

Высокомолекулярные соединения (серия Б), 2021, T. 63, № 6, стр. 383-418
РЕАКЦИЯ ФОТОВОССТАНОВЛЕНИЯ КАРБОНИЛСОДЕРЖАЩИХ СОЕДИНЕНИЙ В СИНТЕЗЕ И МОДИФИКАЦИИ ПОЛИМЕРОВ
Н. А. Леньшина a, *, М. П. Шурыгина a, С. А. Чесноков a
a Институт металлоорганической химии им. Г.А. Разуваева Российской академии наук
603950 Нижний Новгород, ул. Тропинина, 49, Россия
* E-mail: lennin@iomc.ras.ru
Поступила в редакцию 23.08.2021
После доработки 01.09.2021
Принята к публикации 24.09.2021
Аннотация
Фотополимеризация является одним из основных способов синтеза полимеров. Она применяется для получения различных полимерных изделий и покрытий методами стереолитографии и 3D-печати, а также широко используется в стоматологической практике и протезировании. В обзоре рассмотрены основные направления исследований в области применения реакций фотовосстановления карбонилсодержащих соединений для синтеза и функционализации полимеров, включая инициирование радикальной и катионной полимеризации, проведение прививочной полимеризации, получение привитых слоев и сшитых полимеров. Описаны механизмы этих процессов и основные используемые карбонилсодержащие соединения и соинициаторы.
ВВЕДЕНИЕ
Фотохимические реакции широко используются в синтезе и модификации полимеров. Они, в частности, применяются для получения органических стекол, покрытий, изготовления печатных форм, микросхем, различных изделий и их прототипов методами 3D-печати, в стоматологической практике и протезировании. Широкое распространение данные реакции получили из-за простоты их контроля и легкости управления скоростью процесса с помощью источников освещения. Основной используемой в химии полимеров фотохимической реакцией является фотополимеризация (как правило, УФ-инициируемая). На данный момент разработано и исследовано большое число фотоинициаторов и фотоинициирующих систем [1–3]. В общем случае по характеру образующейся инициирующей частицы и инициируемого процесса выделяют две группы фотоинициаторов.
1. Радикальные фотоинициаторы, которые в свою очередь делятся на фотоинициаторы первого типа (генерируют инициирующие радикалы за счет гомолитического распада фотоинициатора) и фотоинициаторы второго типа (генерируют инициирующие радикалы за счет переноса водорода в паре с соинициатором.
2. Катионные фотоинициаторы, которые генерируют инициирующие катионные частицы по различным механизмам.
Большую группу фотоинициаторов радикальной полимеризации первого и второго типа представляют карбонилсодержащие соединения. Многие карбонилсодержащие фотоинициаторы второго типа инициируют радикальные процессы за счет фотовосстановления карбонильного фрагмента в присутствии соинициатора – донора водорода (рис. 1). К таким инициаторам относятся бензофенон (БФ)
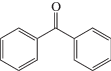 ,
,
камфорохинон (КХ)

и производное тиоксантона (ТК) 2-изопропилтиоксантон (ИТК)

Рис. 1.
Основные используемые карбонилсодержащие фотоинициаторы второго типа и их спектральные характеристики в ближнем УФ- и видимой части спектра [4]. Цветные рисунки можно посмотреть в электронной версии.

Двух- и поликомпонентные фотоинициирующие системы, включающие карбонилсодержащие соединения, как правило, используются для инициирования радикальной фотополимеризации. Но в зависимости от соинициатора (например, ониевые соли или некоторые фосфоросодержащие соединения) они могут инициировать также и катионную фотополимеризацию. Все это позволяет использовать фотоинициирующие системы на основе карбонилсодержащих фотоинициаторов второго типа для полимеризации широкого круга мономеров: (мет)акриловых, виниловых, эпоксидов:

Реакция фотовосстановления карбонилсодержащих соединений также используются для модификации полимеров за счет прививочной полимеризации и сшивания цепей. В обзоре рассмотрены основные направления применения реакций фотовосстановления карбонилсодержащих соединений для инициирования процессов синтеза и функционализации полимеров.
ОБЩИЕ СВЕДЕНИЯ О РЕАКЦИИ ФОТОВОССТАНОВЛЕНИЯ КАРБОНИЛСОДЕРЖАЩИХ СОЕДИНЕНИЙ
Реакция фотовосстановления карбонилсодержащих соединений привлекает к себе внимание исследователей на протяжении почти ста лет. Интерес к этой реакции определяется тем, что она лежит в основе процессов фотоинициирования радикальной полимеризации, выцветания красителей, фотобиохимических процессах, в том числе фотосинтеза. На примере реакции фотовосстановления карбонилсодержащих соединений изучают механизм и кинетические закономерности процессов фотопереноса как атома водорода, так и электрона и протона по отдельности.
Реакция фотовосстановления карбонилсодержащих соединений заключается в отрыве фотовозбужденной молекулой акцептора А* атома водорода от молекулы Н-донора (DH) и присоединении его к атому кислорода карбонильной группы [5, 6] (схема 1а). Показано [7–15], что перенос атома водорода проходит через последовательные стадии переноса электрона и переноса протона (схема 1б). Сначала происходит поглощение кванта света молекулой A; она переходит в низшее возбужденное триплетное состояние A* и затем отрывает атом водорода от молекулы Н-донора. Этот процесс осуществляется в несколько этапов: перенос электрона от молекулы донора водорода на фотовозбужденную молекулу A* с образованием триплетной ион-радикальной пары 3[AH•– + D•+]; перенос протона с образованием триплетной радикальной пары 3[AH•, D•]; выход радикалов AH• и D• из клетки, образованной молекулами растворителя. Наиболее эффективно протекает отрыв атома водорода от атома углерода, находящегося в α-положении по отношению к гетероатому [16].
Перенос электрона и протона происходит в столкновительном комплексе, для обозначения которого в фотохимических исследованиях применяют термин “эксиплекс”. Эксиплекс образуется между фотовозбужденной молекулой карбонилсодержащего соединения и молекулой донора водорода, находящейся в основном состоянии [12, 17, 18]. Константа скорости фотовосстановления kH является комбинацией констант скорости стадий переноса электрона ke и переноса протона kH+.
Скорость реакции фотовосстановления зависит от пространственных характеристик реагентов, влияющих на строение эксиплекса, в том числе на расстояние между реакционными центрами молекул реагентов в нем [19–21]. Влияние стерических факторов на скорость реакции было изучено на примере фотовосстановления п-хинонов в присутствии различных алкилзамещенных аренов в качестве доноров водорода [20]. Ниже приведены примеры моделей столкновительных комплексов п-хлоранила с гексаметилбензолом (а) и гексаэтилбензолом (б).

Замена в алкиларене метильных групп на этильные приводит к увеличению минимального расстояния между плоскостями колец хинона и DH с 3.5 до 4.5 Ǻ. Как следствие, константа скорости переноса электрона уменьшается в 30 раз. При переходе от стерически не затрудненных стирола, мезитилена и т.д. к стерически затрудненным аренам с трет-бутильными заместителями наблюдалось уменьшение константы скорости переноса электрона более чем в 100 раз [20].
На скорость реакции фотовосстановления также оказывает влияние природа растворителя. Это проявляется в зависимости направления трансформации контактной ион-радикальной пары 3[A•–, DH•+]s (схема 1б) от полярности растворителя. А именно, ион-радикальная пара 3[A•–, DH•+]s может переходить в сольватно-разделенную ион-радикальную пару A•– и DH•+, в которой перенос протона уже невозможен из-за увеличения расстояния между частицами [8, 11, 22–24]. С ростом полярности растворителя увеличивается и вероятность выхода ион-радикалов из клетки и образования ион-радикальных продуктов A•– и DH•+. При этом скорость фотовосстановления карбонилсодержащих соединений также снижается [25, 26].
Интересным направлением исследований кинетики реакции фотовосстановления является разработка теорий, предсказывающих зависимость скорости реакции от спектральных и электрохимических характеристик реагентов. Предложена модель, в рамках которой kH представлена как функция свободной энергии переноса электрона ∆Ge , рассчитывающейся по уравнению Рэма–Веллера [27–30].
(1)
$\Delta {{G}_{{\mathbf{e}}}} = - \Delta {{E}_{{00}}} - {{Е}_{{({{{\mathbf{A}}}^{{\centerdot - }}}/{\mathbf{A}})}}} + {{E}_{{(AmH/Am{{H}^{ \bullet }} + )}}} + С,~$Такие исследования важны для предсказания эффективности использования тех или иных комбинаций фотоинициаторов и соинициаторов в фотоинициирующих системах. В основе работы фотоинициирующих систем лежит реакция их фотовосстановления в присутствии доноров водорода. Соответственно кинетика фотоинициирования полимеризации в значительной степени определяется кинетикой фотовосстановления фотоинициаторов.
ИНИЦИИРОВАНИЕ ПОЛИМЕРИЗАЦИИ С ИСПОЛЬЗОВАНИЕМ РЕАКЦИИ ФОТОВОССТАНОВЛЕНИЯ КАРБОНИЛСОДЕРЖАЩИХ СОЕДИНЕНИЙ
Радикальная полимеризация
Реакция фотовосстановления карбонилсодержащих соединений в полимерной химии в основном применяется для инициирования свободнорадикальной полимеризации под воздействием УФ- и видимого излучения. Механизм инициирования фотополимеризации системой карбонилсодержащее соединение–донор водорода представлен на схеме [37, 38]

Инициирование полимеризации происходит за счет образующегося в процессе фотовосстановления радикала D•. Из схемы видно, что на эффективность фотоинициирующих систем влияет скорость генерации радикалов при прохождении реакции фотовосстановления. Кроме того, эффективность фотоинициирующих систем определяется инициирующей способностью образующегося радикала D• и побочными реакциями радикалов AH• и D•. Радикалы D• инициируют полимеризацию мономера М, но могут также вступать в реакцию рекомбинации с образованием различных продуктов, способных, ингибировать полимеризацию, обрывая цепи за счет присоединения к макрорадикалам [25]. Ингибирующей способностью обладают кетильные радикалы AH• бензофенона [39], TК [40, 41] и бензохинонов [42, 43]. Скорость полимеризации также зависит от природы мономера (констант скорости реакций роста, обрыва и передачи цепи) [44–47], от экспериментальных условий (концентраций фотоинициатора и соинициатора, источника освещения, температуры) [48–57], а также от присутствия или отсутствия в системе кислорода [58–61].
К настоящему времени многие карбонилсодержащие соединения изучены на предмет наличия инициирующей способности под действием ближнего УФ- и видимого излучения. Постоянно появляются работы, посвященные синтезу новых фотоинициаторов и разработке фотоинициирующих систем, подходящих для разных типов мономеров. Адекватная систематизация и сравнение результатов по реакционной способности фотоинициаторов и фотоинициирующих систем, полученных разными научными группами, существенно осложняется не только большим количеством информации по данной теме, но и различными условиями проведения фотохимических исследований и полимеризации, применения разного оборудования, источников излучения и самодельных установок. Во многих статьях новые фотоинициаторы сравнивают с аналогичными незамещенными соединениями и с КХ или БФ как с эталонными фотоинициаторами.
Как отмечалось выше, наиболее изученными и часто применяемыми являются фотоинициирующие системы на основе БФ, КХ и их производных, а также производных ТК. При этом исследования по фотоинициирующим системам на основе КХ в основном сконцентрированы на поиске новых эффективных соинициаторов, а не синтезе замещенных производных КХ, в то время как работы по БФ и ТК тяготеют к получению новых фотоинициаторов на их основе.
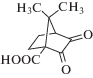
Фотоинициирующие системы на основе КХ инициируют фотополимеризацию под действием видимого излучения. В спектре КХ присутствуют две полосы поглощения: в диапазоне 200–300 нм и в видимой области спектра 400–500 нм с максимумом при ~469 нм [62] (по указанной полосе проводят процесс фотоотверждения материалов). “Рабочая” полоса КХ находится в синей области спектра, поэтому в качестве источников излучения для инициирования часто используют синие LED. Фотоинициирующие системы на основе КХ содержат в качестве соинициаторов третичные амины, особенно этил-4-диметиламинобензоат, диметилбензоат, N,N-диметил-p-толуидин, и аминосодержащий мономер 2-(диметиламино)этил метакрилат [37, 63–65]. Такие фотоинициирующие системы достаточно эффективно полимеризуют композиции на основе ди- и поли(мет)акриловых мономеров [37, 64] и широко применяются для отверждения стоматологических композитов [63, 66–68]. Усовершенствование фотоинициирующих систем на основе КХ – одно из важных направлений в разработке фотоинициирующих систем, чувствительных к видимому излучению. На данный момент такие исследования связаны с изучением влияния добавок солей йодония [69–71] и обладающих антибактериальными и противовоспалительными свойствами природных соединений. Предполагается, что использование последних будет способствовать улучшению биосовместимости получаемых имплантов [72, 73]. К недостаткам фотоинициирующих систем на основе КХ можно отнести небольшую величину коэффициента экстинкции КХ в диапазоне 400–500 нм (40 дм3/моль см) [74], что приводит к потере большой части энергии, излучаемой источниками света. Одним из немногих соединений, полученных модификацией КХ, является водорастворимая камфорохинон-карбоксильная кислота [75, 76]
 ,
,
используемая как фотоинициатор в биомедицинских приложениях [77].
Бензофенон поглощает УФ-излучения с максимумами при ~205, ~250 и ~350 нм [78]. В литературе описано большое количество фотоинициаторов, содержащих БФ, полученных путем введения фрагментов амина [79, 80], в том числе один из часто применяемых аминсодержащих бензофенонов – кетон Михлера

и его производные [81–83]. Структуру БФ модифицировали различными эфирными, гетероциклическими, акрилатными и другими фрагментами. Тиоксантон, фотохимические превращения которого также подробно исследованы [84–86], поглощает электромагнитное излучение с длиной волны до 430 нм с максимумом при ~377 нм. Тиоксантон сам по себе используется реже, чем КХ и БФ. Обычно применяются производные ТК с заместителями в положении 2, такие как ИТК и 2-хлоротиоксантон

Включение в молекулу ТК ароматических и гетероциклических фрагментов, таких как антрацен [87], карбазол [88] и флуоренон [89], позволяет сместить область поглощения в сторону более длинных волн.
Структура некоторых карбонилсодержащих соединений, которые реже используют для фотоинициирования полимеризации и получения новых фотоинициаторов химической модификацией молекулы приведена ниже. Это ксантон
 ,
,
антрахинон
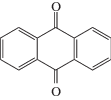 ,
,
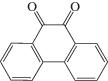

Новые фотоинициаторы получают химической модификацией молекулы кумарина
 ,
,
который сам по себе не применяют в качестве фотоинициатора полимеризации. Многие производные кумарина характеризуются высокими коэффициентами экстинкции в видимом диапазоне и исследуются как фотоинициаторы свободнорадикальной и катионной фотополимеризации для использования в 3D-печати [95–97].
Для фотоинициирования полимеризации ди- и поли(мет)акриловых мономеров под действием видимого света используют производные о-бензохинона [42, 98–100].
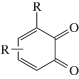
Замещенные о-бензохиноны поглощают УФ- и видимое излучение от 350 до 650 нм, и имеют две полосы поглощения в диапазонах 400–450 нм и 550–650 нм. В качестве соинициаторов для замещенных о-бензохинонов используются третичные амины, из которых наиболее эффективен диметилэтаноламин. Существенным недостатком их использования является трудоемкий синтез пространственно-затрудненных о-бензохинонов.
Некоторые примеры карбонилсодержащих фотоинициаторов на основе описанных структур приведены в табл. 1.
Таблица 1.
Некоторые карбонилсодержащие соединения, используемые в фотоинициирующих системах
| Формула | λmax, нм | Тип инициируемой полимеризации | Литература |
|---|---|---|---|
 |
~250 ~350 |
Радикальная, катионная, прививочная | [39, 101, 102] |
 |
~365 | Радикальная | [103] |
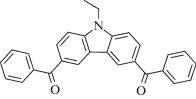 |
~335 | Радикальная, катионная | [104] |
 |
~230 ~298 |
Радикальная | [105] |
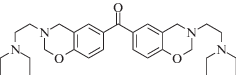 |
~302 | Радикальная | [106] |
 |
~382 ∼418 |
Радикальная, катионная | [107] |
 |
~380 | Радикальная, катионная | [80] |
 |
~286 | Радикальная | [108] |
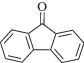 |
~430 | Радикальная | [92] |
 |
~280 ~330 |
Прививочная | [109, 110] |
 |
~378 | Радикальная, катионная, прививочная | [40, 109, 111] |
 |
~384 | Радикальная | [112] |
 |
~386 | Радикальная, катионная, прививочная | [113, 114] |
 |
~385 | Радикальная, катионная, прививочная | [115–117] |
 |
~383 | Радикальная | [118] |
 |
~386 | Радикальная | [119] |
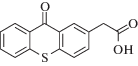 |
~379 ~574 |
Радикальная | [120] |
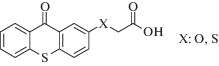 |
~385 ~397 |
Радикальная | [121] |
 |
~392 ~583 |
Радикальная (однокомпонентный) | [122] |
 |
~407 | Радикальная | [123] |
 |
~408 | Радикальная | [123] |
 |
~406 | Радикальная | [123] |
 |
~410 | Радикальная | [124] |
 |
~407 | Радикальная (однокомпонентный) | [125] |
 |
~350 ~450 |
Радикальная | [89] |
 |
~380 | Радикальная | [126] |
 |
~403 ~456 |
||
 |
~425 | ||
 |
~324 ~414 ~434 |
Радикальная, катионная | [127] |
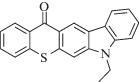 |
450 | Радикальная, катионная | [88] |
 |
~330 ~409 ~431 |
Радикальная | [87] |
 |
~530 ~640 |
Радикальная | [128] |
 |
400 | Радикальная | [129] |
 |
~380 | Радикальная (однокомпонентный, водорастворимый) | [130] |
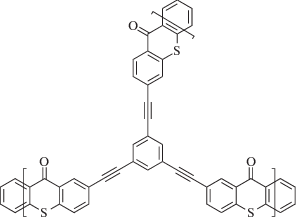 |
~390 | Радикальная и катионная | [131] |
 |
~468 | Радикальная, катионная, прививочная | [118, 132] |
 |
~225 ~460 |
Радикальная и катионная (водорастворимый) | [75] |
 |
~468 | Радикальная | [133] |
 |
~345 | Радикальная | [134] |
 |
~256 ~325 |
Радикальная, катионная, прививочная | [135, 136] |
 |
~477 | Радикальная и катионная | [137] |
 |
~325 | Радикальная и прививочная | [138, 139] |
 |
~407 | Радикальная и катионная | [140] |
 |
~522 | Радикальная и катионная | [141] |
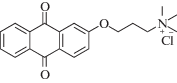 |
~337 | Радикальная | [142] |
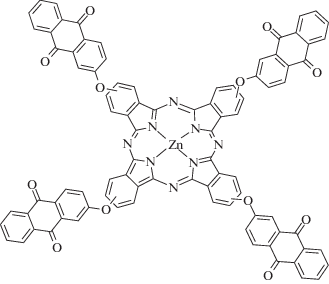 |
~342 ~650 |
Радикальная и катионная | [143] |
 |
~442 | Радикальная и катионная | [144] |
 |
~421 | Радикальная и катионная | [145] |
 |
~372 | Радикальная и катионная | [97] |
 |
~349 | Радикальная и катионная | [95] |
 |
~361 | Радикальная и катионная | [146] |
 |
~330 | Радикальная и катионная | [147] |
 |
~415 | Радикальная и катионная | [148, 149] |
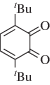 |
~410 ~ 598 |
Радикальная | [42, 98] |
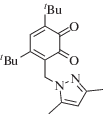 |
~408 ~586 |
Радикальная | [100] |
 |
~403 ~585 |
Радикальная | [99] |
В качестве доноров водорода могут выступать различные соединения, но чаще всего используют амины:

В последние годы выбор соинициаторов существенно расширился за счет тиолов, силанов, германов и станнанов, фосфор- и борорганических соединений
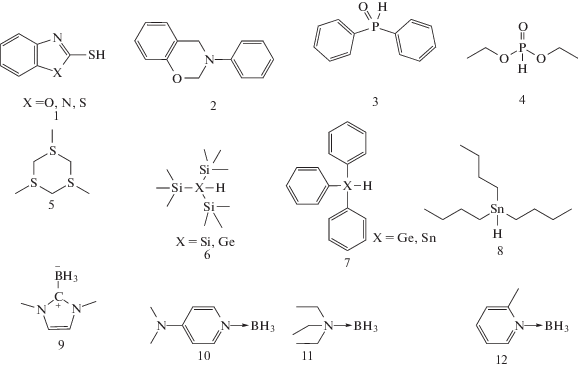
Несмотря на то, что фотоинициирующие системы второго типа подразумевают наличие двух соединений для прохождения инициирования, существуют однокомпонентные системы, не требующие специального добавления соинициатора. Это возможно в двух случаях: 1) карбонилсодержащий фотоинициатор может вступать в реакцию фотовосстановления и отрывать атом водорода от мономера или другого компонента системы, например, растворителя; 2) карбонилсодержащий фотоинициатор может сам служить донором водорода за счет протекания межмолекулярной или внутримолекулярной реакции.
Было получено и протестировано большое количество однокомпонентных фотоинициаторов второго типа, содержащих в одной молекуле карбонильный фрагмент и фрагмент, выступающий донором атома водорода. Также существует направление исследований по получению высокомолекулярных фотоинициаторов для снижения их токсичности, уменьшения миграционной способности для увеличения точности воспроизведения при 3D-печати. Некоторые из них являются однокомпонентными за счет полимерной цепи, содержащей лабильные атомы водорода. Кроме того, существуют трехкомпонентные фотоинициирующие системы второго типа. В качестве третьего компонента часто используются ониевые соли. Трехкомпонентные фотоинициирующие системы с добавками ониевых солей применяются для инициирования как радикальной, так и катионной фотополимеризации.
Катионная полимеризация
Фотоинициируемая катионная полимеризация лактонов, виниловых мономеров, эпоксидов и других циклических простых эфиров широко используется в различных областях УФ-отверждения [150]. В отличие от свободнорадикальной полимеризации катионная полимеризация не чувствительна к ингибированию процесса кислородом и может проводиться на воздухе, но более чувствительна к влаге. Чаще всего для катионной фотополимеризации используют различные ониевые соли [151–153]. Эти фоточувствительные соединения при облучении подвергаются прямому фотолизу через гетеролитический или гомолитический разрыв связи, что приводит к образованию катионов или катион-радикалов соответственно. Ниже приведен общий механизм фотолиза ониевых солей с образованием инициирующей катионную полимеризацию частицы в присутствии доноров водорода на примере соли дифенилйодония [153].

Эффективность фотоинициирования катионной полимеризации значительно увеличивается при использовании ониевых солей совместно с донорами водорода за счет образования инициирующих катионную полимеризацию кислот Бренстеда. Главным недостатком использования ониевых солей является то, что основная полоса поглощения этих соединений находится в среднем УФ-диапазоне, что не позволяет проводить инициирование катионной полимеризации видимым светом. Чтобы сдвинуть поглощение в более длинноволновую область спектра, ониевые соли иногда объединяют со свободнорадикальными фотоинициаторами (рис. 2, путь а) [154]. Карбонилсодержащие фотоинициаторы второго типа [154–156] могут участвовать в инициировании катионной полимеризации совместно с ониевыми солями. При этом инициирующая катионную полимеризацию частица образуется за счет взаимодействия сгенерированного радикала с ониевой солью (рис. 2, путь б). Фотосенсибилизированный таким образом фотолиз ониевых солей широко применяется для инициирования катионной фотополимеризации в более длинноволновом диапазоне и считается эффективной стратегией для разработки фотоинициирующих систем, инициирующих катионную полимеризацию в видимом диапазоне. Использование свободнорадикальных фотоинициирующих систем – одно из направлений катионной полимеризации, стимулируемой свободными радикалами. Обычно для увеличения эффективности образования радикалов в систему также добавляют соинициаторы для карбонилсодержащего фотоинициатора, формируя трехкомпонентную фотоинициирующую систему карбонилсодержащее соединение– донор водорода–ониевая соль.

Сообщается, что БФ [102, 159], КХ [156, 160], ТК [111] и его производные [88, 155, 161]
 ,
,
производные кумарина [148, 162, 163]
 ,
,
9,10-фенантенхинон и некоторые другие о-хиноны [164]
 ,
,
способны инициировать катионную полимеризацию при облучении в присутствии солей дифенилйодония в комбинации с донорами водорода.
Ниже представлен механизм инициирования катионной полимеризации фотоинициирующей системы КХ–соль йодония–этилдиметиламинобензоат. Предлагаемый механизм включает процесс отрыва атома водорода кетоном, после чего инициирующие катионную полимеризацию частицы могут возникать как за счет окисления образовавшегося кетильного радикала солью йодония, так и по реакции аминильного радикала с солью йодония. При этом также образуется радикал, способный инициировать радикальную полимеризацию. Показано, что катионные частицы в основном образуются за счет реакции аминильного радикала [156].

Ониевые соли, чаще всего используемые в инициирующих катионную полимеризацию фотоинициирующих системах совместно с карбонилсодержащими фотоинициаторами второго типа, приведены ниже.

К ним относятся соли диарилйодония (структуры 1–4), трифенилсульфония (структура 5) и N-этокси-2-метилпиридиния (структура 6). Однако в большинстве случаев используются соли дифенилйодония (структура 1).
Ниже представлены эффективные доноры водорода, наиболее часто участвующие в фотоиндуцированной катионной полимеризации, стимулируемой свободными радикалами.

В качестве доноров водорода обычно используют ароматические амины

Несмотря на более низкую чувствительность катионной полимеризации к присутствию кислорода, возбужденное состояние фотосенсибилизатора может быть дезактивировано кислородом. Использование в качестве соинициаторов силанов, станнанов и германов

делает процедуру устойчивее к присутствию кислорода [165–167]. В последнее время фотоинициирующие системы состава производное кумарина–донор водорода–ониевая соль широко исследуются для применения в 3D-печати [95, 144, 145, 168].
Поверхностно-инициируемая прививочная полимеризация
Модификация уже “готовых” полимеров с целью придания им необходимых свойств, представляет собой привлекательный способ получения полимерных материалов для широкого применения [169–175]. Одним из простых вариантов осуществления такой модификации является прививка. Полимеры, состоящие из линейных основных цепей с привитыми боковыми цепочками, могут иметь свои физические свойства, значительно отличающиеся от свойств исходного полимера, улучшенные путем изменения плотности прививки или видов прививки. В литературе выделяют три основных варианта получения привитых полимеров: “прививка от”, “прививка к”/“прививка на” и “прививка через” [176–179]:

“Прививка от” представляет собой прививочную полимеризацию мономера к молекуле полимера, содержащей фрагменты инициатора [101, 180]. “Прививка к” – это присоединение бокового полимерного фрагмента посредством проведения химической реакции между ним и основной полимерной цепью [179]. Данный метод обычно не дает большой плотности привитых цепей из-за стерических препятствий потому, что хемосорбция первой фракции цепей препятствует диффузии последующих цепей к поверхности, образуя макромолекулярный барьер. “Прививка через” представляет собой полимеризацию макромономеров [181].
Такая классификация способов получения привитых полимеров характерна и для прививки на полимерные поверхности. Модификация поверхности полимерных материалов имеет особое значение для хроматографии, микрофлюидных и биомедицинских устройств [182–184]. Вторичная функционализация поверхности полимерных материалов прививочной фотополимеризацией позволяет получать модифицированные поверхности с заданным рисунком различных функциональных зон, что является важной задачей при изготовлении устройств, применяемых в микробиологии и микрофлюидике.

При этом “прививка от” осуществляется с использованием инициаторов, закрепленных на поверхности полимерного материала и преобразуется в поверхностно-инициируемую прививочную полимеризацию функционализирующего мономера. “Прививка через” будет представлять собой сополимеризацию макромономеров с имеющимися на поверхности полимерного материала полимеризационноспособными группами [186–188]. За счет использования реакции фотовосстановления карбонилсодержащих соединений возможно проведение всех трех способов получения привитых полимеров [189].
На реакции фотовосстановления карбонильных соединений основан метод модификации полимерных поверхностей по варианту “прививка от” – фотоиндуцированной прививочной полимеризацией под действием УФ-излучения с использованием БФ в качестве фотоинициатора [190, 191]. Этот способ широко применим для функционализации как пористых, так и непористых полимеров [182, 190–193]. Ключевым этапом в данном методе является получение инициирующих полимеризацию центров на функционализируемой поверхности. Радикальные центры образуются за счет отрыва атома водорода от материала стенки поры при проведении фотоинициируемой реакции восстановления фотоинициатора. Используются двустадийная и одностадийная методики [101, 191, 194, 195]. В одностадийной методике образец заполняют раствором, содержащим мономер и фотоинициатор, после чего облучают через фотошаблон. Плюс одностадийной методики – ее простота и меньшее количество операций, а также однократное воздействие УФ-излучения на полимерный материал. В двустадийной методике полимерный образец сначала насыщают раствором БФ и облучают через фотошаблон, затем промывают, заполняют раствором мономера и снова облучают через тот же фотошаблон. Двустадийная методика убирает конкуренцию между полимеризацией мономера в растворе и на поверхности пор, предотвращая, таким образом, возможность закупоривания пор гомополимером и увеличивая степень прививки [195, 196]. Для систем с БФ в качестве фотоинициатора был предложен “квазиживой” механизм полимеризации за счет обратимого возбуждения БФ [195, 197–199]:

При УФ-облучении молекула БФ поглощает квант света, переходит в возбужденное синглетное состояние (S1), далее в триплетное состояние (T1) и отрывает атомы водорода от полимерного монолита. При этом на поверхности полимера возникают радикальные центры, которые в дальнейшем могут реагировать либо с мономерами, приводя к образованию привитых цепей (одностадийная методика), либо рекомбинировать с радикалами, закрепляя на пористой поверхности БФ (двустадийная методика). Дальнейшее УФ-облучение может расщеплять данную связь, инициируя полимеризацию.
Были проведены многочисленные исследования по влиянию концентрации инициатора [195, 200] и мономера [195, 199], природы растворителей [195, 201], времени облучения и дозы энергии [200], температуры [199, 201] на выход прививки для разных мономеров на различных пористых подложках. Такой метод нашел применение для изменения гидрофильных/ гидрофобных свойств пористых полимеров, а также для синтеза пористых полимеров с заданным распределением гидрофильных и гидрофобных областей. В литературе описан способ получения пористых полимерных слоев с гидрофобной сеткой [202]. Эти структуры использовали как клеточные микрочипы для проведения обратной трансфекции клеток с целью получения функциональной характеристики их генома.
Инициирующие центры на поверхности пор для прививки могут быть получены и другим способом. Так, описан простой и, тем не менее, эффективный метод поверхностного УФ-инициирования полимеризации в котором инициирующие центры формируются иммобилизацией БФ [184, 203, 204] и других карбонильных фотоинициаторов [138] на поверхности полимера с помощью физической адсорбции до полимеризации (схема 2 , путь а). Образец пористого полимера выдерживают в органическом растворе инициатора, высушивают, а затем помещают в водный раствор мономера и проводят функционализацию. Подразумевается, что инициатор полимеризации не растворим в воде, а мономер растворим. Это накладывает ограничения на круг используемых мономеров, так как далеко не все из них растворимы в воде, а использование органического растворителя будет приводить к вымыванию инициатора с поверхности образца, и полимеризация будет происходить в растворе.

Улучшенным вариантом является глубокая адсорбция инициатора в тонком поверхностном слое полимера за счет его набухания (схема 2 , путь б). В зависимости от свойства растворителя, механизм иммобилизации инициатора может быть изменен от адсорбции на более глубокую сорбцию в набухшем слое. Количество инициатора, иммобилизованного на подложке, зависит от времени, сорбционных свойств полимера и концентрации инициатора, а также последующей промывки растворителями.
Полимерные материалы из полиэфирэфиркетона (ПЭЭК), содержащего фрагмент БФ, могут подвергаться модификации прививочной полимеризацией под действием УФ-излучения без использования дополнительного фотоинициатора [180, 205], позволяя прививать широкий спектр мономеров разной химической природы: н-бутилакрилат, стирол, винилфосфоновая кислота, олиго(этиленгликоль)метакрилат, акриловая кислота. Ниже приведен механизм поверхностно-инициируемой прививочной полимеризации [180] и некоторые мономеры, прививаемые на поверхность ПЭЭК.

ПЭЭК широко используется для замены металлических деталей в биомедицине, автомобильной и аэрокосмической отраслях. ПЭЭК как не биодеградируемый материал перспективен для изготовления различных имплантатов. Благодаря достаточно высокой гидрофобности он биоинертен. Недостаток ПЭЭК заключается в том, что он не врастает в окружающую ткань [206]. Также в случае ПЭЭК наблюдается высокое поверхностное трение, что приводит к преждевременному выходу из строя и сокращению срока службы материала в экстремальных условиях [207]. По этой причине основным направлением в исследованиях по модификации поверхности ПЭЭК является повышение биосовместимости и улучшение остеоинтеграции материала.
Поверхность ПЭЭК функционализировали УФ-инициируемой прививочной полимеризацией 2-метакрилоилоксиэтилфосфорилхолина (структура 1) [180, 205, 208] и его сополимером (структура 2) [209], винилфосфорной кислотой (структура 5) [210], натриевой солью винилсульфоновой кислоты (структура 6) [211]. Полимеры 2-метакрилоилоксиэтилфосфорилхолина демонстрируют отличную устойчивость к адсорбции белка и ингибируют клеточную адгезию [212, 213], что важно для антитромбогенных покрытий на медицинских устройствах и сердечно-сосудистых стентах. Покрытия из поли(2-метакрилоилоксиэтилфосфорилхолина) используются для имплантов [214, 215]. Модификация ПЭЭК улучшает смачиваемость поверхности водой. Функционализация ПЭЭК полимером или сополимером 2-метакрилоилоксиэтилфосфорилхолина снижает активацию тромбоцитов при контакте с плазмой, богатой тромбоцитами человека [209, 216], а также значительно уменьшает бактериальную адгезию и адсорбцию белков плазмы независимо от типа белка [180, 205]. Данная модификация приводит к существенному улучшению совместимости с кровью. Также была проведена модификация поверхности ПЭЭК с помощью фотополимеризации ониевых солей 2-(метакрилоилокси) этилтриметиламмонийхлорида (структура 3) и калиевой соли 3-сульфопропилметакрилата (структура 4), снижающих перикапсулярное обрастание и обеспечивающих низкий коэффициент трения для смоченных водой поверхностей [217].
Помимо большого количества работ с использованием БФ в качестве УФ-инициатора, описаны примеры проведения прививочной полимеризации, инициируемой за счет фотовосстановления других карбонилсодержащих соединений, таких как ксантон [110], ИТК, производные антрахинона, 9,10-фенантренхинон и даже ацетон [218].
Авторы [109] на примере прививочной полимеризации акриловых и метакриловых мономеров (λ > 300 нм, ртутная лампа высокого давления) на полиэтиленовую пленку исследовали активность ряда фотоинициаторов в прививочной полимеризации. Высокий процент привитого полимера (более 80 мас. %) был зарегистрирован для ароматических кетонов (БФ, ксантон и ТК) и для хинонов (2-хлороантрахинон, 2-метилантрахинон и 9,10-фенантрахинон). В работе [189] также проведено сравнение способности ряда известных карбонилсодержащих фотоинициаторов вести прививочную полимеризацию акриловой кислоты на поверхности ПЭ под действием УФ-излучения. Авторами было показано, что БФ, ксантон, ацетофенон и 9-флуоренон являются эффективными инициаторами поверхностно-инициируемой фотополимеризации для получения толстых привитых слоев, в то время как антрахинон, 4,4-дихлорбензофенон, 4-бензилбензофенон, бензоилформиновая кислота и биацетил служат подходящими фотоинициаторами для получения тонких привитых слоев.
Антрахинон и его производные применяются для инициирования прививочной фотополимеризации и модификации свойств полимеров [138, 219] и полисахаридов [135, 220]. Так, авторами [135] была продемонстрирована способность 2-этилантрахинона инициировать прививку к хлопковым волокнам по механизму радикальной полимеризации под действием ближнего ультрафиолета в инертной атмосфере. Процедуру проводили в две стадии: вымачивание хлопковой ткани в растворе фотоинициатора с последующей промывкой от несорбированного пигмента, а затем облучение полимера в растворе мономера (также с последующей промывкой). При повышении концентрации фотоинициатора в прививочном растворе от 1 до 9 мас. % степень прививки увеличивалась от 5 до 25%. Степень прививки также возрастала с увеличением продолжительности облучения. При этом введение в систему кислорода снижает эффективность прививки до нуля. Предполагаемый механизм прививочной полимеризации на поверхности хлопковых волокон с использованием 2-этилантрахинона в качестве фотоинициатора [135] приведен ниже

Отрыв водорода предпочтительно идет из положения 1 гетероцикла гексозы в целлюлозе. Полимеризационную цепочку начинает радикал на молекуле целлюлозы, поскольку радикалы, образующиеся из хинона, менее реакционноспособны по отношению к мономеру из-за стерических препятствий и работают в основном как ингибиторы полимеризации. Авторы утверждают, что по сравнению с существующими методами прививки к поверхности данный метод кубового крашения с последующим фотографтингом более практичен и экологичен. Как пропитанная 2-этилантрахиноном ткань, так и ткань после прививки демонстрируют инициируемую светом антимикробную активность против грамположительных и грамотрицательных бактерий.
Прививочную полимеризацию могут на полимерной поверхности, содержащей соинициаторы для карбонильного фотоинициатора, вести, например, амины. Авторами [219] была осуществлена двустадийная модификация поверхности полиэтилентерефталата мономером на основе ванилина: сначала поверхность полиэтилентерефталата была модифицирована N,N-диэтилэтилендиамином посредством аминолиза, затем была проведена прививочная фотополимеризация акриламидного мономера, инициируемая антрахиноном.

Такая модификация поверхности придала полимеру антибактериальные свойства за счет снижения адгезии грамположительных и грамотрицательных штаммов.
Способностью инициировать прививочную фотополимеризацию также обладают 2-хлорантрахинон [117], водорастворимые натриевые соли антрахинон-2-сульфоновой [138] и антрахинон-2,6-дисульфоновой кислот [221], которые используют для проведения прививочной полимеризации в водной среде, и некоторые другие производные антрахинона [109].
Для ИТК также показана способность инициировать прививочную полимеризацию к аминсодержащей гидрогелевой подложке:

В работе [222] были получены гидрогели с сайт-контролируемым (анизотропным) высвобождением вещества.
Для этого поверхность гидрогеля на основе 2-(N,N-диметил)этилметакрилата модифицировали фотоинициируемой прививочной полимеризацией глицидилметакрилата с использованием ИТК в качестве фотоинициатора (λмакс = = 365 нм) и катионной фотополимеризацией с раскрытием эпоксидного цикла глицидилметакрилата для сшивки, инициируемой ониевой солью. В результате на поверхности гидрогеля образовывался сшитый слой, текстура которого отличалась от текстуры немодифицированной поверхности. Было исследовано высвобождение из гидрогеля Родамина В. Время высвобождения красителя из модифицированного гидрогеля возрастало с увеличением времени проведения прививки и катионной полимеризации. Для фрагмента гидрогеля, у которого вся поверхность за исключением нижней стороны была модифицирована, наблюдалось анизотропное высвобождение красителя.
Помимо большого количества публикаций по УФ-инициируемой прививочной полимеризации к настоящему времени также имеется несколько работ по инициируемым видимым излучением поверхностно-инициируемой полимеризации с использованием карбонилсодержащих фотоинициаторов. Такой тип фотоинициирования в перспективе имеет ряд преимуществ перед УФ-инициированием, например, отсутствие необходимости использования специального оборудования и УФ-прозрачных матриц; и возможность применения перестраиваемых компьютерных масок, генерируемых стандартными проекционными аппаратами.
Описан способ прививки полимера на поверхность полиуретана с использованием в качестве фотоинициатора КХ под действием видимого излучения [132]. Для ИТК представлена возможность проведения прививочной полимеризации под действием видимого излучения (λмакс = = 385 нм [114], λ = 380–700 нм [223]) с преоблучением УФ-светом с λмакс = 254 нм для фотовосстановления инициатора и его закреплением рекомбинацией на модифицируемой поверхности:

Показана возможность инициируемой видимым светом прививочной полимеризации гидрофобизирующих мономеров с использованием в качестве фотоинициатора 9,10-фенантренхинона [224, 225]. За счет смещения рабочей области фотоинициатора в видимый диапазон получены пористые полимеры с гидрофобным рисунком по поверхности и объему с применением нарисованных на компьютере виртуальных фотошаблонов и стандартной проекционной оптики. Полученный рисунок гидрофобной области визуализировали вымачиванием образца в водном растворе тиоцианата железа(III) [224].

Описанная методика фотогидрофобизации представляет собой быстрый и простой способ получения как полностью, так и частично гидрофобизированных пористых полимеров с использованием недорогого оптического оборудования, полностью исключающего использование материальных фотошаблонов при проведении локальной засветки.
МОДИФИКАЦИЯ ПОЛИМЕРОВ МЕТОДОМ ФОТОСШИВАНИЯ И “ПРИВИВКИ К” С ИСПОЛЬЗОВАНИЕМ РЕАКЦИИ ФОТОВОССТАНОВЛЕНИЯ КАРБОНИЛСОДЕРЖАЩИХ СОЕДИНЕНИЙ
Радикалы, формирующиеся в результате фотоиндуцированного переноса водорода при фотовосстановлении карбонилсодержащего соединения, могут рекомбинировать с образованием ковалентных связей между реагентами. За счет этого некоторые карбонилсодержащие соединения можно использовать в качестве якорной группы для функционализации полимерными слоями поверхностей, которые могут служить донорами водорода, по варианту “прививка к” за счет внедрения карбонилсодержащего соединения по связи C–H. Как и в случае прививочной полимеризации, основным применяемым для этого фотоинициатором является БФ. Фотоиндуцируемое внедрение бензофенонового фрагмента давно используется в биохимии при функционализации определенных удаленных связей C–H для получения фотоаффинных меток в белках, нуклеиновых кислотах, липидах и стероидах и для картирования гибких цепей с целью изучения их конформаций и взаимодействий в растворе, мицеллах и мембранах [226–229].
Такой способ функционализации поверхности применим для модификации как полимерных, так и стеклянных [230], кремневых [231] и металических [232] подложек, а также оксидов металлов [233]. При этом в случае неорганических подложек, не содержащих связей C–H, на них первоначально фиксируется содержащий терминальный бензофеноновый фрагмент алкилоксисилан. А затем при УФ-облучении происходит формирование полимерного слоя на подложке, который может содержать или не содержать бензофеноновые фрагменты. На основе этого принципа к поверхностям прикреплялись полимерные монослои различных полимеров из поливинилового спирта, полистирола, полиакриловой и полиметакриловой кислот [234, 235]. Ниже приведена общая схема получения привитого несшитого (а) и сшитого (б) полимерного слоя на неорганических подложках путем внедрения бензофенонового фрагмента по связям C–H.

В случае полимерных подложек бензофенон на функционализируемой поверхности не фиксируют, а используют полимер, содержащий БФ:

Обычно для этого подложку покрывают тонким слоем содержащего бензофеноновые фрагменты полимера путем аэрозольного распыления или спин-коутинга, а затем облучают. Отрыв атома водорода фотовозбужденным фрагментом БФ может проходить как от поверхности подложки, так и от самого прививаемого полимера, и, если полимер содержит несколько бензофеноновых фрагментов, то рекомбинация образовавшихся радикалов может приводить не только к прививке полимера к поверхности, но и к его сшиванию с образованием сеток и гидрогелей, ковалентно связанных с подложкой [236, 237]. Таким образом, за счет введения в полимер бензофенонового фрагмента можно получать как привитые полимеры, так и привитые сшитые гидрогели [238]. Использование бензофенон-содержащих полимеров или сшивателей для получения сшитых полимеров является одним из способов сшивания за счет внедрения по связям C–H [239].
Ниже представлены структуры некоторых бензофенон-содержащих полимеров, закрепленных таким образом на полимерных подложках.
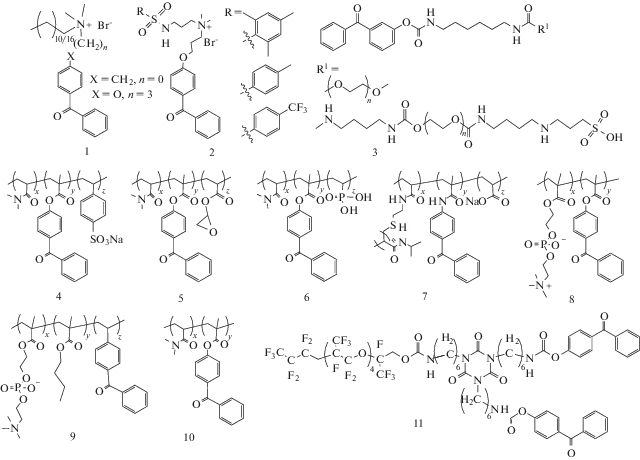
Для получения несшитых слоев используются соединения с одним терминальным бензофеноновым фрагментом. Этот метод часто используется для прививки полимеров с аммониевыми и фосфониевыми фрагментами (для которых была показана антибактериальная активность) на различные полимерные подложки, включая ПП, ПЭ, ПС, и хлопок [240, 241] (структуры 1–3).
Для получения сшитых слоев гидрогелей обычно применяются сополимеры с бензофеноновым фрагментом. Включение этого фрагмента в полимер может осуществляться за счет сополимеризации с БФ-содержащим мономером [242, 243] или модификацией готового полимера [244]. Такие полимеры, содержащие аммониевые и фосфониевые и некоторые другие фрагменты, используются для модификации поверхности различных полимерных подложек для получения тонких привитых слоев гидрогелей, которые улучшают адгезию клеток к поверхности. Также получают слои гидрогелей-субстратов с имобилизироваными в них биомолекулами (ДНК, олигонуклеотиды, белки и белковые антитела), в том числе с целью создания микропланшетов для аналитического обнаружения белков и других биополимеров (структуры 4–10) [245–247]. Гидрофобное соединение (структура 11) было взято для получения высокопрочного антибликового и предотвращающего загрязнения покрытия на полиэтилентерефталате [248].
Авторами [249] был предложен и осуществлен быстрый, эффективный и простой метод функционализации поверхности ПВХ путем комбинирования реакции сера (VI)–фторидного обмена с фотовнедрением бензофенонового фрагмента по связям C–H в поливинилхлориде:

С помощью этого метода на поверхности ПВХ были иммобилизованы различные функциональные соединения, включая RAFT-агент для проведения прививочной RAFT-полимеризации; ионную жидкость, придающую антибактериальные свойства; тиол/алкинильные группы для проведения последующей модификации.
Бензофенонсодержащие молекулы могут служить сшивателями [250] и без прививки к поверхности. Например, был разработан и синтезирован симметричный бензофенонсодержащий тритиокарбонат [251], который может действовать не только как RAFT-агент для полимеризации, но также как сшивающий агент под действием УФ-облучением
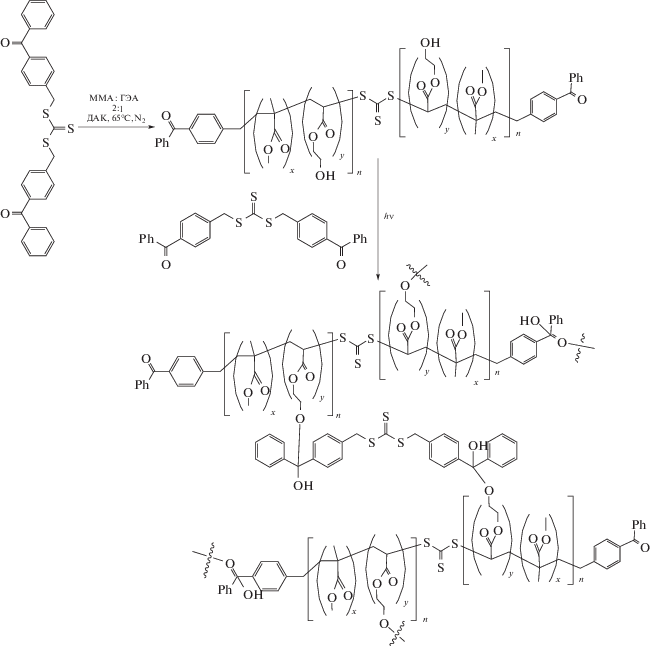
Полученный таким образом полимер способен самовосстанавливаться за счет перетасовки связей C–S тритиокарбоната при облучении.
Предложен метод синтеза блок-сополимеров, основанный на сочетании кетильных макрорадикалов [252].

Полимеры с концевыми бензофеноновыми группами были синтезированы радикальной полимеризацией с переносом атома с использованием бензофенонсодержащего инициатора. Затем полученные концевые функциональные полимеры смешивали в соотношении 1 : 1 и облучали. Методами ЯМР- и УФ-спектроскопии было доказано эффективное связывание между кетильными радикалами. Термическая лабильность такого соединения может обеспечить возможность разделения сегментов блока при облучении.
Описаны примеры использования других карбонилсодержащих фотоинициаторов. В работе [253] предложен способ прививки декстрана на матрицу из микроволокна поли(3-гидроксибутират-со-3-гидроксивалерата), полученную электроспиннингом:

Тиоксантоновые фрагменты могут быть включены в полимерные цепи [254] для производства функциональных сополимеров, которые можно быстро и эффективно сшивать с помощью низкоэнергетического УФ-света (λ = = 365 нм) за счет CHiC
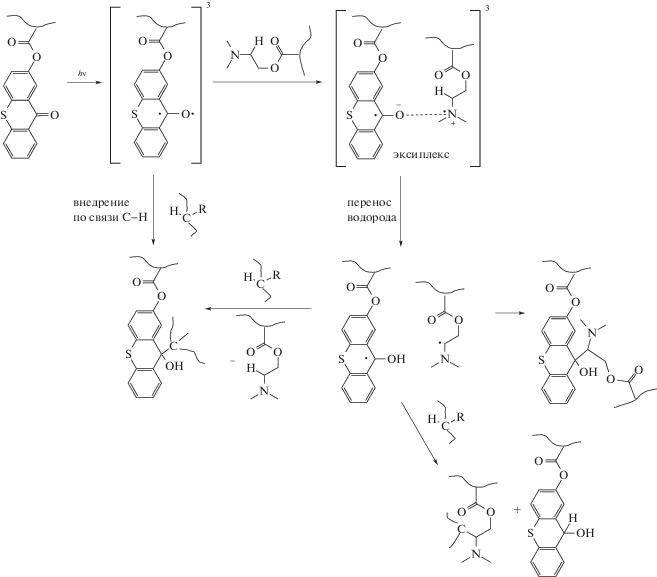
Использование тиоксантонового фрагмента может значительно смягчить условия получения гидрогелей методом CHiC по сравнению с системами на основе бензофенона, которые требуют либо жесткого УФ-излучения, либо длительного облучения длинноволновым излучением. Расширенное спектральное окно позволяет также включать в состав полимеров светочувствительные соединения. Внедрение фрагмента амина положительно сказывается на растворимости полимера в воде, что может сделать такой метод применимым для бимедицинских целей.
ЗАКЛЮЧЕНИЕ
Реакция фотовосстановления карбонилсодержащих светочувствительных соединений – один из самых давно используемых как в исследованиях, так и в промышленности методов инициирования фотополимеризации. За счет этой реакции работает большое количество карбонилсодержащих фотоинициаторов второго типа, позволяющих проводить фотополимеризацию под действием УФ- и видимого излучения, осуществлять инициирование радикальной, катионной и прививочной полимеризации и полимеризовать разные типы мономеров. Усовершенствование фотоинициирующих систем на основе карбонилсодержащих фотоинициаторов второго типа происходит по двум направлениям: получение новых фотоинициаторов и подбор новых соинициаторов. Синтез новых фотоинициаторов в основном осуществляется введением заместителей в ранее известные молекулы. Основными модифицируемыми таким образом молекулами являются БФ, ТК и кумарин. В последние два десятилетия применение обоих вариантов привело к значительному прогрессу в области разработки эффективных фотоинициирующих систем, позволяющих осуществлять синтез и модификацию различных полимеров, подходящих для промышленного использования в сфере аддитивных технологий. Это удобный и простой способ синтеза и модификации полимерных материалов с пространственным контролем.
По универсальности применения для инициирования при синтезе и модификации полимеров особо выделяется БФ. На основе БФ синтезировано много фотоинициаторов, используемых в фотоинициирующих системах, инициирующих как радикальную, так и катионную фотополимеризацию. БФ является основным фотоинициатором, применяемым для проведения поверхностно-инициируемой прививочной полимеризации и наиболее часто используемым фрагментом для модификации подложек и получения сшитых полимерных гелей за счет фотоиндуцированного внедрения по связи C–H.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 20-13-50428).
Список литературы
Garra P., Fuassier J.P., Lakhdar S., Yagci Y., Lalevée J. // Prog. Polym. Sci. 2020. V. 107. P. 101277.
Pigot C., Noirbent G., Brunel D., Dumur F. // Eur. Polym. J. 2020. V. 133. P. 109797.
Fouassier J.-P., Morlet-Savary F., Lalevée J., Allonas X., Ley C. // Materials. 2010. V. 3. № 12. V. 5130.
Allushi A., Kutahya C., Aydogan C., Kreutzer J., Yilmaz G., Yagci Y. // Polym. Chem. 2017. V. 8. № 12. P. 1972.
Cohen S.G., Chao H.M. // J. Am. Chem. Soc. 1968. V. 90. № 1. P. 165.
Coyle J., Carless H. // Chem. Soc. Rev. 1972. V. 1. № 4. P. 465.
Hiroshi M., Kazuhiro M., Kenji K., Noboru M. // Bul. Chem. Soc. Jpn. 1990. V. 63. № 12. P. 3385.
Devadoss C., Fessenden R.W. // J. Phys. Chem. 1991. V. 95. № 19. P. 7253.
Dreyer J., Peters K.S. // J. Phys. Chem. 1996. V. 100. № 50. P. 19412.
Tarasyuk A.Y., Granchak V.M., Dilung I.I. // J. Photochem. Photobiol. A. 1995. V. 85. № 1. P. 39.
Peters K.S., Cashin A. // J. Phys. Chem. A. 2000. V. 104. № 21. P. 4833.
Cossy J., Belotti D. // Tetrahedron. 2006. V. 62. № 27. P. 6459.
Yamaji M., Oshima J., Hidaka M. // Chem. Phys. Lett. 2009. V. 475. № 4. P. 235.
Morozova O.B., Panov M.S., Fishman N.N., Yurkov-skaya A.V. // Phys. Chem. Chem. Phys. 2018. V. 20. № 32. P. 21127.
Chesnokov S.A., Shurygina M.P., Abakumov G.A. // High Energy Chem. 2011. V. 45. № 4. P. 287.
Chesnokov S.A., Zakharina M.Y., Shaplov A.S., Lozinskaya E.I., Malyshkina I.A., Abakumov G.A. // J. Polym. Sci., Polym. Chem. 2010. V. 48. № 11. P. 2388.
Peters K.S., Lee J. // J. Phys. Chem. 1993. V. 97. № 15. P. 3761.
Peters K.S., Kim G. // J. Phys. Chem A. 2004. V. 108. № 14. P. 2598.
Rathore R., Hubig S.M., Kochi J.K. // J. Am. Chem. Soc. 1997. V. 119. № 47. P. 11468.
Hubig S.M., Rathore R., Kochi J.K. // J. Am. Chem. Soc. 1999. V. 121. № 4. P. 617.
Rosokha S.V., Kochi J.K. // Acc. Chem. Res. 2008. V. 41. № 5. P. 641.
Cohen S.G., Parola A., Parsons G.H. // Chem. Rev. 1973. V. 73. № 2. P. 141.
Levin P.P., Kuz’min V.A. // Russ. Chem. Rev. 1987. V. 56. № 4. P. 307.
Ci X., Da Silva R.S., Goodman J.L., Nicodem D.E., Whitten D.G. // J. Am. Chem. Soc. 1988. V. 110. № 25. P. 8548.
Chesnokov S.A., Cherkasov V., Chechet Y.V., Nevodchikov N., Abakumov G.A., Mamysheva O.N. // Russ. Chem. Bull. 2000. V. 49. № 9. P. 1506.
Shurygina M.P., Chesnokov S.A., Abakumov G.A. // High Energy Chem. 2016. V. 50. № 3. P. 196.
Leonhardt H., Weller A. // Berichte Bunsengesellschaft Phys. Chem. 1963. V. 67. № 8. P. 791.
Knibbe H., Rehm D., Weller A. // Berichte Bunsengesellschaft Phys. Chem. 1969. V. 73. № 8–9. P. 839.
Sarkar S., Pramanik R., Seth D., Setua P., Sarkar N. // Chem. Phys. Lett. 2009. V. 477. № 1. P. 102.
Yamaji M., Aoyama Y., Furukawa T., Itoh T., Tobita S. // Chem. Phys. Lett. 2006. V. 420. № 1. P. 187.
Левин П. // Хим. физика. 1989. Т. 8. № 6. P. 752.
Cohen S.G., Parsons G. // J. Am. Chem. Soc. 1970. V. 92. № 26. P. 7603.
Чесноков С.А, Черкасов В.К., Чечет Ю.В.. Неводчиков В.И., Абакумов Г.А., Мамышева О.Н. // Изв. РАН. Сер хим. 2000. Т. 49. № 9. С. 1506.
Чесноков С.А., Шурыгина М.П., Абакумов Г.А. // Химия высоких энергий. 2011. V. 45. № 4. P. 319.
Hubig S.M., Kochi J.K. // J. Am. Chem. Soc. 1999. V. 121. № 8. P. 1688.
Shurygina M.P., Chesnokov S.A., Lopatin M.A., Cherkasov V.K., Abakumov G.A. // Russ. Chem. Bull. 2004. V. 53. № 11. P. 2485.
Jakubiak J., Allonas X., Fouassier J.P., Sionkowska A., Andrzejewska E., Linden L.Å. // Polymer. 2003. V. 44. № 18. P. 5219.
Гранчак В.М., Чемерская З.Ф., Дилунг И.И. // Высомолек. соед. А. 1985. Т. 27. № 2. С. 276.
Block H., Ledwith A., Taylor A.R. // Polymer. 1971. V. 2. № 4. P. 271.
Anderson D.G., Davidson S.R., Elvery J.J. // Polymer. 1996. V. 37. № 12. P. 2477.
Christmann J., Allonas X., Ley C., Croutxé-Barghorn C. // Polym. Chem. 2019. V. 10. № 9. P. 1099.
Chesnokov S.A., Cherkasov V.K., Abakumov G.A., Mamysheva O.N., Zakharina M.Y., Shushunova N.Y., Chechet Yu.V., Kuropatov V.A. // Polymer Science B. 2014. V. 56. № 1. P. 11.
Shushunova N.Y., Chesnokov S.A. // Polymer Science B. 2009. V. 51. № 11–12. P. 427.
Lee T.Y., Roper T.M., Jönsson E.S., Guymon C.A., Hoyle C.E. // Macromolecules. 2004. V. 37. № 10. P. 3659.
Hoyle C.E., Chawla C.P., Griffin A.C. // Mol. Cryst. Liq. Cryst. 1988. V. 157. № 1. P. 639.
Zubov V.P., Kumar M.V., Masterova M.N., Kabanov V.A. // J. Macromol. Sci. A. 1979. V. 13. № 1. P. 111.
Andrzejewska E. // Prog. Polym. Sci. 2001. V. 26. № 4. P. 605.
Emami N., Söderholm K.J.M. // J. Mater. Sci.: Mater. Med. 2005. V. 16. № 1. P. 47.
Broer D.J., Mol G.N., Challa G. // Polymer. 1991. V. 32. № 4. P. 690.
Ito K. // J. Polym. Sci., Polym. Chem. Ed. 1975. V. 13. № 2. P. 401.
Ito K. // J. Polym. Sci., Polym. Chem. Ed. 1977. V. 15. № 7. P. 1759.
Corcione C.E., Greco A., Maffezzoli A. // Polymer. 2005. V. 46. № 19. P. 8018.
Lecamp L., Youssef B., Bunel C., Lebaudy P. // Polymer. 1997. V. 38. № 25. P. 6089.
Price R.B., Whalen J.M., Price T.B., Felix C.M., Fahey J. // Dent. Mater. 2011. V. 27. № 10. P. 983.
Lovelh L.G., Newman S.M., Bowman C.N. // J. Dent. Res. 1999. V. 78. № 8. P. 1469.
Lalevée J., Allonas X., Jradi S., Fouassier J.-P. // Macromolecules. 2006. V. 39. № 5. P. 1872.
Dietlin C., Schweizer S., Xiao P., Zhang J., Morlet-Savary F., Graff B., Fouassier J.-P., Lalevée J. // Polym. Chem. 2015. V. 6. № 21. P. 3895.
Cao H., Currie E., Tilley M., Jean Y.C. // Am. Chem. Soc. 2003. V. 847. P. 152.
Bhanu V.A., Kishore K. // Chem. Rev. 1991. V. 91. № 2. P. 99.
O'Brien A.K., Bowman C.N. // Macromolecules. 2006. V. 39. № 7. P. 2501.
Yeow J., Chapman R., Gormley A.J., Boyer C. // Chem. Soc. Rev. 2018. V. 47. № 12. P. 4357.
Chen Y.-C., Ferracane J.L., Prahl S.A. // Dent. Mater. 2007. V. 23. № 6. P. 655.
Asmusen S., Arenas G., Cook W.D., Vallo C. // Dent. Mater. 2009. V. 25. № 12. P. 1603.
Asmussen S., Arenas G., Cook W.D., Vallo C. // Eur. Polym. J. 2009. V. 45. № 2. P. 515.
Morlet-Savary F., Klee J.E., Pfefferkorn F., Fouassier J.P., Lalevée J. // Macromol. Chem. Phys. 2015. V. 216. № 22. P. 2161.
Nie J., Lindén L.Å., Rabek J.F., Fouassier J.P., Morlet-Savary F., Scigalski F. et al. // Acta Polymerica. 1998. V. 49. № 4. P. 145.
Potts T.V., Petrou A. // J. Endodontics. 1990. V. 16. № 6. P. 265.
Leprince J.G., Palin W.M., Hadis M.A., Devaux J., Leloup G. // Dent. Mater. 2013. V. 29. № 2. P. 139.
Ogliari F.A., Ely C., Petzhold C.L., Demarco F.F., Piva E. // J. Dent. 2007. V. 35. № 7. P. 583.
Verzola K.C., Dressano D., Saraceni C.H.C., Gonçalves L.S., Hadis M., Watts D.C., Palin W.M., Lima A.F. // Dent. Mater. 2020. V. 36. № 10. P. 1282.
Kirschner J., Paillard J., Bouzrati-Zerelli M., Becht J.-M., Klee J.E., Chelli S., Lakhdar S., Lalevée J. // Molecules. 2019. V. 24. № 16. P. 2913.
Sprick E., Becht J.-M., Graff B., Salomon J.-P., Tigges T., Weber C., Lalevée J. // Dent. Mater. 2021. V. 37. № 3. P. 382.
Sprick E., Graff B., Becht J.-M., Tigges T., Neuhaus K., Weber C., Lalevée J. // Eur. Polym. J. 2020. V. 134. P. 109794.
Neumann M.G., Miranda W.G., Schmitt C.C., Rueggeberg F.A., Correa I.C. // J. Dent. 2005. V. 33. № 6. P. 525.
Kamoun E.A., Winkel A., Eisenburger M., Menzel H. // Arab. J. Chem. 2016. V. 9. № 5. P. 745.
Matsuda T., Magoshi T. // Biomacromolecules. 2002. V. 3. № 5. P. 942.
Tomal W., Ortyl J. // Polymers. 2020. V. 12. № 5. P. 1073.
Castro G., Blanco S., Giordano O. // Molecules. 2000. V. 5. № 3. P. 424.
Temel G., Enginol B., Aydin M., Balta D.K., Arsu N. // J. Photochem. Photobiol. A. 2011. V. 219. № 1. P. 26.
Liu S., Brunel D., Sun K., Zhang Y., Chen H., Xiao P., Dumur F., Lalevée J. // Macromol. Rapid Commun. 2020. V. 41. № 23. P. 2000460.
Huang Q.H., Dou X.Q., Bai X.X., Hu Z.Y., Cheng C.J. // Adv. Mater. Res. 2013. V. 818. P. 54.
Wen Y., Jiang X., Liu R., Yin J. // Polym. Adv. Technol. 2011. V. 22. № 5. P. 598.
Wen Y., Jiang X., Yin J. // Prog. Org. Coat. 2009. V. 66. № 1. P. 65.
Allonas X., Ley C., Bibaut C., Jacques P., Fouassier J.P. // Chem. Phys. Lett. 2000. V. 322. № 6. P. 483.
Corrales T., Peinado C., Catalina F., Neumann M.G., Allen N.S., Rufs A.M., Encinas M.V. // Polymer. 2000. V. 41. № 26. P. 9103.
Ley C., Morlet-Savary F., Jacques P., Fouassier J.P. // Chem. Phys. 2000. V. 255. № 2. P. 335.
Balta D.K., Arsu N. // J. Photochem. Photobiol. A. 2013. V. 257. P. 54.
Tunc D., Yagci Y. // Polym. Chem. 2011. V. 2. № 11. P. 2557.
Karaca N., Balta D.K., Ocal N., Arsu N. // J. Polym. Sci., Polym. Chem. 2016. V. 54. № 7. P. 1012.
Albuquerque P.A.C., Bertolo M.L., Cavalcante L.M.A., Pfeifer C., Schneider L.F.S. // J. Esthet. Restor. Dent. 2015. V. 27. № S1. P. S49.
Zakharina M.Y., Chechet Yu.V., Shurygina M.P., Chesnokov S.A. // Polymer Science B. 2018. V. 60. № 6. P. 708.
Encinas M.V., Lissi E.A., Rufs A.M., Previtali C.M. // J. Polym. Sci., Polym. Chem. 1994. V. 32. № 9. P. 1649.
Kubota H., Ogiwara Y. // J. Appl. Polym. Sci. 1982. V. 27. № 7. P. 2683.
Allen N.S., Hardy S.J., Jacobine A., Glaser D.M., Catalina F. // Eur. Polym. J. 1989. V. 25. № 12. P. 1219.
Abdallah M., Dumur F., Hijazi A., Rodeghiero G., Gualandi A., Cozzi P.G., Lalevée J. // J. Polym. Sci. 2020. V. 58. № 8. P. 1115.
Salmi H., Tar H., Ibrahim A., Ley C., Allonas X. // Eur. Polym. J. 2013. V. 49. № 8. P. 2275.
Rahal M., Graff B., Toufaily J., Hamieh T., Dumur F., Lalevée J. // Eur. Polym. J. 2021. V. 154. P. 110559.
Chesnokov S.A., Cherkasov V.K., Abakumov G.A., Mamysheva O.N., Chechet Y.V., Nevodchikov V.I. // Russ. Chem. Bull. 2001. V. 50. № 12. P. 2366.
Zhiganshina E.R., Arsenyev M.V., Konev A.N., Chechet Y.V., Chesnokov S.A. // Key Eng. Mater. 2020. V. 869. P. 129.
Shurygina M.P., Zakharina M.Y., Baten’kin M.A., Ko-nev A.N., Shavyrin A.S., Chelnokov E.A., Shushunova N.Yu., Arsenyev M.V., Chesnokov S.A., Abakumov G.A. // Eur. Polym. J. 2020. V. 127. P. 109573.
Rånby B. // Makromol. Chem. Macromol. Symp. 1992. V. 63. № 1. P. 55.
Kahveci M.U., Tasdelen M.A., Yagci Y. // Polymer. 2007. V. 48. № 8. P. 2199.
Schroeder W.F., Asmussen S.L., Cook W.D., Vallo C.I. // Polym. Int. 2011. V. 60. № 9. P. 1362.
Liu S., Brunel D., Sun K., Xu Y., Morlet-Savary F., Graff B., Xiao P., Dumur F., Lalevée J. // Polym. Chem. 2020. V. 11. № 21. P. 3551.
Wang K., Jiang S., Liu J., Nie J., Yu Q. // Progr. Org. Coat. 2011. V. 72. № 3. P. 517.
Bai J., Shi Z. // J. Appl. Polym. Sci. 2013. V. 128. № 3. P. 1785.
Xiao P., Dumur F., Graff B., Gigmes D., Fouassier J.P., Lalevée J. // Macromolecules. 2013. V. 46. № 19. P. 7661.
Wang J., Cheng J., Liu J., Gao Y., Sun F. // Green Chem. 2013. V. 15. I. 9. P. 2457.
Kubota H., Kobayashi K., Ogiwara Y. // Polym. Photochem. 1986. V. 7. № 5. P. 379.
Kubota H., Suka I.G., Kuroda S., Kondo T. // Eur. Polym. J. 2001. V. 37. № 7. P. 1367.
Rodrigues M.R., Neumann M.G. // Macromol. Chem. Phys. 2001. V. 202. № 13. P. 2776.
Encinas M.V., Rufs A.M., Corrales T., Catalina F., Peinado C., Schmith K., Neumann M.G., Allen N.S. // Polymer. 2002. V. 43. № 14. P. 3909.
Wu C.-C., Hsu S.L.-C., Liao W.-C. // Microelectron. Eng. 2009. V. 86. № 3. P. 325.
Balasubramaniam A., Manderfeld E., Krause L.M.K., Wanka R., Schwarze J., Beyer C.D., Rosenhahn A. // Polym. Chem. 2021. V. 12. № 4. P. 618.
Catalina F., Peinado C., Mateo J.L., Bosch P., Allen N.S. // Eur. Polym. J. 1992. V. 28. № 12. P. 1533.
Manivannan G., Fouassier J.P., Crivello J.V. // J. Polym. Sci., Polym. Chem. 1992. V. 30. № 9. P. 1999.
Kubota H., Fukushima Y., Kuwabara S. // Eur. Polym. J. 1997. V. 33. № 1. P. 67.
Cokbaglan L., Arsu N., Yagci Y., Jockusch S., Turro N.J. // Macromolecules. 2003. V. 36. № 8. P. 2649.
Allen N.S., Salleh N.G., Edge M., Corrales T., Shah M., Catalina F. // J. Photochem. Photobiol. A. 1997. V. 103. № 1. P. 185.
Esen D.S., Temel G, Balta D.K., Allonas X., Arsu N. // Photochem. Photobiol. 2014. V. 90. № 2. P. 463.
Aydin M., Arsu N., Yagci Y. // Macromol. Rapid Commun. 2003. V. 24. № 12. P. 718.
Tar H., Sevinc Esen D., Aydin M., Ley C., Arsu N., Allonas X. // Macromolecules. 2013. V. 46. № 9. P. 3266.
Lougnot D.J., Turck C., Fouassier J.P. // Macromolecules. 1989. V. 22. № 1. P. 108.
Dogruyol S.K., Dogruyol Z., Arsu N. // J. Polym. Sci., Polym. Chem. 2011. V. 49. № 18. P. 4037.
Doğruyol S.K., Doğruyol Z., Arsu N. // J. Lumin. 2013. V. 138. P. 98.
Karaca N., Ocal N., Arsu N., Jockusch S. // J. Photochem. Photobiol. A. 2016. V. 331. P. 22.
Karaca N., Karaca Balta D., Ocal N., Arsu N. // J. Lumin. 2014. V. 146. P. 424.
Valandro S.R., Poli A.L., Venâncio T., Pina J., de Melo J.S., Burrows H.D. et al. // J. Photochem. Photobiol. A. 2016. V. 327. P. 15.
Yilmaz G., Acik G., Yagci Y. // Macromolecules. 2012. V. 45. № 5. P. 2219.
Akat H., Gacal B., Balta D.K., Arsu N., Yagci Y. // J. Polym. Sci., Polym. Chem. 2010. V. 48. № 10. P. 2109.
Dadashi-Silab S., Bildirir H., Dawson R., Thomas A., Yagci Y. // Macromolecules. 2014. V. 47. № 14. P. 4607.
Ziani-Cherif H., Abe Y., Imachi K., Matsuda T. // J. Biomed. Mater. Res. 2002. V. 59. № 2. P. 386.
Ullrich G., Herzog D., Liska R., Burtscher P., Moszner N. // J. Polym. Sci., Polym. Chem. 2004. V. 42. № 19. P. 4948.
Encinas M.V., Majmud C., Lissi E.A. // J. Polym. Sci., Polym. Chem. 1990. V. 28. № 9. P. 2465.
Zhuo J., Sun G. // Carbohydrate Polymers. 2014. V. 112. P. 158.
Crivello J.V., Sangermano M. // J. Polym. Sci., Polym. Chem. 2001. V. 39. № 3. P. 343.
Zhang J., Lalevée J., Zhao J., Graff B., Stenzel M.H., Xiao P. // Polym. Chem. 2016. V. 7. № 47. P. 7316.
Geuskens G., Etoc A., Di Michele P. // Eur. Polym. J. 2000. V. 36. № 2. P. 265.
Anwaruddin Q., Santappa M. // J. Polym. Sci., Polym. Lett. 1967. V. 5. № 5. P. 361.
Sautrot-Ba P., Jockusch S., Malval J.-P., Brezová V., Rivard M., Abbad-Andaloussi S., Blacha-Grzechnik A., Versace D.-L. // Macromolecules. 2020. V. 53. № 4. P. 1129.
Zhang J., Lalevée J., Hill N.S., Launay K., Morlet-Savary F., Graff B., Stenzel M.H., Coote M.L., Xiao P. // Macromolecules. 2018. V. 51. № 20. P. 8165.
Catalina F., Peinado C., Blanco M., Corrales T., Allen N.S. // Polymer. 2001. V. 42. № 5. P. 1825.
Breloy L., Brezová V., Blacha-Grzechnik A., Presset M., Yildirim M.S., Yilmaz I., Yagci Y., Versace D.-L. // Macromolecules. 2020. V. 53. № 1. P. 112.
Rahal M., Graff B., Toufaily J., Hamieh T., Noirbent G., Gigmes D., Dumur F., Lalevée J. // Molecules. 2021. V. 26. № 6. P. 1753.
Abdallah M., Hijazi A., Graff B., Fouassier J.-P., Rodeghiero G., Gualandi A., Dumur F., Cozzi P.G., Lalevée J. // Polym. Chem. 2019. V. 10. № 7. P. 872.
Abdallah M., Hijazi A., Lin J.-T., Graff B., Dumur F., Lalevée J. // ACS Appl. Polym. Mater. 2020. V. 2. № 7. P. 2769.
Xiao P., Wang Y., Dai M., Shi S., Wu G., Nie J. // Polym. Eng. Sci. 2008. V. 48. № 5. P. 884.
Zhu Q.Q., Schnabel W. // Polymer. 1996. V. 37. № 18. P. 4129.
Kamińska I., Ortyl J., Popielarz R. // Polym. Test. 2015. V. 42. P. 99.
Sangermano M., Razza N., Crivello J.V. // Macromol. Mater. Eng. 2014. V. 299. № 7. P. 775.
Crivello J.V. // J. Polym. Sci., Polym. Chem. 1999. V. 37. № 23. P. 4241.
Michaudel Q., Kottisch V., Fors B.P. // Angew. Chem. Int. Ed. 2017. V. 56. № 33. P. 9670.
Photochemistry and Photophysics of Polymer Materials / Ed. by N.S. Allen. New Jersey: Willey, 2010.
Yagci Y. // Macromol. Symp. 2004. V. 215. № 1. P. 267.
Yilmaz G., Beyazit S., Yagci Y. // J. Polym. Sci., Polym. Chem. 2011. V. 49. № 7. P. 1591.
Schroeder W.F., Asmussen S.V., Sangermano M., Vallo C.I. // Polym. Int. 2013. V. 62. № 9. P. 1368.
Shi S., Croutxé-Barghorn C., Allonas X. // Progr. Polym. Sci. 2017. V. 65. P. 1.
Sari E., Mitterbauer M., Liska R., Yagci Y. // Progr. Org. Coat. 2019. V. 132. P. 139.
Pappas S.P., Gatechair L.R., Jilek J.H. // J. Polym. Sci., Polym. Chem. Ed. 1984. V. 22. № 1. P. 77.
Yagci Y., Sangermano M., Rizza G. // Polymer. 2008. V. 49. № 24. P. 5195.
Golaz B., Michaud V., Leterrier Y., Månson J.A.E. // Polymer. 2012. V. 53. № 10. P. 2038.
Ortyl J., Topa M., Kamińska-Borek I., Popielarz R. // Eur. Polym. J. 2019. V. 116. P. 45.
Abdallah M., Hijazi A., Dumur F., Lalevée J. // Molecules. 2020. V. 25. № 9. P. 2063.
Zhang J., Wang S., Lalevée J., Morlet-Savary F., Lam E.S.-H., Graff B., Liu J., Xing F., Xiao P. // J. Polym. Sci. 2020. V. 58. № 6. P. 792.
Lalevée J., El-Roz M., Allonas X., Pierre Fouassier J. // J. Polym. Sci., Polym. Chem. 2008. V. 46. № 6. P. 2008.
Lalevée J., Tehfe M.-A., Morlet-Savary F., Graff B., Allonas X., Fouassier J.P. // Progr. Org. Coat. 2011. V. 70. № 1. P. 23.
Lalevée J., Dirani A., El-Roz M., Allonas X., Fouassier J.P. // J. Polym. Sci., Polym. Chem. 2008. V. 46. № 9. P. 3042.
Abdallah M., Hijazi A., Graff B., Fouassier J.-P., Dumur F., Lalevée J. // J. Photochem. Photobiol. A. 2020. V. 400. P. 112698.
Pakula T., Zhang Y., Matyjaszewski K., Lee H.-I., Boerner H., Qin S., Berry G.C. // Polymer. 2006. V. 47. № 20. P. 7198.
Johnson J.A., Lu Y.Y., Burts A.O., Xia Y., Durrell A.C., Tirrell D.A., Grubbs R.H. // Macromolecules. 2010. V. 43. № 24. P. 10326.
Liu X., Tian Z., Chen C., Allcock H.R. // Macromolecules. 2012. V. 45. № 3. P. 1417.
Heinrich C.D., Thelakkat M. // J. Mater. Chem. C. 2016. V. 4. № 23. P. 5370.
Hu Y., Li Y., Xu F.-J. // Acc. Chem. Res. 2017. V. 50. № 2. P. 281.
Feng C., Huang X. // Acc. Chem. Res. 2018. V. 51. № 9. P. 2314.
Singh B., Kumar A. // Carbohydr. Res. 2020. V. 489. P. 107949.
Conjugted Polymers for Biological and Biomedical Applications / Ed. by B. Liu. Weinheim: Wiley, 2018.
Zhang S., Tezuka Y., Zhang Z., Li N., Zhang W., Zhu X. // Polym. Chem. 2018. V. 9. № 6. P. 677.
Feng C., Li Y., Yang D., Hu J., Zhang X., Huang X. // Chem. Soc. Revs. 2011. V. 40. № 3. P. 1282.
Handbook of Polymer Synthesis, Characterization, and Processing / Ed. by E. Saldivar-Guerra, E. Vivaldo-Lima. New Jersey: Wiley, 2013.
Kyomoto M., Ishihara K. // ACS Appl. Mater. Interfaces. 2009. V. 1. № 3. P. 537.
Enright T.P., Hagaman D., Kokoruz M., Coleman N., Sidorenko A. // J. Polym. Sci., Polym. Phys. 2010. V. 48. № 14. P. 1616.
Bhattacharya A., Misra B. // Progr. Polym. Sci. 2004. V. 29. № 8. P. 767.
Ulbricht M., Yang H. // Chem. Mater. 2005. V. 17. № 10. P. 2622.
Korolkov I.V., Gorin Y.G., Yeszhanov A.B., Kozlovskiy A.L., Zdorovets M.V. // Mater. Chem. Phys. 2018. V. 205. P. 55.
Zahner D., Abagat J., Svec F., Fréchet J.M.J., Levkin P.A. // Adv. Mater. 2011. V. 23. № 27. P. 3030.
Henze M., Mädge D., Prucker O., Rühe J. // Macromolecules. 2014. V. 47. № 9. P. 2929.
Chu E., Sidorenko A. // Langmuir. 2013. V. 29. № 40. P. 12585.
Datta P., Genzer J. // J. Polym. Sci., Polym. Chem. 2016. V. 54. № 2. P. 263.
Yang W., Rånby B. // Eur. Polym. J. 1999. V. 35. № 8. P. 1557.
Rånby B. // Polym. Eng. Sci. 1998. V. 38. № 8. P. 1229.
Deng J., Wang L., Liu L., Yang W. // Progr. Polym. Sci. 2009. V. 34. № 2. P. 156.
Kato K., Uchida E., Kang E.-T., Uyama Y., Ikada Y. // Progr. Polym. Sci. 2003. V. 28. № 2. P. 209.
Zhou T., Zhu Y., Li X., Liu X., Yeung K.W., Wu S., Wang X., Cui Z., Yang X., Chud P.K. // Progr. Mater. Sci. 2016. V. 83. P. 191.
Currivan S., Jandera P. // Chromatography. 2014. V. 1. № 1. P. 24.
Ma H., Davis R.H., Bowman C.N. // Macromolecules. 2000. V. 33. № 2. P. 331.
Stachowiak T.B., Svec F., Fréchet J.M.J. // Chem. Mater. 2006. V. 18. № 25. P. 5950.
Yang W., Rånby B. // Macromolecules. 1996. V. 29. № 9. P. 3308.
Rånby B. // Int. J. Adhes. Adhes. 1999. V. 19. № 5. P. 337.
Yang W.T., Rånby B. // J. Appl. Polym. Sci. 1997. V. 63. № 13. P. 1723.
Deng J.P., Yang W.T., Rånby B. // J. Appl. Polym. Sci. 2000. V. 77. № 7. P. 1522.
Ma H., Davis R.H., Bowman C.N. // Polymer. 2001. V. 42. № 20. P. 8333.
Geyer F.L., Ueda E., Liebel U., Grau N., Levkin P.A. // Angew. Chem. Int. Ed. 2011. V. 50. № 36. P. 8424.
Ulbricht M., Oechel A., Lehmann C., Tomaschewski G., Hicke H.-G. // J. Appl. Polym. Sci. 1995. V. 55. № 13. P. 1707.
Ulbricht M. // React. Funct. Polym. 1996. V. 31. № 2. P. 165.
Kyomoto M., Moro T., Takatori Y., Kawaguchi H., Nakamura K., Ishihara K. // Biomaterials. 2010. V. 31. № 6. P. 1017.
Almasi D., Iqbal N., Sadeghi M., Sudin I., Abdul Kadir M.R., Kamarul T. // Int. J. Biomater. 2016. V. 2016. P. 8202653.
Mir A.H., Charoo M.S. // Mater. Sci. Eng. 2019. V. 561. P. 012051.
Kyomoto M., Moro T., Yamane S., Hashimoto M., Takatori Y., Ishihara K. // Biomaterials. 2013. V. 34. № 32. P. 7829.
Kawasaki Y., Iwasaki Y. // J. Biomater. Sci. Polym. Ed. 2014. V. 25. № 9. P. 895.
Zheng Y., Liu L., Xiao L., Zhang Q., Liu Y. // Colloids Surf. B. 2019. V. 173. P. 591.
Zheng Y., Liu L., Ma Y., Xiao L., Liu Y. // Ind. Eng. Chem. Res. 2018. V. 57. № 31. P. 10403.
Iwasaki Y., Ishihara K. // Sci. Technol. Adv. Mater. 2012. V. 13. № 6. P. 064101.
Ishihara K. // J. Biomed. Mater. Res. A. 2019. V. 107. № 5. P. 933.
Abe Y., Ishii K., Isoyama T., Saito I., Inoue Y., Sato M., Hara S., Hosoda K., Ariyoshi K., Nakagawa H., Ono T., Fukazawa K., Ishihara K., Imachiet K. // 35 Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC)”. Osaka, Japan, 2013.
Ham J., Kim Y., An T., Kang S., Ha C., Wufue M., Kim Y., Jeon B., Kim S., Kim J., Choi T.H., Seo J.-H., Kim D.W., Park J.-U., Lee Y. // ACS Appl. Mater. Interfaces. 2020. V. 2. № 27. P. 30198.
Tateishi T., Kyomoto M., Kakinoki S., Yamaoka T., Ishihara K. // J. Biomed. Mater. Res. A. 2014. V. 102. № 5. P. 1342.
Chouwatat P., Hirai T., Higaki K., Higaki Y., Sue H.-J., Takahara A. // Polymer. 2017. V. 116. P. 549.
Liqun Z., Irwan G.S., Kondo T., Kubota H. // Eur. Polym. J. 2000. V. 36. № 8. P. 1591.
Salmi-Mani H., Terreros G., Barroca-Aubry N., Aymes-Chodur C., Regeard C., Roger P. // Eur. Polym. Jl. 2018. V. 103. P. 51.
Davis N.P., Garnett J.L., Urquhart R. // J. Polym. Sci., Polym. Lett. Ed. 1976. V. 14. P. 537.
Geacintov N., Stannett V., Abrahamson E.W. // Makromol. Chem. 1960. V. 36. № 1. P. 52.
Zhang J., Nie J., Zhu X. // Macromol. Rapid Commun. 2018. V. 39. № 20. P. 1800144.
Zhu X., He B., Zhao C., Ma Y., Yang W. // Langmuir. 2017. V. 33. № 22. P. 5577.
Lenshina N.A., Arsenyev M.V., Baten’kin M.A., Polushtaytsev Y.V., Chesnokov S.A. // Polym. Eng. Sci. 2020. V. 60. № 4. P. 682.
Lenshina N.A., Arsenyev M.V., Baten’kin M.A., Chesnokov S.A. // Russ. Chem. Bull. 2019. V. 68. № 9. P. 1741.
Tao T., Scheiner C.J., Lamkin M. // Biochemistry. 1986. V. 25. № 23. P. 7633.
Williams N., Ackerman S.H., Coleman P.S. // Meth. Enzymol. 1986. V. 126. P. 667.
Ross G.F., Smith P.M., McGregor A., Turnbull D.M., Lightowlers R.N. // Bioconjug. Chem. 2003. V. 14. № 5. P. 962.
Bechtella L., Kirschbaum C., Cosset M., Clodic G., Matheron L., Bolbach G., Sagan S., Walrant A., Sachon E. // Anal. Chem. 2019. V. 91. № 14. P. 9102.
Schenderlein H., Voss A., Stark R.W., Biesalski M. // Langmuir. 2013. V. 29. № 14. P 4525.
Pérez-Perrino M., Navarro R., Prucker O., Rühe J. // Macromolecules. 2014. V. 47. № 8. P. 2695.
Pahnke J., Rühe J. // Macromol. Rapid Commun. 2004. V. 25. № 15. P. 1396.
Kim S.-H., Ohtsuka H., Tria M.C.R., Tanaka K., Advincula R.C., Usui H. // Thin Solid Films. 2014. V. 554. P. 78.
Prucker O., Naumann C.A., Rühe J., Knoll W., Frank C.W. // J. Am. Chem. Soc. 1999. V. 121. № 38. P. 8766.
Murata H., Chang B.J., Prucker O., Dahm M., Rühe J. // Surf. Sci. 2004. V. 570. I. 1. P. 111.
Schuler A.-K., Prucker O., Rühe J. // Macromol. Chem. Phys. 2016. V. 217. № 13. P. 1457.
Toomey R., Freidank D., Rühe J. // Macromolecules. 2004. V. 37. № 3. P. 882.
Czech Z., Kowalczyk A., Kabatc J., Świderska J. // Eur. Polym. J. 2012. V. 48. № 8. P. 1446.
Prucker O., Brandstetter T., Rühe J. // Biointerphases. 2018. V. 13. № 1. P. 010801.
Gao J., Huddleston N.E., White E.M., Pant J., Handa H., Locklin J. // ACS Biomat. Sci. Eng. 2016. V. 2. № 7. P. 1169.
Porosa L., Caschera A., Bedard J., Mocella A., Ronan E., Lough A.J., Wolfaardt G., Foucher D.A. // ACS Appl. Mater. Interfaces. 2017. V. 9. № 33. P. 27491.
Kecici Z., Babaoglu S., Temel G. // Prog. Org. Coat. 2018. V. 115. P. 138.
Christensen S.K., Chiappelli M.C., Hayward R.C. // Macromolecules. 2012. V. 45. № 12. P. 5237.
Li Y., Vergaelen M., Schoolaert E., Hoogenboom R., De Clerck K. // Eur. Polym. J. 2019. V. 112. P. 24.
Rendl M., Bönisch A., Mader A., Schuh K., Prucker O., Brandstetter T., Rühe J. // Langmuir. 2011. V. 27. № 10. P. 6116.
Moschallski M., Evers A., Brandstetter T., Rühe J. // Anal. Chim. Acta. 2013. V. 781. P. 72.
Pidhatika B., Zhao N., Zinggeler M., Rühe J. // J. Polym. Res. 2019. V. 26. № 3. P. 69.
Lim S.M., Lee M.S., Sohn E.-H., Lee S.-G., Park I.J., Kang H.S. // Sci. Rep. 2020. V. 10. № 1. P. 15121.
Liu W., Dong Y., Zhang S., Wu Z., Chen H. // ChemComm. 2019. V. 55. № 6. P. 858.
Mukherjee S., Xie R., Reynolds V.G., Uchiyama T., Levi A.E., Valois E., Wang H., Chabinyc M.L., Bates C.M. // Macromolecules. 2020. V. 53. № 3. P. 1090.
Cheng C., Bai X., Zhang X., Li H., Huang Q., Tu Y. // J. Polym. Res. 2015. V. 22. № 4. P. 46.
Taskin O.S., Temel B.A., Tasdelen M.A., Yagci Y. // Eur. Polym. J. 2015. V. 62. P. 304.
Versace D.-L., Ramier J., Babinot J., Lemechko P., Soppera O., Lalevee J., Albanese P., Renard E., Langlois V. // J. Mater. Chem. B. 2013. V. 1. № 37. P. 4834.
Zunker S., Rühe J. // Macromolecules. 2020. V. 53. № 5. P. 1752.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)