поли(2-алкил-2-оксазолин)ов
В ампулу помещали инициатор и мономер в мольном соотношении 1 : 240 соответственно, добавляли сульфолан. Смесь нагревали при 100°С в течение 24 ч, затем вводили 1 мл пирролидина и полученный раствор перемешивали в течение 1 ч при 50°С. Продукт очищали диализом относительно воды в течение 24 ч, лиофильно сушили.
Полимер I. 2 г (0.0235 моля) 2-метил-2-оксазолина, 0.3358 г (9.79 × 10–5 моль) инициатора, 2 г (0.017 моля) сульфолана. Выход продукта составил 1.9 г (82%). Спектр ЯМР 1H (400 MГц, СDCl3, 20°С): δ (м.д.) 3.48 (m), 2.13 (m).
Полимер II. 2 г (0.0201 моля) 2-этил-2-оксазолина, 0.2872 г (8.38 × 10–5 моль) инициатора, 2 г (0.017 моля) сульфолана. Выход продукта составил 1.8 г (79%). Спектр ЯМР 1H (400 MГц, СDCl3, 20°С): δ (м.д.) 3.47 (m, 4H), 2.50–2.21 (m, 2H), 1.13 (m, 3H).
Полимер III. 2 г (0.0177 моля) 2-изопропил-2-оксазолина, 0.2527 г (7.37 × 10–5 моль) инициатора, 2 г (0.017 моля) сульфолана. Выход продукта составил 1.8 г (80%). Спектр ЯМР 1H (400 MГц, СDCl3, 20°С): δ (м.д.) 3.47 (m, 4H), 3.09‒2.55 (m, 1H), 1.13 (m, 6H).
Синтез звездообразного блок-сополимера
Полимер IV. В ампулу помещали 1 г (0.0101 моля) 2-этил-2-оксазолина, 0.2887 г (8.41 × 10–5 моль) инициатора, 2 г (0.017 моля) сульфолана. Смесь нагревали при 100°С в течение 24 ч, затем добавляли 1.14 г (0.0101 моля) 2-изопропил-2-оксазолина и продолжали нагревать в течение 24 ч при той же температуре. Полимеризацию обрывали пирролидином. Продукт очищали диализом относительно воды в течение 24 ч, лиофильно сушили. Выход продукта составил 2.0 г (82%). Спектр ЯМР 1H (400 MГц, СDCl3, 20°С): δ (м.д.) 3.47 (m), 2.92 (m), 2.70 (m), 2.51–2.22 (m), 1.13 (s).
Отделение поли(2-оксазолин)овых лучей от каликсаренового ядра звездообразного полимера
Навеску 0.1 г звездообразного поли(2-алкил-2-оксазолин)а растворяли в 5 мл этилцеллозольва, добавляли 0.28 г (0.005 моля) гидроксида калия. Смесь нагревали при 120°С в течение 10 мин. Раствор очищали диализом относительно воды в течение 24 ч, лиофильно сушили.
Методы характеризации химических соединений
Спектры ЯМР 1H растворов образцов в дейтерированных растворителях снимали на приборе “Bruker AC400” (400 MГц), спектры УФ-видимого излучения − на спектрофотометре “ЛОМО ФОТОНИКА СФ-256”. Диализ растворов полимеров проводили с использованием диализных мешков “CellaSep” (MWCO 3500 D). Образцы поли(2-оксазолин)овых лучей исследовали методом ГПХ на приборе “Shimadzu Prominence LC-20”, оснащенном рефрактометрическим детектором RID-10A и хроматографической колонкой “Shim-pack GPC-80M”. Измерения осуществляли при температуре термостата 40°C и скорости потока элюента (ТГФ) 1.0 мл/мин. Калибровочную кривую рассчитывали с использованием полистирольных стандартов.
Изучение фазовых переходов растворов полимеров
Термочувствительность полимеров исследовали методом турбодиметрии на модифицированном спектрофотометре “Specord UV-VIS” (“Carl Zeiss Jena”). Пропускание света (λ = 625 нм) измеряли в водных растворах соответствующих полимеров с массовой концентрацией с = 0.5% в слое толщиной 1 см при постоянном нагревании со скоростью 0.5 град/мин.
Определение критической концентрации мицеллообразования
К растворам полимера объемом 2 мл с массовой концентрацией в диапазоне 0.0003–0.5% добавляли 20 мкл раствора куркумина в метаноле с концентрацией 1 × 10–4 г/мл. Смеси выдерживали 24 ч при комнатной температуре в темноте. Спектры поглощения полученных растворов измеряли в области длины волн λ = 300–500 нм.
Определение солюбилизирующей способности
К водным растворам полимера объемом 2 мл и концентрацией 0.5% прибавляли навески куркумина массой 0.0005–0.0040 г. Растворы выдерживали в ультразвуковой ванне в течение 15 мин, затем перемешивали 3–4 ч. Избыток куркумина отделяли от растворов центрифугированием. Измеряли спектры поглощения полученных растворов в области длины волн λ = 300–500 нм. Для определения концентрации солюбилизата в мицеллах каждый раствор упаривали до полного высыхания, затем остаток растворяли в 2 мл метанола и устанавливали оптическую плотность в максимуме поглощения куркумина. Концентрацию рассчитывали с использованием предварительно построенной калибровочной зависимости.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез инициатора полимеризации
Использование каликс[n]аренсульфонилхлоридов в качестве инициаторов полимеризации 2-алкил-2-оксазолинов предполагает введение сульфогрупп в структуру каликсаренового макроцикла, при этом возможна модификация как в верхнем, так и в нижнем кольцевых ободах макроцикла. Исходя из поставленных задач, были рассмотрены возможные пути синтеза октафункционального инициатора на основе окта-(трет-бутил)каликс[8]арена с инициирующими группами в нижнем кольцевом ободе. В частности, один из предложенных способов заключался в активации фенольных гидроксогрупп сильными основаниями и последующей реакцией с 1,3-пропан-, 1,4-бутансультонами [14–16].
В рамках работы сделан пробный синтез инициатора с использованием 1,4-бутансультона, однако выделить целевой продукт, а также установить его присутствие в спектрах ЯМР 1H реакционной смеси не удалось:

В связи с этим, была предложена принципиально новая методика введения сульфонилхлоридных групп в нижний обод. Известно, что скорость алкоголиза хлорангидридов карбоновых кислот и сульфонилхлоридов существенно различается в отсутствие основного катализа [17, 18]. Такое различие позволяет селективно присоединить спейсер с сульфонилхлоридной группой к нижнему ободу каликс- [8]арена:

Для установления возможности синтеза инициатора по предложенной методике проведена модельная реакция фенола с хлорангидридом 11-хлорсульфонилундекановой кислоты. Методом спектроскопии ЯМР 1H показано, что в реакционной смеси присутствует исключительно продукт взаимодействия карбонилхлоридных и гидроксильных групп, в то время, как сульфонилхлоридная группа остается в неизменном виде (рис. 1). Спейсер (хлорангидрид 11-хлорсульфонилундекановой кислоты) был синтезирован из коммерчески доступной 11-бромундекановой кислоты с заимствованием методики получения сульфокислот по Вейганду [19], а также методики получения хлорангидрида, описанной W. Rigby [20]. В качестве оптимального растворителя для проведения синтеза инициатора был выбран хлорбензол.
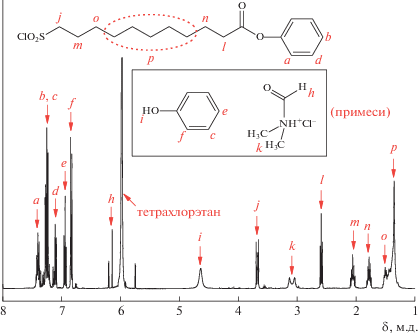
Исследование кинетики полимеризации
В целях установления возможности контролируемого синтеза полимеров с использованием полученного октафункционального инициатора исследована кинетика полимеризации 2-этил-2-оксазолина в сульфолане при температуре 100°С. Условия проведения полимеризации были выбраны с учетом опыта предшествующих работ по синтезу поли(2-алкил-2-оксазолинов) на основе сульфонилхлоридов [21, 22]. Количественный анализ проведен методом спектроскопии ЯМР 1H: конверсия мономера рассчитана по интегральной интенсивности сигнала протонов оксазолинового гетероцикла (4.20 м.д.) с использованием сигнала протонов сульфолана (3.0 м.д.) в качестве внутреннего стандарта. На рис. 2 приведена зависимость логарифма обратной конверсии мономера от продолжительности полимеризации. Установлено, что на начальном этапе полимеризации присутствует индукционный период, характерный для инициирования алкилсульфонилгалогенидами [21]. Далее наблюдается линейный участок, подтверждающий, что рост цепи происходит в соответствии с кинетикой первого порядка, и необратимый обрыв цепей отсутствует.

Аппроксимация линейного участка кривой в соответствии с кинетическим уравнением первого порядка (1) позволила рассчитать кажущуюся константу скорости роста цепей, которая составила kp = 1.20 × 10–3 л/моль с. В уравнении (1) концентрация растущих макрокатионов [M*] заменена на концентрацию макроинициатора [I0] из предположения, что все сульфонилхлоридные группы прошли стадию инициирования:
(1)
$\ln \left( {\frac{{{{{\left[ {\text{M}} \right]}}_{0}}}}{{\left[ {\text{M}} \right]}}} \right) = {{\left[ {\text{I}} \right]}_{0}}{{k}_{p}}t,$Вид полученной кинетической зависимости не дает однозначного ответа о наличии реакций передачи цепей в процессе полимеризации, однако их отсутствие может быть подтверждено построением графической зависимости степени полимеризации (среднечисленной молекулярной массы) от конверсии мономера [23]. В случае отсутствия процессов передачи цепи среднечисленная молекулярная масса Mn полимера должна линейно зависеть от степени превращения мономера x:
(2)
${{M}_{n}} = {{M}_{1}}\left( {D{{P}_{n}}} \right) = \frac{{{{M}_{1}}\left( {\left[ {{{{\text{M}}}_{0}}} \right] - \left[ {\text{M}} \right]} \right)}}{{\left[ {{{{\text{I}}}_{0}}} \right]}} = {{M}_{1}}x\frac{{[{{{\text{M}}}_{0}}]}}{{\left[ {{{{\text{I}}}_{0}}} \right]}}$Здесь M1 – молекулярная масса мономерного звена, DPn – среднечисленная степень полимеризации, х – конверсия мономера.
На рис. 3 представлена зависимость среднечисленной молекулярной массы растущих цепей поли(2-этил-2-оксазолин)а от конверсии мономера, линейный характер которой указывает на отсутствие процесса передачи цепи. Суммарная среднечисленная молекулярная масса растущих цепей и их степень полимеризации были рассчитаны методом спектроскопии ЯМР 1H по соотношению интенсивностей сигналов протонов основной цепи поли(2-этил-2-оксазолин)а (3.6–3.2 м.д.) и протонов метиленовых групп спейсера в структуре макроинициатора (1.40–1.15 м.д.).
Рис. 3.
Зависимость среднечисленной молекулярной массы растущих цепей поли(2-этил-2-оксазолин)а от конверсии мономера.

Сравнение кинетических данных макроинициаторов на основе каликс[8]арена с разной конфигурацией и типом сульфонилхлоридных групп показывает, что кажущаяся константа скорости роста цепей для алифатического сульфонилхлорида (1.20 × 10–3 л/моль с) значительно выше по сравнению с ароматическим сульфонилхлоридом (2.12 × 10–4 л/моль с) [12]. Однако в случае ароматического сульфонилхлорида замедленного инициирования полимеризации не наблюдается, что позволяет синтезировать полимеры с потенциально более узким молекулярно-массовым распределением. Разница в значениях скорости полимеризации для сравниваемых макроинициаторов предположительно связана со стерическим фактором: в нижнем ободе каликс[8]арена растущие цепи дистанцированы путем присоединения через спейсер, в то время как в верхнем ободе они присоединены непосредственно к ароматическим кольцам и их плотность повышена.
Синтез звездообразных поли(2-алкил-2-оксазолин)ов
С использованием полученного инициатора синтезированы звездообразные поли(2-алкил-2-оксазолин)ы с различной длиной и строением алкильного заместителя (схема 3). Соотношение значений концентрации мономера и инициатора было равным [M]/[I] = 240, чтобы теоретическая степень полимеризации каждого полимерного луча составляла DPn = 30. Для проведения полимеризации 2-оксазолинов был выбран сульфолан [24], так как он обеспечивал хорошую растворимость каликсаренового инициатора. Полимеризация проводилась при температуре 100°С, после чего реакция обрывалась добавлением в реакционную смесь пирролидина, обеспечивающего быстрый и необратимый обрыв цепей [25].

Структура синтезированных поли(2-алкил-2-оксазолин)ов подтверждена методами спектроскопии ЯМР 1H и УФ. В УФ-спектрах спиртовых растворов образцов наблюдается поглощение в области 250–290 см–1 с характерной для ароматических фрагментов колебательной структурой полосы поглощения.
Методом пост-полимеризации был синтезирован звездообразный поли(2-этил-2-оксазолин)-блок-поли(2-изопропил-2-оксазолин). Известно, что при отсутствии необратимого обрыва цепи полимеризация после исчерпания первого мономера может быть реинициирована добавлением в реакционную смесь мономера другой структуры [26]. Мономеры 2-этил- и 2-изопропил-2-оксазолин были добавлены последовательно в эквимольном количестве, чтобы теоретическая степень полимеризации каждого блока составляла DPn = 15. В спектре ЯМР 1H образца (рис. 4) были обнаружены сигналы протонов этильных (2.5–2.2 м.д.) и изопропильных (3.0–2.6 м.д.) боковых групп, что подтверждает образование сополимера. На основании расчетов интегральных интенсивностей сигналов протонов установлено, что соотношение мономерных звеньев 2-этил- и 2-изопропил-2-оксазолинов в лучах блок-сополимера близко к 1 : 1.

Определение молекулярных масс полимеров
Для определения молекулярно-массовых характеристик синтезированных образцов была проведена селективная деструкция звездообразных полимеров с сохранением структуры и первоначальной длины поли(2-алкил-2-оксазолин)овых лучей. Данный подход успешно применялся ранее для установления молекулярных масс звездообразных полимеров с каликсареновым центром ветвления [27]. Отделение поли(2-алкил-2-оксазолин)овых лучей от каликс[8]аренового ядра проводилось путем расщепления сложноэфирной группы в составе спейсера в безводной сильнощелочной среде:

В табл. 1 приведены молекулярно-массовые характеристики образцов линейных поли(2-алкил-2-оксазолин)ов, исследованных методом гель-проникающей хроматографии. Среднечисленные молекулярные массы синтезированных полимеров были определены альтернативным способом: на основании спектров ЯМР 1H образцов с использованием описанной выше методики.
Исследование термочувствительности полимеров
Поли(2-оксазолин)ы, содержащие алкильные заместители в боковых цепях (за исключением метильного), проявляют термочувствительные свойства в водных растворах. Температура их фазового расслоения варьируется в зависимости от длины алкильного заместителя. Кроме того, изменение гидрофобно-гидрофильного баланса макромолекулы может происходить при введении массивного центра ветвления в структуру звездообразных поли(2-оксазолин)ов, а также функциональных концевых групп. В общем случае показано, что температуры фазового расслоения для растворов звездообразных поли(2-оксазолин)ов, как правило, на 10–20°С ниже по сравнению с линейными аналогами, имеющими идентичную молекулярную массу [28–30].
В настоящей работе фазовые переходы в водных растворах синтезированных полимеров массовой концентрацией 0.5% исследовались турбидиметрическим методом. Полученная зависимость светопропускания растворов полимеров от температуры изображены на рис. 5. Точки помутнения растворов tcp были определены в соответствии с предложенными рекомендациями [31] как значения температуры, при которых светопропускание водного раствора снижается до 80 и 50% (табл. 2).
Рис. 5.
Зависимость светопропускания растворов полимеров I, II и III от температуры для водных растворов поли(2-алкил-2-оксазолин)ов.

Таблица 2.
Характеристики температурно-индуцированных фазовых переходов в растворах синтезированных полимеров
| Образец | tcp, °C | Δt, °C | |
|---|---|---|---|
| Светопропу-скание 80% | Светопропу-скание 50% | ||
| I | – | – | – |
| II | 68.0 | 69.0 | 3.5 |
| III | 38.9 | 39.4 | 2.0 |
| IV | 45.2 | 46.3 | 6.0 |
Установлено, что звездообразный поли(2-метил-2-оксазолин) вследствие высокой гидрофильности полимерных цепей не проявляет термочувствительности, несмотря на наличие гидрофобного каликсаренового ядра и углеводородных спейсеров. Для полимеров на основе 2-этил- и 2-изопропил-2-оксазолинов (образцы II–IV) точки помутнения растворов tcp были обнаружены в температурном диапазоне 38–69°C. Полученные значения близки к показателям температуры фазовых переходов для звездообразных поли(2-оксазолин)ов с каликс[8]ареновым центром ветвления иной конфигурации, которые обсуждались в предыдущей работе [12]. Довольно широкие температурные интервалы фазовых переходов для образцов II–IV, превышающие 1°C, обусловлены большими по величине показателями дисперсности Đ, не характерными для полимеризации, протекающей по живому механизму (Đ = 1.1–1.4). Причиной таких отклонений предположительно является замедленное инициирование полимеризации.
Определение критической концентрации мицеллообразования
Подобно линейным аналогам звездообразные амфифильные полимеры способны самоорганизовываться в разбавленных водных растворах. Ассоциация звездообразного поли(2-изопропил-2-оксазолин)а в водных растворах рассмотрена методом солюбилизации гидрофобного красителя, светопоглощение которого чувствительно к микроокружению [32]. В ряде исследований это свойство использовалось для определения критической концентрации мицеллообразования (ККМ) низкомолекулярных и полимерных поверхностно-активных веществ [33–36]. В качестве гидрофобного красителя чаще всего использовался 1,6-дифенил-1,3,5-гексатриен, степень светопоглощения которого (λmax = 356 нм) минимальна в воде, но увеличивается в гидрофобных средах. Однако сообщалось и об успешном применении других красителей [37].
В настоящей работе для этой цели был использован органический краситель куркумин 1,7-бис-(4-гидрокси-3-метоксифенил)-1,6-гептадиен-3,5-дион – аналог синтетических азокрасителей, обладающий сходным с дифенилгексатриеном строением (максимум поглощения λmax = 340 нм) и коэффициентом экстинкции [38]. Для установления процессов агрегации и мицеллообразования макромолекул исследовались спектры поглощения водных растворов, содержащих одинаковую концентрацию 2.7 × 10–6 моль/л куркумина и варьируемую в диапазоне 3 × 10–6‒7.5 × 10–3 г/мл концентрацию поли(2-изопропил-2-оксазолин)а (рис. 6а). Как видно на рис. 6б, на зависимости интенсивности светопоглощения от концентрации полимера в растворе могут быть условно выделены три области. В первой области, соответствующей сильно разбавленным растворам полимера, прослеживается снижение интенсивности поглощения красителя. Известно, что такой характер изменения спектра свидетельствует о формировании молекулярных комплексов между красителем и амфифильным полимером, выполняющим роль поверхностно-активного вещества [39–43]. Начиная с определенной концентрации макромолекул в растворе, происходит увеличение интенсивности поглощения, свидетельствующее о том, что молекулы красителя находятся в другом микроокружении (сольватохромный эффект). Предположительно в этой концентрационной области (область II) в растворе начинается образование мицелл [44–46]. Данное утверждение хорошо согласуется с результатами исследования водных растворов звездообразных поли(2-оксазолин)ов и поли(2-оксазин)ов методом статического светорассеяния [47, 48]. Известно, что в умеренно разбавленных растворах подобных амфифильных полимеров присутствуют ассоциаты с гидродинамическими радиусами в диапазоне 10−100 нм. В третьей области возникает насыщение кривой зависимости, но дальнейшего увеличения интенсивности поглощения не происходит, что свидетельствует об окончании процесса солюбилизации красителя мицеллами и завершении процесса формирования мицелл.
Рис. 6.
Cпектры поглощения водных растворов с содержанием полимера в диапазоне 6 × 10–5–5 × 10−3 г/мл (а) и зависимость интенсивности светопоглощения при λmax = 430 нм от концентрации полимера в растворе (б).
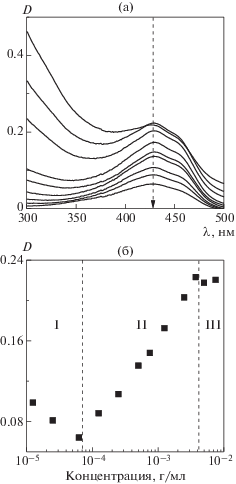
В соответствии с предложенной методикой [32], значение ККМ было определено как точка пересечения касательных к кривой в областях I и II − СККМ = 6.25 × 10–5 г/мл.
Определение солюбилизирующей способности полимера
Как было показано выше, макромолекулы поли(2-изопропил-2-оксазолин)а находятся в агрегированном мицеллярном состоянии в широкой области концентрации водных растворов, ограниченной ККМ снизу и пределом растворимости полимера сверху (0.01–0.015%). Известно, что амфифильные полимеры и другие поверхностно-активные вещества способны солюбилизировать нерастворимые в воде липофильные вещества при концентрации выше ККМ, увеличивая их растворимость в водных средах. Это свойство может быть использовано в фармакологии для повышения растворимости гидрофобных лекарственных веществ. К таким веществам, в частности, относится куркумин, который обладает потенциальными противовоспалительным и противоопухолевым действиями, однако имеет ограниченную применимость вследствие низкой водорастворимости [49, 50].
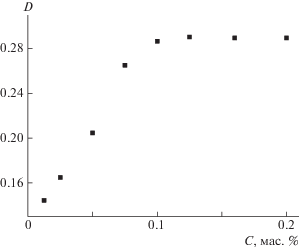
В связи с этим, в настоящей работе была проведена оценка солюбилизирующей способности звездообразного поли(2-изопропил-2-оксазолин)а по отношению к куркумину, растворимость которого в воде не превышает 0.6 мкг/мл [51]. На рис. 7 видно, что при растворении куркумина в водном растворе полимера наблюдается область насыщения, позволяющая рассчитать предельную концентрацию солюбилизированного куркумина.
Рис. 7.
Зависимость интенсивности светопоглощения в максимуме λmax = 368 нм от массовой доли куркумина, добавленного в раствор полимерного ПАВ.
Солюбилизирующую способность поверхностно-активных веществ обычно характеризуют мольным коэффициентом солюбилизации при насыщении Rm, который расчитывается по формуле:
где ns, nw – полная растворимость солюбилизата и его растворимость в воде соответственно, с – общая концентрация ПАВ в водном растворе, Сk – критическая концентрация мицеллообразования ПАВ [52].Другим существенным параметром является мольный коэффициент распределения солюбилизата между мицеллами и дисперсионной средой Km:
Отметим, что расчеты солюбилизирующей способности в настоящей работе носят лишь оценочный характер, так как не представляется возможным определить точную молекулярную массу полимерного ПАВ. Тем не менее было использовано расчетное значение, взятое на основе средневесовой молекулярной массы лучей звездообразного поли(2-изопропил-2-оксазолин)а. Приведенные в табл. 3 значения Rm и Km в сравнении c солюбилизацией куркумина классическими катионными ПАВ [53], позволяют сделать вывод об эффективности применения звездообразного поли(2-изопропил-2-оксазолин)а в качестве солюбилизатора.
ЗАКЛЮЧЕНИЕ
Синтетический потенциал химии поли(2-алкил-2-оксазолин)ов может быть существенно дополнен использованием мультифункциональных инициаторов на основе каликсареновых макроциклов. При этом инициирующие полимеризацию сульфонилхлоридные группы могут быть введены не только в верхний, но и в нижний кольцевой обод каликсарена, позволяя изменять конфигурацию центра ветвления звездообразного полимера.
Синтезированные амфифильные звездообразные поли(2-оксазолин)ы проявляют термочувствительные свойства в водных растворах и могут быть использованы для солюбилизации гидрофобных органических соединений, что делает перспективным их применение в различных сферах деятельности.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований в рамках научного проекта (код проекта 19-33-90002).
Список литературы
Goddard P., Hutchinson L.E., Brown J., Brookman L.J. // J. Controlled Release. 1989. V. 10. P. 5.
Maechling-Strasser C., Dejardin P., Galin J.C., Schmitt A., House-Ferrari V., Sebille B., Mulvihill J.N., Cazenave J.P. // J. Biomed. Mater. Res. 1989. V. 23. P. 1395.
Weber C., Hoogenboom R., Schubert U.S. // Prog. Polym. Sci. 2012. V. 37. P. 686.
Hoogenboom R. // Angew. Chem. Int. Ed. 2009. V. 48. P. 7978.
Sadamatsu H., Hanada T., Morisada S., Kawakita H., Ohto K. // J. Incl. Phenom. Macrocycl. Chem. 2016. V. 84. P. 87.
Izatt R.M., Lamb J.D., Hawkins R.T., Brown P.R., Izatt S.R., Christensen J.J. // J. Am. Soc. 1983. V. 105. P. 1782.
Da Silva E., Lazar A.N., Coleman A.W. // J. Drug Delivery Sci. Technol. 2004. V. 14. P. 3.
Ten’kovtsev A.V., Trofimov A.E., Shcherbinskaya L.I. // Polymer Science B. 2012. V. 54. № 3–4. P. 142.
Amirova A.I., Dudkina M.M., Tenkovtsev A.V., Filippov A.P. // Coll. Polym. Sci. 2015. V. 293. № 1. P. 239.
Amirova A.I., Golub O.V., Kirila T.U., Razina A.B., Tenkovtsev A.V., Filippov A.P. // Soft Materials. 2016. V. 14. № 1. P. 15.
Nekrasova T.N., Ananeva T.D., Pautov V.N., Tenkov-tsev A.V. // Polymer Science A. 2017. V. 59. № 3. P. 324.
Blokhin A.N., Razina A.B., Ten’kovtsev A.V. // Polymer Science B. 2018. V. 60. № 3. P. 307.
Munch J.H., Gutsche C.D. // Org. Syn. 1990. V. 68. P. 243.
Shinkai S., Arimura T., Araki K., Kawabata H., Satoh H., Tsubaki T., Manabe O., Sunamoto J. // J. Chem. Soc., Perkin Trans. 1989. V. 1. P. 2039.
Hwang K.M., Qi Y.M., Liu S.-Y., Lee T.C., Choy W., Chen J. Pat. 54099959 USA. 1995.
Rekkab S., Lahouel M., Ben Hadda T., Felix C., Kabouche Z. // C. R. Chimie. 2013. V. 16. P. 672.
Kabalka G.W., Varma M., Varma R.S. // J. Org. Chem. 1986. V. 51. № 12. P. 2386.
Ballistreri F.P., Cantone A., Maccarone E., Tomaselli G.A., Tripolone M. // J. Chem. Soc., Perkin Trans. 1981. V. 2. P. 438.
Organisch-Chemische Experimentierkunst / Eds. by G. Hilgetag, A. Martini. Leipzig: Verlag, 1964.
Rigby W. // J. Chem. Soc. 1956. P. 2550.
Blokhin A.N., Razina A.B., Parilova E.V., Tenkovtsev A.V. // Vestn. SPbSU. Ser. 4. 2015. V. 2. № 4. P. 357.
Blokhin A.N., Kurlykin M.P., Razina A.B., Dudkina M.M., Ten’kovtsev A.V. // Polymer Science B. 2018. V. 60. № 4. P. 421.
Matyjaszewski K. // Chem. Rev. 2001. V. 101. P. 2921.
Vergaelen M., Verbraeken B., Monnery B.D., Hoogenboom R. // ACS Macro Lett. 2015. V. 4. P. 825.
Verbraeken B., Monnery B.D., Lava K., Hoogenboom R. // Eur. Polym. J. 2017. V. 88. P. 451.
Hoogenboom R., Fijten M.W.M., Brandli C., Schroer J., Schubert U.S. // Macromol. Rapid Commun. 2003. V. 24. № 1. P. 98.
Angot S., Murthy K.S., Taton D., Gnanou Y. // Macromol. 2000. V. 33. P. 7261.
Lambermont-Thijs H.M.L., Fijten M.W.M., Schubert U.S., Hoogenboom R. // Aust. J. Chem. 2011. V. 64. P. 1026.
Brummelhuis N., Schlaad H. // Polym. Chem. 2011. V. 2. P. 1180.
Kowalczuk A., Kronek J., Bosowska K., Trzebicka B., Dworak A. // Polym. Int. 2011. V. 60. P. 1001.
Zhang Q., Weber C., Schubert U.S., Hoogenboom R. // Mater. Horiz. 2017. V. 4. P. 109.
Momekova D., Budurova D., Drakalska E., Shenkov S., Momekov G., Trzebicka B., Lambov N., Tashev E., Rangelov S. // Int. J. Pharmaceut. 2012. V. 436. P. 410.
Alexandridis P., Holzwarth J.F., Hatton T.A. // Macromolecules. 1994. V. 27. P. 2414.
Chattopadhyay A., London E. // Anal. Biochem. 1984. V. 139. P. 408.
Halacheva S., Rangelov S., Tsvetanov C.B. // Macromolecules. 2006. V. 39. P. 6845.
Scherlund M., Brodin A., Malmsten M. // Int. J. Pharm. 2000. V. 211. P. 37.
Lebed A.S., Yefimova S.L., Guralchuk G.Ya., Sorokin A.V., Borovoy I.A., Malyukin Yu.V. // Biophysichny Vestn. 2010. V. 24. P. 46.
Nong H.V., Hung L.X., Thang P.N., Chinh V.D., Vu L.V., Dung P.T., Trung T.V., Nga P.T. // SpringerPlus. 2016. V. 5. P. 1147.
Diaz Garcia M.E., Sanz-Medel A. // Talanta. 1986. V. 33. № 3. P. 255.
Forte-Tavcer P. // Dyes Pigments. 2004. V. 63. P. 181.
Göktürk S., Tuncay M. // Spectrochim. Acta. A. 2003. V. 59. P. 1857.
Göktürk S. // J. Photochem. Photobiol. A. 2005. V. 169. P. 115.
Ishchenko A.A., Shapovalov S.A. // J. Appl. Spectroscopy. 2004. V. 71. № 5. P. 605.
Bielska M., Sobczynska A., Prochaska K. // Dyes Pigments. 2009. V. 80. P. 201.
Akbas H., Taner T. // Spectrochim. Acta. A. 2009. V. 73. P. 150.
Sato H., Kawasaki M., Kasatani K., Nakashima N., Yoshihara K. // Bull. Chem. Soc. Jpn. 1983. V. 56. P. 3588.
Amirova A., Blokhin A., Razina A., Tenkovtsev A., Filippov A. // Mendeleev Commun. 2019. V. 29. № 4. P. 472.
Kirila T.U., Smirnova A.V., Filippov A.S., Razina A.B., Tenkovtsev A.V., Filippov A.P. // Eur. Polym. J. 2019. V. 120. P. 109215.
Aggarwal B.B., Harikumar K.B. // Int. J. Biochem. Gell Biology. 2009. V. 41. № 1. P. 40.
Patel V.B., Misra S., Patel B.B. // Nutrition Cancer. 2010. V. 62. № 7. P. 958.
Kurien B.T., Singh A., Matsumoto H., Scofield R.H. // ASSAY Drug Development Technol. 2007. V. 5. № 4. P. 567.
Bhat P.A., Dar A.A., Rather G.M. // J. Chem. Eng. Data. 2008. V. 53. P. 1271.
Kumar A., Kaur G., Kansal S.K., Chaudhary G.R., Mehta S.K. // Food Chem. 2016. V. 199. P. 660.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)