

Высокомолекулярные соединения (серия Б), 2021, T. 63, № 4, стр. 258-270
ЭЛЕКТРОХИМИЧЕСКАЯ ПОЛИМЕРИЗАЦИЯ ДИФЕНИЛАМИН-2-КАРБОНОВОЙ КИСЛОТЫ НА СТЕКЛОУГЛЕРОДЕ И АКТИВИРОВАННОЙ ГРАФИТОВОЙ ФОЛЬГЕ
В. В. Абаляева a, *, Н. Н. Дремова a, Е. Н. Кабачков a, О. Н. Ефимов a, Ю. В. Баскакова a, Г. П. Карпачева b
a Институт проблем химической физики Российской академии наук
142432 Черноголовка, Московской обл., просп. Академика Н.Н. Семенова, 1, Россия
b Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
* E-mail: avva@icp.ac.ru
Поступила в редакцию 23.03.2021
После доработки 13.04.2021
Принята к публикации 30.04.2021
Аннотация
Изучены особенности электрохимической полимеризации дифениламин-2-карбоновой кислоты в щелочном электролите на анодированной графитовой фольге в сравнении со стеклоуглеродной подложкой. Исследованы физико-химические свойства и электрохимические характеристики электроактивных полимерных покрытий (полидифениламин-2-карбоновая кислота) в 1 М растворе H2SO4 в интервале потенциалов –1 …+1 В и показано образование на изученных подложках двух разных полимеров. Результаты электрохимических, электронно-микроскопических и рентгенофотоэлектронных исследований позволили предположить возможный путь электрополимеризации на поверхностях анодированной графитовой фольги и стеклоуглерода. Рассчитанные значения электрохимической емкости, кулоновской эффективности и устойчивости полимерных покрытий на основе полидифениламин-2-дикарбоновой кислоты на подложке из анодированной графитовой фольги при многоцикличных исследованиях показали хорошие характеристики для разработки электродов для суперконденсаторов.
ВВЕДЕНИЕ
Быстрый рост генерации электроэнергии, растущее использование ее в транспорте, популярность носимых электронных устройств создают растущий интерес к разработке более эффективных систем запасания энергии. Суперконденсаторы как один из основных видов таких систем в последнее время привлекают особое внимание [1]. Хотя суперконденсаторы уступают аккумуляторам по количеству запасенной энергии (плотность энергии), но из- за высоких зарядо-разрядных токов (плотность мощности), они имеют преимущество особенно в условиях неравномерной генерации энергии (солнечные батареи, ветроэлектростанции) или разницы в потреблении энергии ночью и днем. В перспективе предполагается, что характеристики двух указанных видов устройств для запасания энергии по мере совершествования электродных материалов будут сближаться, что позволит создавать гибридные электрохимические ультраконденсаторы [2]. Исходя из этого, актуальным является создание инновационных электродных материалов для суперконденсаторов с улучшенными энергетическими характеристиками, близкими к аккумуляторным. На сегодняшний день наилучшие результаты достигаются в гибридных суперконденсаторах, где запасание энергии происходит по механизму электростатического заряжения двойного электрического слоя (двойнослойная емкость) и протекания быстрых и обратимых фарадеевских реакций (псевдоемкость) на границе раздела электрод–электролит. Этого удается достичь благодаря выбору и оптимальному сочетанию компонентов электродного материала, в качестве которых используют углеродные наноматериалы и проводящие полимеры [3]. Акценты делаются на дизайне композита, особенно на создании иерархической пористости, методе синтеза, емкостных характеристиках и возможности применения в гибких конструкциях. Большинство разработчиков предпочитают использовать наноматериалы с высоким соотношением поверхности к объему наночастиц. При этом важная роль отводится оптимальной пористости и электропроводности композитного электродного материала. Система пор должна обеспечивать быстрый транспорт ионов электролита, что необходимо для достижения высокой плотности токов заряда и разряда [4]. В проводящих полимерах, используемых в гибридных суперконденсаторах, зарядо-разрядные процессы ассоциированы с допированием–дедопированием полимерных цепей. При p-допировании происходит частичное окисление полимера и интеркаляция анионов, при n-допировании – частичное восстановление и интеркаляция катионов. Вместе с тем данный процесс сопровождается заряжением двойного электрического слоя, так как противоионы электростатически связаны с заряженной полимерной цепью. Благодаря присутствию сольватной оболочки наблюдается значительное изменение объема. Это является существенным недостатком электродных материалов, поскольку протекающие релаксационные явления замедляют скорость электродной реакции [5]. Снижение релаксационного вклада достигается за счет взаимодействия с наноуглеродными материалами (нанотрубки, оксид и восстановленный оксид графена, тонко диспергированный углерод, графитовые материалы). Такое взаимодействие обусловлено преимущественно π-стекингом (взаимодействием ароматических систем) и присутствием полярных групп на поверхности наноуглеродных частиц. Углеродный компонент, таким образом, играет роль высокопроводящего каркаса, наполненного ориентировнными к нему полимерными цепями. Другое важное следствие образования такого нанокомпозита – противодействие агрегации наночастиц и проводящие полимеры этому препятствуют. Более того, суммарно структура композита имеет систему пор разного диаметра, что обеспечивает высокую ионную проводимость и электронную проводимость за счет углеродного каркаса [6–8].
Ранее мы описали возможность создания гибких электродов нанесением электроактивного полимерного покрытия на основе полианилина и его N-замещенных на тонкую графитовую фольгу, поверхность которой была подвергнута анодной обработке [9–11]. В результате поверхность фольги становилась пористой и на ней возрастала концентрация кислородсодержащих групп. Это в свою очередь приводило к хорошей смачиваемости электрода электролитом и высокой адгезии полимерного покрытия.
В настоящей работе в качестве мономера использовали дифениламин-2-карбоновую кислоту (ДФАК), химическая окислительная полимеризация которой была исследована в работах [12, 13]. Интерес к полимерам и композитам на ее основе вызван возможностью включения в состав композита углеродных нанотрубок и наночастиц металлов и их оксидов за счет координации с аминными и карбоксильными группами [14]. Композитные покрытия на основе ПДФАК помимо использования в суперконденсаторах могут иметь потенциал использования в качестве электромагнитных экранов, сенсоров, сорбентов, электрокатализаторов и т.п. Литературные данные об электрополимеризации ДФАК ограничены всего одной статьей [15], в которой представлены результаты электросинтеза полимера ДФАК на гладкой стеклоуглеродной подложке. Задачей настоящей работы является исследование электрополимеризации ДФАК на рыхлой поверхности анодированной графитовой фольги (АГФ) и электрохимического поведения сформированного на ней электроактивного полимерного покрытия.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходные реактивы и материалы
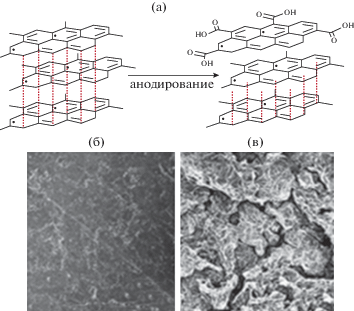
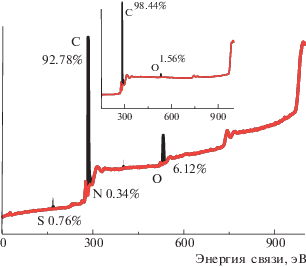
Исходные реактивы ДФАК, гидроокись калия квалификации ч.д.а дополнительной очистке не подвергали. Для электрохимического синтеза и электрохимических исследований готовили 1 × 10–3 М раствор ДФАК в 0.2 М КОН и 1 М раствор H2SO4 в дистиллированной воде. Пластину стеклоуглерода (СУ) размером 35 мм × 10 мм × × 1 мм обрабатывали азотной кислотой, водой и спиртом. Пластину графитовой фольги размером 50 мм × 10 мм × 0.6 мм активировали по описанной методике [16, 17] в 0.1% (NH4)2SO4 при 3 В в течение 3 мин (рис. 1а). В данном процессе рабочим анодом служила пластина анодируемой графитовой фольги, катодом – пластина из нержавеющей стали. После анодирования пластину АГФ осторожно вынимали из раствора, промывали дистиллированной водой, высушивали при 60°С до постоянного веса, после чего использовали в качестве рабочего электрода в электрохимическом синтезе. Разрыхление поверхности графитовой фольги начинается с первой минуты анодирования (рис. 1б), а после 5 мин наблюдается разрушение (отслоение) поверхностных слоев. Время анодирования определяет степень разрыхления. В настоящей работе использовали АГФ, полученную после 3 мин разрыхления. Она имеет плотно упакованную чешуйчатую структуру (рис. 1в). По сравнению с графитовой фольгой поверхность разрыхленной АГФ характеризуется возросшей концентрацией кислородсодержащих групп (рис. 2) и повышенной гидрофильностью поверхности, что приводит к улучшению смачивания электрода электролитом.
Условия электрохимического синтеза и электрохимических исследований
Электрохимический синтез полимера ДФАК проводили в потенциостатическом режиме из 1 × × 10–3 М раствора ДФАК в 0.2 М КОН. Электрохимические исследования проводили в потенциодинамическом или гальваностатическом режимах в интервале потенциалов –1 …+1 В (относительно Ag/AgCl) в трехкамерной стеклянной электрохимической ячейке (используемый объем электролита 15 мл) при комнатной температуре в 1 М H2SO4 на потенциостате PS-7 (фирма “Элинс”, Россия) с программным обеспечением. Все потенциалы приведены относительно указанного электрода сравнения. Пространство рабочего и вспомогательного электродов было разделено пористой стеклянной перегородкой. Рабочим электродом служила пластина из АГФ или СУ-2000, вспомогательным электродом – чистая пластина СУ-2000. Удельную электрохимическую емкость (Ф/см2) рассчитывали по формуле CS = jt/ΔV, где j – удельная плотность тока (А/см2), рассчитанная из площади композитной пленки на электроде; t – время разряда (с); ΔV – область потенциалов (В), в которой происходит разряд. Кулоновскую эффективность находили по формуле µ = = tразр/tзар, где t – времена разряда и заряда.
Физико-химические исследования композитных материалов
Электронно-микроскопические исследования осуществляли на растровом электронном автоэмиссионном микроскопе “Supra 25 производства “Zeiss” с рентгеноспектральной энергодисперсионной приставкой “INCA Energy” производства “Oxford Instruments”. Разрешение на получаемых изображениях составляет 1–2 нм.
Рентгеновские фотоэлектронные спектры высокого разрешения С1s, O1s и N1s регистрировали на электронном спектрометре для химического анализа “Specs PHOIBOS 150 MCD”, рентгеновская трубка с магниевым анодом (MgKα-излучение 1253.6 эВ). При съемке спектров вакуум в камере спектрометра не превышал 3 × 10–9 мбар. Мощность источника составляла 225 Вт. Спектры регистрировали в режиме постоянной энергии пропускания (40 эВ для обзорного спектра и 10 эВ для отдельных линий). Обзорный спектр записывали с шагом 1 эВ, спектры отдельных линий – с шагом 0.05 эВ.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Характеристика электродов
СУ получают путем карбонизации высокомолекулярных углеводородов при температуре порядка 2000°С. Он состоит из чистого углерода с внедрениями высокомолекулярных углеводородов. На поверхности СУ в основном присутствует углерод sp3 (с небольшой примесью углерода sp2) и поверхностные кислородные соединения различной природы. Поверхность графитовой фольги составляет углерод в sp2-гибридизации, соединенный с соседними атомами посредством σ- и π-связей в гексагональную двумерную кристаллическую решетку с небольшим содержанием углерода sp3-гибридизации, который локализуется в основном на торцах разорванных структур (особенно на АГФ). Как видно на рис. 2, после активации поверхности графитовой фольгой (рис. 1а) на образующейся поверхности АГФ содержание кислорода увеличивается с 1.6 до 6.12% в виде всевозможных кислотных, фенольных и других групп. В отличие от гладких электродов СУ и неактивированной исходной графитовой фольги полученная АГФ может поглощать значительное количество наносимого раствора и стабильно удерживать его. Этот факт определяется, во-первых, доступом для впитывания наносимой суспензии химически полученного полимера внутрь подложки за счет активного разрыва внешних слоев графитовой фольги при анодировании. Во-вторых, при электрохимическом синтезе активное взаимодействие групп-доноров N-содержащего мономера с группами, содержащими кислород на акцепторной графитовой поверхности, способствует образованию координационных связей между N-центром мономера (донора) и поверхностью АГФ (акцептора) на внешних и внутренних поверхностях графитовых пластин. На примере мономера анилина нами методом рентгенофотоэлектронной спектроскопии подробно проанализировано поведение в молекуле анилина атома N соли (сульфат анилина) с последующей адсорбцией раствора анилин сульфата на поверхность АГФ и дальнейшего процесса полимеризации [16]. Анализ полученных экспериментальных данных позволил предположить, что на поверхности АГФ уже на стадии адсорбции раствора сульфата анилина в результате координационного взаимодействия акцепторной поверхности АГФ и мономера анилина (в которой и бензольное кольцо и N-центр ведут себя как доноры) происходит окисление молекулы мономера с образованием сначала поляронной структуры, а затем димера. Аналогичная активация молекулы ДФАК на поверхности АГФ могла ускорить электрохимическую полимеризацию ДФАК с образованием активного полимерного слоя.
Электросинтез и электрохимическое поведение покрытия ПДФАК/СУ
Последовательные ЦВА, снятые в ходе электросинтеза ПДФАК на СУ электроде в 1 × 10–3 М растворе ДФАК в 0.2 М КОН представлены на рис. 3. В начале электросинтеза на ЦВА1 наблюдается резкий рост тока в области +0.6…+0.8 В, что соответствует окислению ДФАК до катион-радикала ДФАК [12] и образованию зародышей полимерного покрытия подобно тому, как это имеет место при электроокислении дифениламина [17]. На катодной ветви ЦВА появляется пик EК = 0.43 В, соответствующий восстановлению катион-радикального состояния растущего полимера. Проведено 300 циклов электросинтеза в интервале потенциалов –0.85…+0.8 В (рис. 3), которые были разделены на три серии по 100 циклов и соответственно обозначены как ЦВА1, ЦВА2, ЦВА3. После 200 циклов на ЦВА5 наблюдаются пики EА = 0.5 В и EК = –0.43 В. Характерно, что в ходе циклирования площадь ЦВА не растет, а уменьшается, что связано с замедлением роста полимерного слоя, обусловленного низкой электропроводностью.
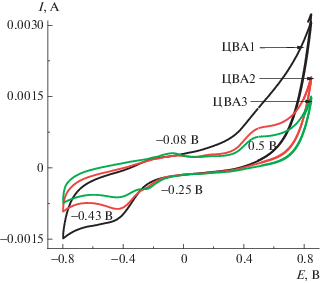
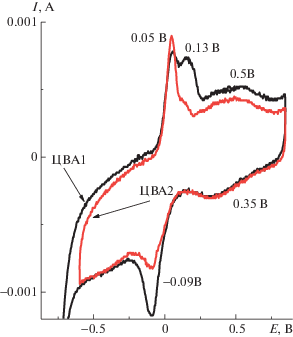
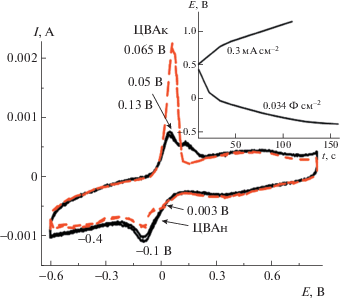
Исследование электрохимического поведения ПДФАК/СУ проводили в 1 М H2SO4. При смене электролита вследствие протонирования полимерного покрытия и повышения его проводимости вид ЦВА резко изменяется, и на исходной ЦВА1 (рис. 4) наблюдаются три анодных пика EА = 0.05 В, EА = 0.13 В и широкий EА = 0.5 В. В катодной области первым двум пикам соответствовали только два пика EК = –0.09 В и широкий пик EК = 0.35 В. При потенциалах отрицательнее –0.6 В наблюдалось резкое возрастание тока, которое, вероятно, обусловлено выделением водорода. В связи с этим катодный предел ЦВА был ограничен –0.6 В. При циклировании второй анодный пик EА = 0.13 В исчезал и возрастала интенсивность пика EА = 0.05 В. В результате исходная ЦВА1 приобретала стабильную форму ЦВА2 (рис. 4). Интерпретация ЦВА ПДФАК/СУ представляет трудную задачу. Можно думать, что первому редокс-переходу EА = 0.05 В соответствует обратимая электродная реакция с участием электроактивных центров в полидифениаминовой цепи [17], возможно, адсорбированных на электроде, на что указывает минимальное значение ∆ЕА-К = 40 мВ. Второй редокс-переход EА = 0.5 В также является обратимым, но его происхождение требует дополнительного исследования.
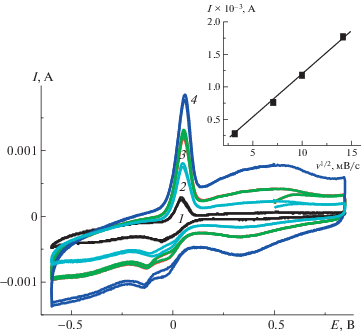
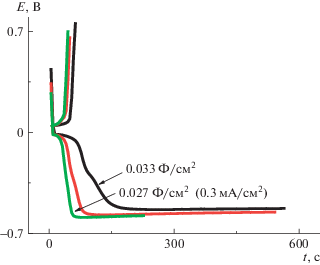
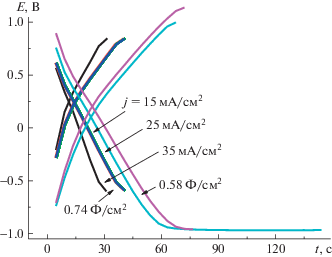
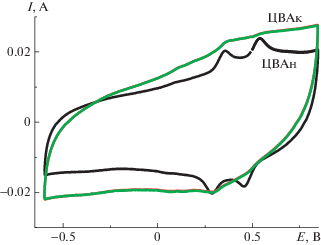
Спрямление зависимости величины анодного тока от корня квадратного из скорости циклирования v (вставка на рис. 5) в координатах $i{\text{/}}{{v}^{{1/2}}}$ для EА = 0.05 В указывает на наличие (присутствие) диффузионных ограничений при допировании–дедопировании ПДФАК [18]. Вид гальваностатических зарядно-разрядных зависимостей для ПДФАК/СУ существенно отличается по сравнению с полианилином на СУ [19]. Как видно на рис. 6, в исследуемом интервале потенциалов при разной плотности тока происходит мгновенный заряд до потенциала 0.7 В и значительно более медленный разряд до –0.65 В с дальнейшим выходом на плато. С учетом этой особенности электрохимическая емкость Суд была рассчитана по активной части разрядной кривой. После проведения зарядно-разрядных исследований практически в два раза выросли анодные токи. Такой вид ЦВА сохранился и после 300 циклирований в исследуемом интервале потенциалов. На рис. 7 представлены начальная ЦВАн и конечная ЦВАк при исследовании ПДФАК/СУ в 1 М H2SO4. Их сравнение показывает, что при длительном циклировании и снятии гальваностатических зарядно-разрядных кривых в 1 М H2SO4 увеличивается вклад псевдоемкости, который, возможно, связан с ростом электроактивных центров. После всех исследований электрода ПДФАК/СУ величина СУД составила 0.034 Ф/см2 при j = 0.3 мА/см2 против 0.027 Ф/см2 при j = 0.3 мА/см2 в начале исследований.
Электросинтез ПДФАК на АГФ-электроде и электрохимическое поведение ПДФАК/АГФ
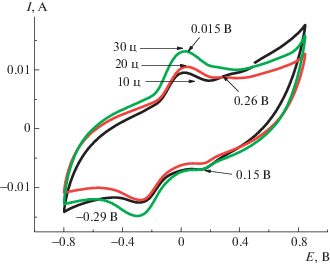
Электросинтез ПДФАК на АГФ-электроде (как описано выше) из 1 × 10–3 М ДФАК в 0.2 М КОН приведен на рис. 8. Электросинтез начинали при потенциале разомкнутой цепи 0.48 В и проводили в интервале потенциалов –0.8….+0.8 В в течение 30 циклов, при которых происходит рост полимерного покрытия. 30 циклов электросинтеза в указанном интервале потенциалов были разделены на три серии по 10 циклов и соответственно на рис. 8 обозначены как 10ц, 20ц и 30ц. При электрохимическом циклировании наблюдался рост тока для редокс-пары ЕА = 0.015 В и EК = –0.29 В. Эти анодный и катодный пики, по-видимому, являются членами редокс-пары, соответствующей окислению и восстановлению ПДФАК, поскольку в каждой серии ЦВА интенсивность катодного и анодного пиков увеличивается с ростом числа циклов. Едва заметный анодный пик при 0.26 В может соответствовать катион-радикалу мономера, который расходуется в процессе электросинтеза. Наличие соответствующего ему катодного пика 0.15 В может свидетельствовать о том, что катион-радикалы не полностью расходуются в последующих электрохимических реакциях.
Рис. 8.
Электросинтез ПДФАК на анодированной графитовой фольге. 1 × 10–3 М раствор ДФАК в 0.2 М КОН, 100 мВ/с.
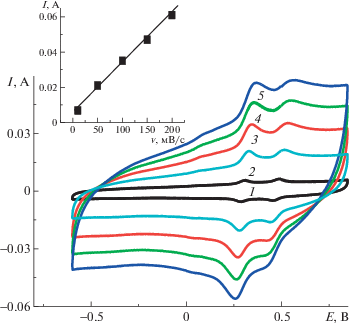
Исследование электрохимического поведения покрытия ПДФАК на АГФ проводили в 1 М H2SO4. Сокращение интервала циклирования от –0.85 В (рис. 9, ЦВА1) до –0.65 В (рис. 9, ЦВА2) не изменяло положение анодных и катодных пиков и приводило к стабильной ЦВА2, которая имела квазипрямоугольную форму, характерную для двойнослойных суперконденсаторов. На ее фоне видны две пары редокс-пиков ЕК = 0.32 В, EК = = 0.26 В и EА = 0.51 В, EК = 0.42 В, причем ∆ЕА-К для соответствующих переходов составляет 60 и 90 мВ, что отвечает обратимому электродному процессу в адсорбированном состоянии. Этот вывод согласуется с линейной зависимостью анодного тока от скорости сканирования (рис. 10, вставка), которая указывает на отсутствие диффузионных ограничений при окислительно-восстановительном циклировании [18]. Значения анодных и катодных потенциалов на ветвях ЦВА практически сохраняют свои значения (рис. 10) при увеличении скорости циклирования во всем интервале от 10 до 200 мВ/с. В данном случае мы имеем классическую картину обратимой реакции [20]. Это означает, что на АГФ в отличие от СУ образуется полимерное покрытие с низким сопротивлением.
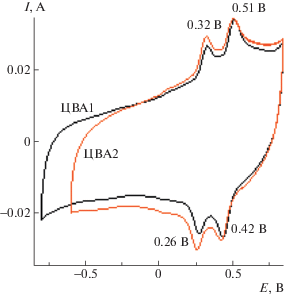
Рис. 10.
ЦВА электрода ПДФАК/АГФ при скорости циклирования 10 (1), 50 (2), 100 (3), 150 (4) и 200 мВ/с (5). На вставке показана зависимость анодного тока от скорости циклирования.
Гальваностатические зарядно-разрядные кривые (рис. 11) показали высокие значения величины поверхностной электрохимической емкости, достигающие 0.74 Ф/см2 при j = 35 мА/см2, а также возможность работы ПДФАК/АГФ в широком интервале потенциалов (–1 В…+1 В). При разной плотности тока и в различных интервалах циклирования кулоновская эффективность ПДФАК/АГФ составляет 98–100%. После 300 циклирований при $v$ = 200 мВ/с (ЦВАк, рис. 12) редокс-переходы практически исчезают, и ЦВА приобретает характерную для суперконденсаторов квазипрямоугольную форму. Таким образом, основной вклад в электрохимическую емкость ПДФАК/АГФ вносит двойнослойное заряжение в отличие от покрытия ПДФАК на СУ, где наблюдается значительный вклад фарадеевской псевдоемкости (рис. 7). Это может быть связано с внедрением полимера в поры АГФ и с образованием кооординационных связей между полимером и графеновыми нанолистами в порах [21, 22].
Физико-химическое изучение полученных полимерных покрытий ПДФАК/СУ и ПДФАК/АГФ
Морфология электросинтезированных композитных электродов. На рис. 13 приведены СЭМ-микрофотографии исследуемых электродных подложек. В отличие от гладкой поверхности СУ на шероховатой поверхности АГФ хорошо видны складки графеновых нанолистов. Такое отличие проявляется в дальнейшем и в морфологии поверхности электродов после проведения электросинтеза полимерных покрытий на АГФ (рис. 14). На микрофотографии поверхности электрода, снятой сразу после электросинтеза ПДФАК на СУ, (рис. 14а) видно, что покрытие ПДФАК/СУ имеет достаточно плотную грубую глобулярную морфологию с размером глобул 200–400 нм.


Покрытие ПДФАК на АГФ (рис. 14б), полученное после электросинтеза в щелочной среде (до помещения образца в кислоту), характеризуется однородной глобулярной морфологией, с размером глобул 20–50 нм. При этом глобулы отделены друг от друга. Отсюда следует, что рост покрытия происходит на активных центрах на графеновых нанолистах АГФ. В покрытии ПДФАК/АГФ после циклирования в 1 М H2SO4 размер глобул (рис. 14в, 14г) значительно увеличивается до 100–150 нм. Хорошо видно, что они расположены равномерно на графеновых нанолистах и разделены значительными промежутками. Электрохимическому окислению мономера предшествует его адсорбция на поверхности АГФ за счет π-стекинга дифениламиновых фрагментов мономера с ароматическими звеньями графеновых нанолистов, а также за счет образования водородных связей с участием аминогрупп ДФАК и кислородсодержащих групп АГФ. Такое взаимодействие происходит уже на стадии адсорбции мономера на поверхности графеновых нанолистов. Этот факт доказан нами в работе [23] для случая полимеризации анилина на поверхности графеновой фольги с включением в процесс комплексобразования азотных и кислородных центров мономера с поверхностью АГФ. Заключение относительно предварительной адсорбции мономера на активных центрах АГФ согласуется с малой величиной EA-K. Увеличение размеров глобул до 100–150 нм при длительном циклировании и зарядно-разрядных исследованиях может быть связано с конденсацией олигомеров в более длинные фрагменты, о чем свидетельствует и исчезновение второго анодного и значительное уменьшение первого анодного пиков на ЦВАк (рис. 12). Это способствует снятию диффузионных ограничений для электрохимических процессов и двойнослойному заряжению благодаря свободному доступу электролита в поры покрытия. Высокопроводящий графеновый каркас обеспечивает высокую электропроводность покрытия.
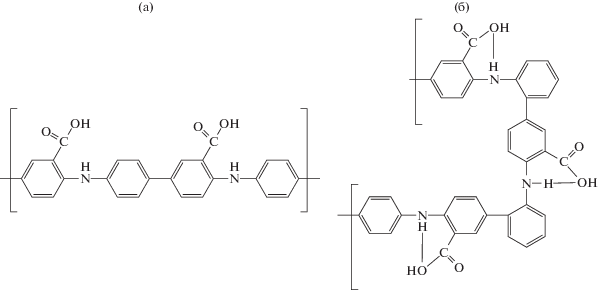
Рентгенофотоэлектронные спектры электросинтезированных покрытий ПДФАК на СУ и АГФ. Рентгеновская фотоэлектронная спектроскопия в данном исследовании была использована для того чтобы проследить изменения характеристических энергий связи C1s, N1s и O1s пиков, соответствующих функциональным группам ПДФАК после электросинтеза. При интерпретации рентгеновских фотоэлектронных спектров были использованы данные работ [24–28]. Известно, что химическая окислительная полимеризация ДФАК может протекать по разным направлениям в кислотной и щелочной средах [13, 14]. Рост цепи в кислотной среде происходит путем С–С-присоединения в пара-положениях фенильных колец (схема 1а), тогда как в щелочной среде рост полимерной цепи осуществляется путем С–С-присоединения в 2- и 4- положениях по отношению к азоту с образованием внутримолекулярных водородных связей вдоль всей полимерной цепи (схема 1б).
При полимеризации на подложке АГФ важным фактором является предварительная адсорбция мономера ДФАК, например, за счет водородных связей с кислородсодержащими (эпоксидными, гидроксильными) группами, которые создают островковое покрытие на поверхности АГФ [29]. Такая координация мономера может ориентировать рост цепи ПДФАК параллельно плоскости графенового нанолиста. В результате образующийся полимер, скорее всего, будет иметь “плоскую” структуру без внутримолекулярных водородных связей. По аналогии с электрохимической полимеризацией дифениламина в ацетонитриле [17] в условиях дефицита протонов возможную последовательность реакций при электрохимическом синтезе ПДФАК в NaOH можно представить следующим образом:
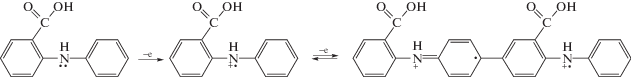
Однако в этих условиях карбоксильные группы должны быть в виде карбоксилат-ионов, присутствие которых обнаруживается только на спектре С1s для электрода ПДФАК/СУ, судя по появлению пиков невысокой интенсивности в области энергий связей 287–289 эВ (рис. 15, С1s, спектр 1). На поверхности электродов ПДФАК/АГФ после исследований в 1 М Н2SO4 (рис. 15, С1s, спектр 2) и ПДФАК/АГФщ после электросинтеза в щелочной среде (рис. 15, С1s, спектр 3) в отличие от ПДФАК/СУ полимер взаимодействует с активированной поверхностью АГФ, в результате которого складываются благоприятные условия для образования водородных связей. В связи с этим на втором и третьем спектрах С1s отсутствуют пики энергий свободных связей 287–289 эВ.
Рис. 15.
Рентгенофотоэлектронные спектры ПДФАК/СУ после циклирования в 1 M H2SO4 (1), ПДФАК/АГФ после электросинтеза и циклирования в 1 M H2SO4 (2) и ПДФАК/АГФЩ сразу после электросинтеза (2Щ).
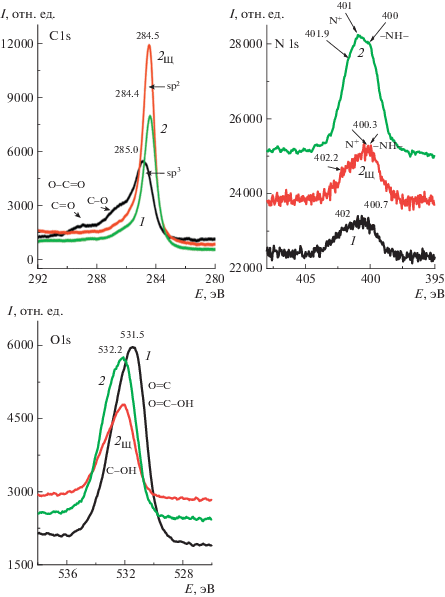
Наиболее информативными оказались спектры N1s, на которых можно проследить влияние взаимодействия аминогрупп полимера с поверхностью АГФ, покрытой кислородсодержащими группами. Образование N-центром водородных связей снимает с атома N часть электронной плотности на образование этой связи. Как следствие, в спектрах N1s полимерного покрытия ПДФАК/АГФ (рис. 15, N1s, спектр 2) локализовано сразу несколько вершин с энергиями связей 400, 401 и 401.7 эВ, что соответствует увеличению доли положительно заряженного азота по сравнению со спектрами 1 и 2щ (ПДФАК/СУ и ПДФАК/АГФЩ). По аналогии с полианилином можно отнести два последних пика 401 и 401.7 эВ к положительно заряженному имину в биполяронном состоянии (дикатион-радикал) и протонированному амину в поляроном состоянии (катион-радикал) [27]. Однако следует иметь в виду, что в среде 1 M H2SO4 амино-группы протонированы, что вносит вклад в пик 401.9 эВ. Пик 400 эВ связан с фениленаминовой группой –NH–, участвующей в образовании внутримолекулярных водородных связей.
Спектры O1s ПДФАК/СУ (спектр 1) свидетельствуют о наличии свободных групп C=O и O=C–OH . На O1s спектрах ПДФАК/АГФЩ (2Щ), ПДФАК/АГФ (2) можно зафиксировать только наличие групп С–ОН. Присутствие на спектрах ПДФАК/АГФЩ, ПДФАК/АГФ пиков с более высокими энергиями связи указывает на наличие связанной воды в образцах [30].
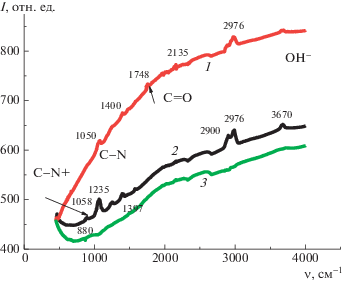
ИК-спектры электросинтезированных покрытий. На рис. 16 представлены ИК-спектры двух образцов ПДФАК/СУ (спектр 1) и ПДФАК/АГФ (спектр 2). Для сравнения приведен ИК-спектр чистой АГФ (спектр 3). Полосы на спектрах 1 и 2 при 1397 и 1400 см–1 относятся к аниону HSO$_{4}^{ - }$, который присутствует после циклирования в 1 М H2SO4 [31]. В области 2700–3300 см–1 расположен целый ряд полос, характеризующих наличие и состояние ОН–, включая их участие в межцепных и внутримолекулярных водородных связях, 1397 и 1400 см–1 свидетельствуют о наличии группы С–N [32]. Наличие группы C–N•+, соответствующей поляронному состоянию, подтверждает пик 880 см–1 [33].
ЗАКЛЮЧЕНИЕ
Показано, что при электрополимеризации дифениламин-2-карбоновой кислоты на двух подложках – стеклоуглероде и анодированной графитовой фольге в щелочной среде образуются разные полимерные покрытия, которые различаются по морфологии (данные СЭМ) и содержанию функциональных групп (данные рентгенофотоэлектронных спектров) на поверхности. Исследование электрохимических свойств покрытий в 1 М H2SO4 показало что ПДФАК/АГФ имеет достаточно высокую поверхностную емкость 0.7 Ф/см2 при j = 35 мА/см2 и обеспечивает длительное циклирование в расширенном окне потенциалов от –1 до +1 В без существенного снижения емкости. Таким образом, электрод ПДФАК/АГФ представляется перспективным при создании стабильно работающих гибких суперконденсаторов.
Работа выполнена в соответствии с Госзаданием ИПХФ РАН (АААА-А19-119071190044-3 и ААА-А19-119061890019-5) и Госзаданием ИНХС РАН.
Работа выполнена с использованием оборудования Аналитического центра коллективного пользования ИПХФ РАН и Научного центра Черноголовка РАН.
Список литературы
Shen F., Pankratov D., Chi Q. // Curr. Opinion Electrochem. 2017. V. 4. № 1. P. 133.
González A., Goikole E., Barren J.A., Mysyk R. // Renewable Sustainable Energy Revs. 2016. V. 58. P. 1189.
Shao H., Wu Y-Ch., Lin Z., Tabernand P.-Lm., Simon P. // Chem. Soc. Rev. 2020. V. 49. P. 3005.
Vadali V.S.S., Ramana G.V., Kumar P.S. // J. Nanosci. Nanotechnol. 2016. V. 16. P. 2418.
Kim B.C., Hong J.Y., Wallace G.G., Park H.S. // Adv Energy Mater. 2015. V. 5 (22). P. 1500959-1.
Zhang J., Zhao X.S. // Chem. Sus. Chem. 2012. V. 5. P. 818.
Gupta S., Price C. // Composites B. 2016. V. 105. P. 46.
Wang M., Xu Y.X. // Chin. Chem. Lett. 2016. V. 27. P. 1437.
Abalyaeva V.V., Nikolaeva G.V., Kabachkov E.N., Kiseleva S.G., Orlov A.V., Efimov O.N., Karpacheva G.P. // Polymer Science B. 2018. V. 60. № 6. P. 777.
Abalyaeva V.V., Nikolaeva G.V., Kabachkov E.N., Efimov O.N. // Prot. Met. Phys. Chem. Surf. 2020. V. 56. P. 493.
Abalyaeva V.V., Efimov M.N., Efimov O.N., Karpacheva G.P., Dremova N.N., Kabachkov E.N., Muratov D.G. // Electrochim. Acta. 2020. V. 354. P. 136671.
Ozkan S.Zh., Eremeev I.S., Karpacheva G.P., Bondarenko G.N. // Open J. Polym. Chem. 2013. V. 3. P. 63.
Ozkan S.Z., Eremeev I.S., Karpacheva G.P., Prudskova T.N., Veselova E.V., Bondarenko G.N., Shandryuk G.A. // Polymer Science B. 2013. V. 55. № 3–4. P. 107.
Ozkan S.Zh., Kostev A.I., Karpacheva G.P., Chernavskii P.A., Vasilev A.A., Muratov D.G. // Polymers. 2020. V. 12. P. 1568.
Nassa H.R., Souri A., Javadian A., Aminia M.K. // Sensors Actuators B. 2015. V. 215. P. 360.
Abalyaeva V.V., Nikolaeva G.V., Dremova N.N., Kne-rel’man E.I., Davydova G.I., Efimov O.N., Ionov S.G. // Prot. Metals Phys. Chem. Surf. 2019. V. 55. № 2. P. 321.
Yang H., Bard A.J. // J. Electroanal. Chem. Interfacial Electrochem. 1991. V. 306. P. 87.
Elgrishi N., Rountree K.J., McCarthy B.D., Rountree E.S., Eisenhart T.T., Dempsey J.L. // J. Chem. Educ. 2018. V. 95. P. 197.
Abalyaeva V.V., Nikolaeva G.V., Kabachkov E.N., Efimov O.N. // Russ. J. Electrochem. 2019. V. 55. № 8. P. 745.
Rodrigues Sh., Munichadrain N.A., Shukla K. // J. Appl. Electrochem. 1998. V. 28. P. 1235.
Salavagione H.J., Martinez G., Ellis G. // Macromol. Rapid Commun. 2011. V. 32 № 22. P. 1771.
Salvatierra R.V., Zitzer G., Savu S.-A., Alves A.P., Zarbin A.J.G., Chassé T., Casu M.B., Rocco M.L.M. // Synth. Met. 2015. V. 203. P. 16.
Abalyaeva V.V., Dremova N.N., Kabachkov E.N., Efimov O.N. // Prot. Met. Phys. Chem. Surf. 2020. V. 56. P. 944.
Neoh K.G., Kang E.T., and Tan K.L. // J. Phys. Chem. 1992. V. 96. № 16. P. 6777.
Yanchun Z., Miao Ch. M., Chen X. // Mater. Chem. Phys. 2005. V. 91. № 2–3. P. 518.
Lee T., Yun T., Park B., Sharma Bh., Song H.-K., Kim B.-S. // J. Mater. Chem. 2012. V. 22. P. 21092.
Kim M., Lee Ch., Jang J. // Adv. Funct. Mater. 2014. V. 24. P. 2489.
Moyseowicz A., Gryglewicz G. // Composites B. 2019. V. 159. P. 4.
Shin D., Kim H.G., Ahn H., Jeong H., Kim Y.-J., Odkhuu D., Tsogbadrakh N., Lee H.-B.-R., Kim B. // RSC Adv. 2017. V. 7. P. 13979.
Al-Mashat L., Shin K., Kalantar-zadeh K., Plessis J.D., Han S.H., Kojima R.W., Kaner R.B., Li D., Gou X., Samuel J., Wlodarsk W. // J. Phys. Chem. C. 2010. V. 114. № 39. P. 16168.
Trchova M., Stejskal J. // Pure Appl. Chem. 2011. V. 83. № 10. P. 1803.
Wang Y., Li H., Xia Y. // Adv. Mater. 2006. V. 18. P. 2619.
Janošević A., Ćirić-Marjanović G., Marjanović B., Holler P., Trchová M., Stejskal J. // Nanotechnology. 2008. V. 19. P. 135606.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)